Photo Protection of Haematococcus pluvialis Algae by Astaxanthin: Unique Properties of Astaxanthin Deduced by EPR, Optical and Electrochemical Studies



Abstract
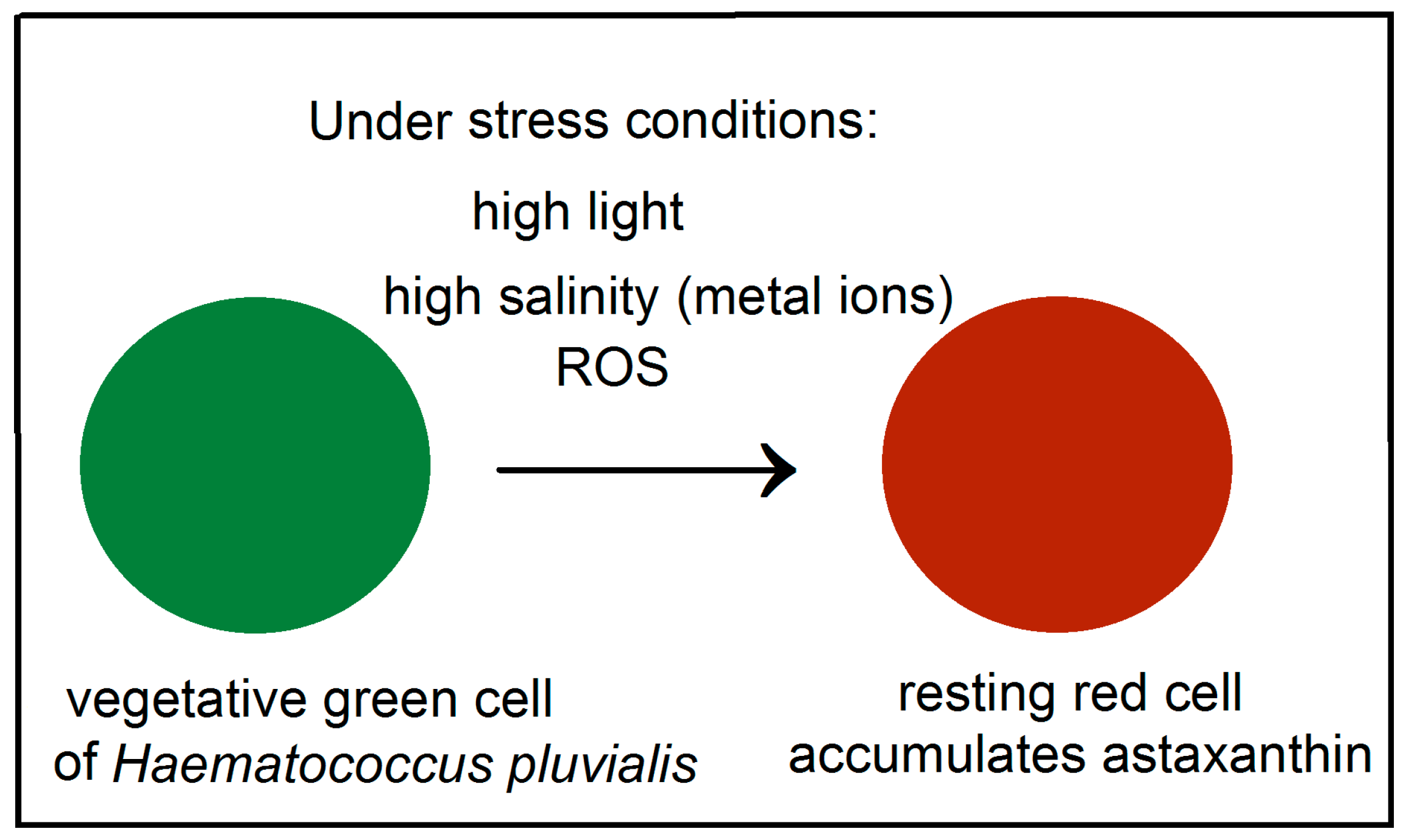
:1. Introduction
2. Unique Properties of Astaxanthin
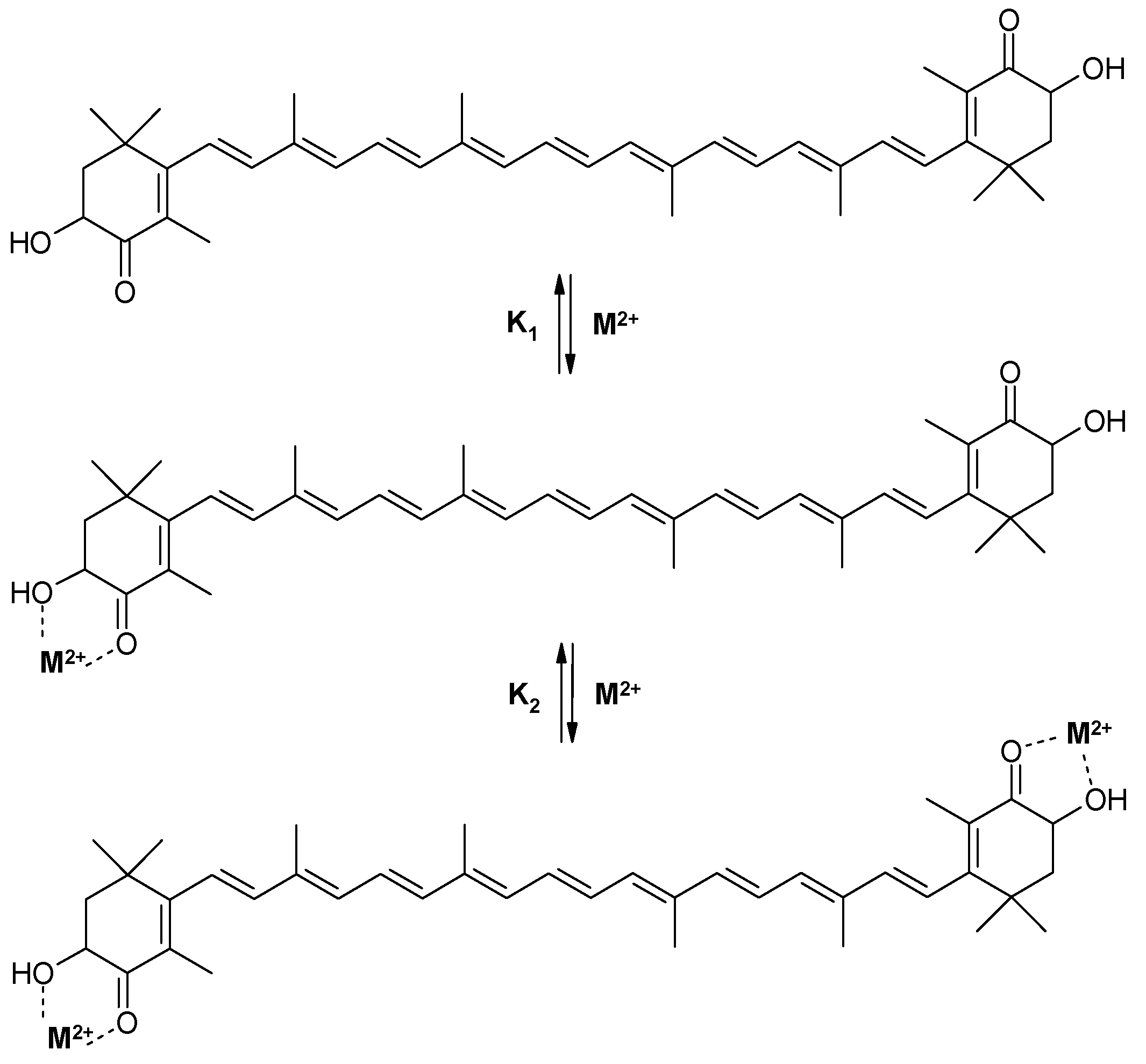
2.1. The Ability of Astaxanthin to Form Chelate Complexes with Metals
2.2. The Ability of Astaxanthin to Esterify and Inability to Aggregate in the Ester Form
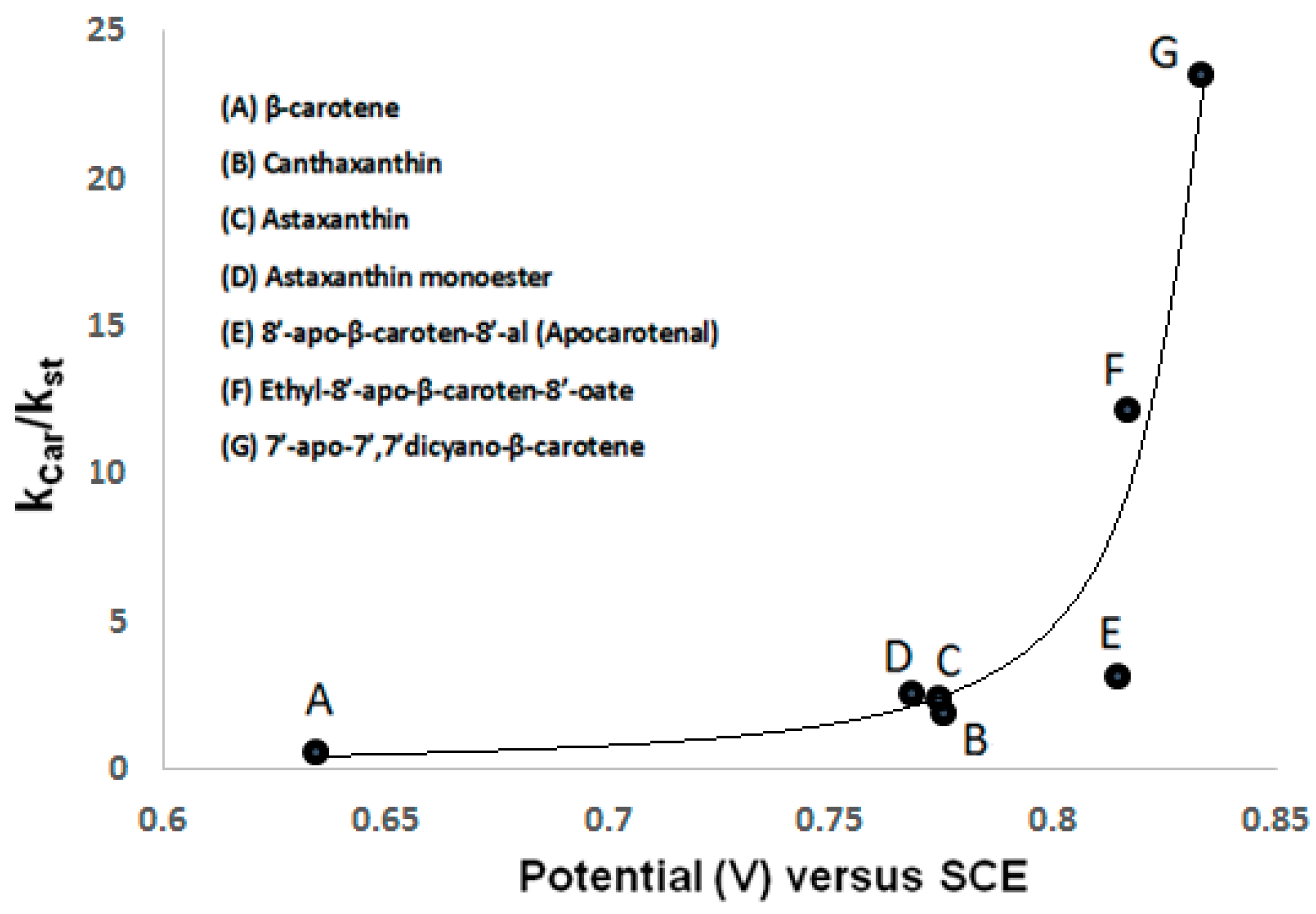
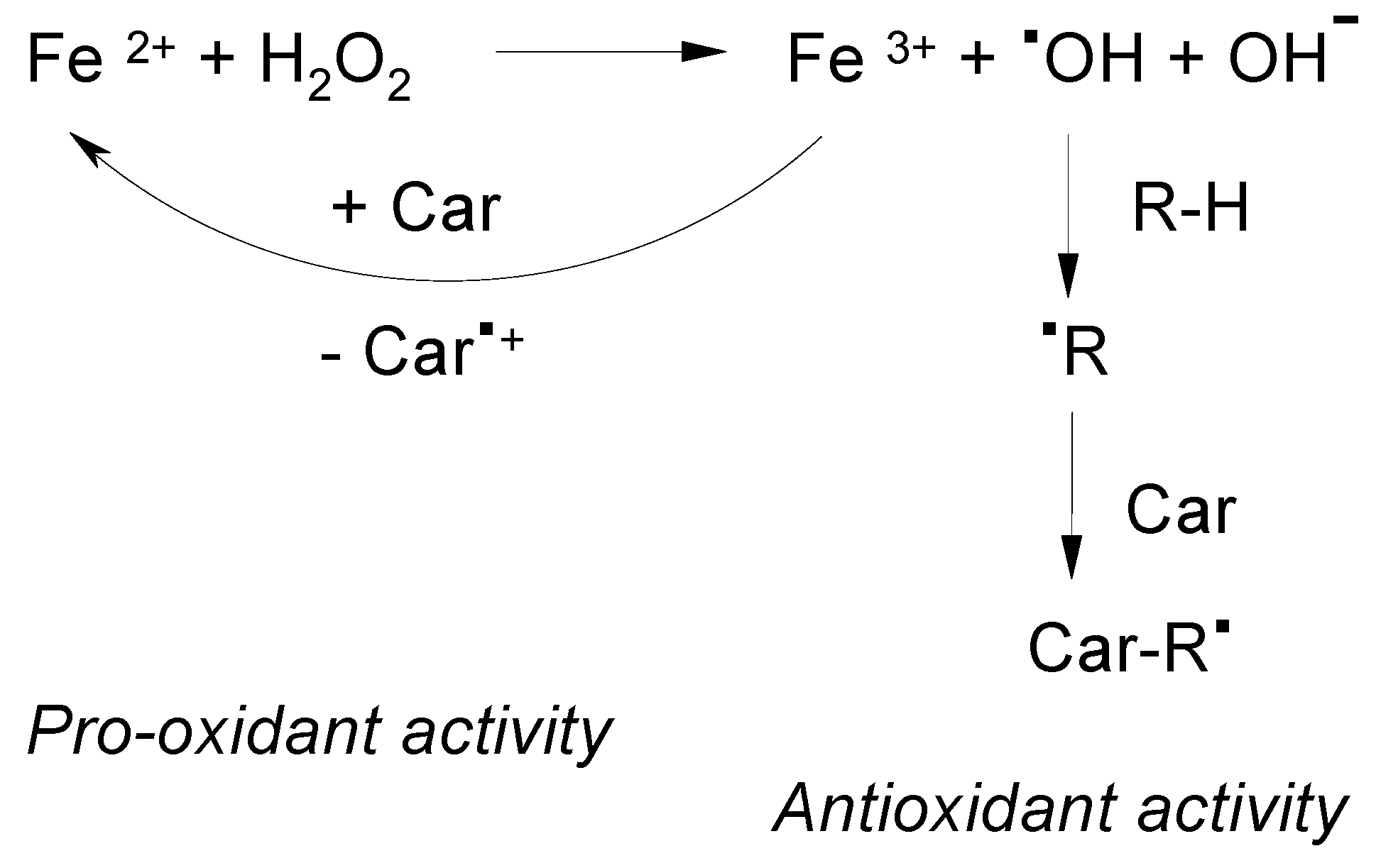
2.3. ROS Scavenging Ability of Astaxanthin. The High Oxidation Potential
- ▪
- Radical addition to the polyene chainR• + Car → (Car–R)•
- ▪
- Hydrogen Abstraction from the CarR• + Car → (Car–H)• + RH
- ▪
- Electron Transfer ReactionR• + Car → R− + Car•+
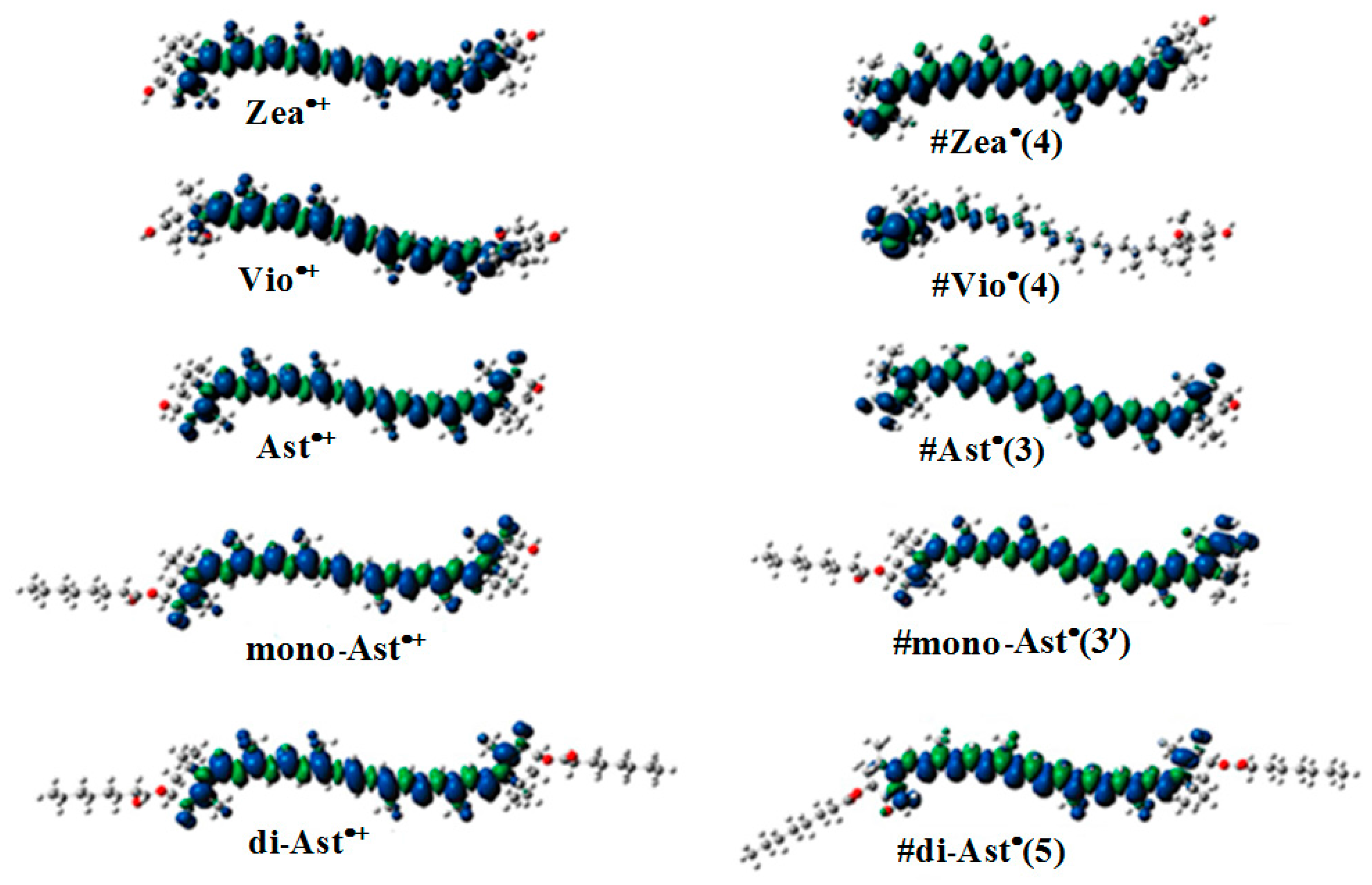
2.4. The Ability of Carotenoids to Form Proton Loss Neutral Radicals under High Illumination
2.4.1. In Arabidopsis thaliana Plant
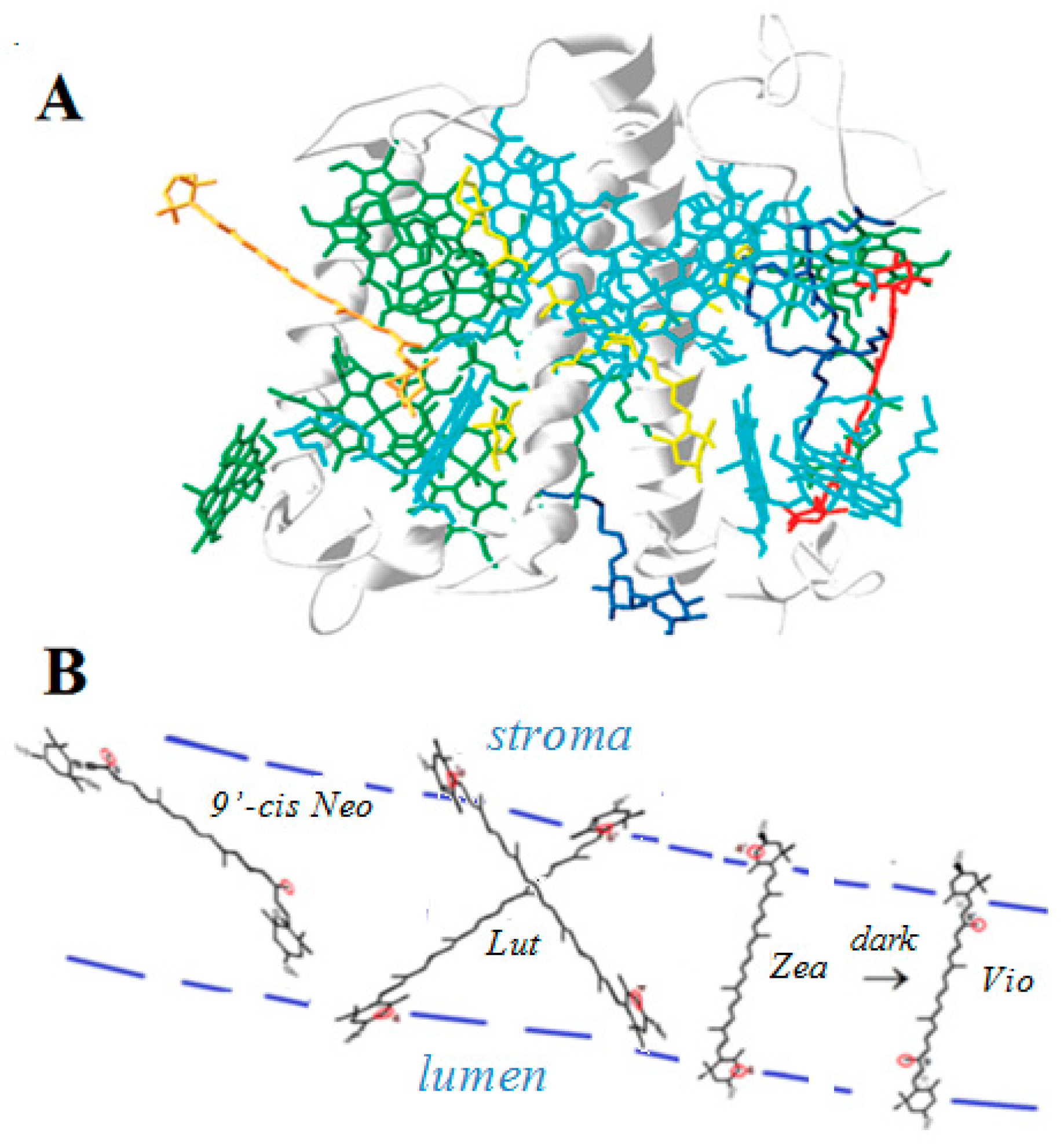
2.4.2. In Light Harvesting Complex II (LHCII)
2.4.3. In Solid Supports: Silica-Alumina, Silica Gel, MCM-41, Metal-Substituted MCM-41 and TiO2
Mechanism of Proton Loss Neutral Radical Formation in an Irradiated TiO2 Matrix. Sources of O
How do Proton Loss Neutral Radicals Form in a Siliceous MCM-41 Matrix Containing a Metal-Oxygen (M-O) Bond (M=Al, Fe, Ti)?
3. Discussion
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Yu, X.; Chen, L.; Zhang, W. Chemicals to enhance microalgal growth and accumulation of high-value bioproducts. Front. Microbiol. 2015, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Harker, M.; Tsavalos, A.J.; Young, A.J. Factors responsible for astaxanthin formation in the chlorophyte Haematococcus pluvialis. Bioresour. Technol. 1996, 55, 207–214. [Google Scholar] [CrossRef]
- Shah, M.M.R.; Liang, Y.; Cheng, J.J.; Daroch, M. Astaxanthin-producing green microalga Haematococcus pluvialis: From single cell to high value commercial products. Front. Plant Sci. 2016, 7, 531. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, T. A Technical Review of Haematococcus Algae, Cyanotech Corporation. Available online: http://www.cyanotech.com/pdfs/bioastin/axbul60.pdf (accessed on 19 October 2017).
- Zhang, L.; Su, F.; Zhang, C.; Liu, J. Changes of photosynthetic behaviors and photoprotection during cell transformation and astaxanthin accumulation in Haematococcus pluvialis grown outdoors in tubular photobioreactors. Int. J. Mol. Sci. 2016, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Zaks, J.; Amarnath, K.; Sylak-Glassman, E.J.; Fleming, G.R. Models and measurements of energy-dependent quenching. Photosynth. Res. 2013, 116, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, X.; Sun, Y.; Lin, W. Antioxidative capacity and enzyme activity in Haematococcus pluvialis cells exposed to superoxide free radicals. Chin. J. Oceanol. Limnol. 2010, 28, 1–9. [Google Scholar] [CrossRef]
- Kobayashi, M. Astaxanthin biosynthesis enhanced by reactive oxygen species in the green alga Haematococcus pluvialis. Nature 2003, 8, 322. [Google Scholar]
- Timoshnikov, V.A.; Kobzeva, T.V.; Polyakov, N.E.; Kontoghiorghes, G.J. Inhibition of Fe2+- and Fe3+- induced hydroxyl radical production by the iron-chelating drug deferiprone. Free Radic. Biol. Med. 2015, 78, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Timoshnikov, V.A.; Klimentiev, V.K.; Polyakov, N.E.; Kontoghiorghes, G.J. Photoinduced transformation of iron chelator deferiprone: Possible implications in drug metabolism and toxicity. J. Photochem. Photobiol. A Chem. 2014, 289, 14–21. [Google Scholar] [CrossRef]
- Hernandez-Marin, E.; Barbosa, A.; Martinez, A. The metal cation chelating capacity of astaxanthin. Does this have any influence on antiradical activity? Molecules 2012, 17, 1039–1054. [Google Scholar] [CrossRef] [PubMed]
- Polyakov, N.P.; Focsan, A.L.; Bowman, M.K.; Kispert, L.D. Free radical formation in novel carotenoid metal ion complexes of astaxanthin. J. Phys. Chem. B 2010, 114, 16968–16977. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tauber, M.J. High-yield singlet fission in a zeaxanthin aggregate observed by picosecond resonance Raman spectroscopy. J. Am. Chem. Soc. 2010, 132, 13988–13991. [Google Scholar] [CrossRef] [PubMed]
- Polyakov, N.E.; Magyar, A.; Kispert, L.D. Photochemical and optical properties of water-soluble xanthophyll antioxidants: Aggregation vs complexation. J. Phys. Chem. B 2013, 117, 10173–10182. [Google Scholar] [CrossRef] [PubMed]
- Young, A.J.; Lowe, G.M. Antioxidant and prooxidant properties of carotenoids. Arch. Biochem. Biophys. 2001, 385, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Gao, Y.; Kispert, L.D. Electrochemical properties of natural carotenoids. J. Electroanal. Chem. 2000, 488, 140–150. [Google Scholar] [CrossRef]
- Liu, D.; Kispert, L.D. Electrochemical aspects of carotenoids. Recent Res. Dev. Electrochem. 1999, 2, 139–157. [Google Scholar]
- Focsan, A.L.; Pan, S.; Kispert, L.D. Electrochemical study of astaxanthin and astaxanthin n-octanoic monoester and diester: Tendency to form radicals. J. Phys. Chem. B 2014, 118, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Hapiot, P.; Kispert, L.D.; Konovalov, V.V.; Savéant, J.-M. Single two-electron transfers vs successive one-electron transfers in polyconjugated systems illustrated by the electrochemical oxidation and reduction of carotenoids. J. Am. Chem. Soc. 2001, 123, 6669–6677. [Google Scholar] [CrossRef] [PubMed]
- Polyakov, N.E.; Kruppa, A.I.; Leshina, T.V.; Konovalova, T.A.; Kispert, L.D. Carotenoids as antioxidants: Spin trapping EPR and optical study. Free Radic. Biol. Med. 2001, 1, 43–52. [Google Scholar] [CrossRef]
- Polyakov, N.E.; Leshina, T.V.; Konovalova, T.A.; Kispert, L.D. Carotenoids as scavengers of free radicals in a Fenton reaction: Antioxidants or pro-oxidants? Free Radic. Biol. Med. 2001, 31, 398–404. [Google Scholar] [CrossRef]
- Enami, S.; Sakamoto, Y.; Colussi, A.J. Fenton chemistry at aqueous interfaces. Proc. Natl. Acad. Sci. USA 2014, 111, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.L.; Shinopoulos, K.E.; Tracewell, C.A.; Focsan, A.L.; Brudvig, G.W.; Kispert, L.D. Formation of carotenoid neutral radicals in photosystem II. J. Phys. Chem. B 2009, 113, 9901–9908. [Google Scholar] [CrossRef] [PubMed]
- Magyar, A.; Bowman, M.K.; Molnár, P.; Kispert, L. Neutral carotenoid radicals in photoprotection of wild-type Arabidopsis thaliana. J. Phys. Chem. B 2013, 117, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Focsan, A.L.; Magyar, A.; Kispert, L. Chemistry of carotenoid neutral radicals. Arch. Biochem. Biophys. 2015, 572, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Focsan, A.L.; Kispert, L.D. Radicals formed from proton loss of carotenoid radical cations: A special form of carotenoid neutral radical occurring in photoprotection. J. Photochem. Photobiol. B 2017, 166, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Hideg, E.; Barta, C.; Kalai, T.; Vass, I.; Hideg, K.; Asada, K. Detection of singlet oxygen and superoxide with fluorescent sensors in leaves under stress by photoinhibition or UV radiation. Plant Cell Physiol. 2002, 43, 1154–1164. [Google Scholar] [CrossRef]
- Holt, N.E.; Zigmantas, D.; Valkunas, L.; Li, X.-P.; Niyogi, K.K.; Fleming, G.R. Carotenoid cation formation and the regulation of photosynthetic light harvesting. Science 2005, 307, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Ahn, T.K.; Avenson, T.J.; Ballottari, M.; Cheng, Y.C.; Niyogi, K.K.; Bassi, R.; Fleming, G.R. Architecture of a charge-transfer state regulating light harvesting in a plant antenna protein. Science 2008, 320, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Avenson, T.J.; Ahn, T.K.; Zigmantas, D.; Niyogi, K.K.; Li, Z.; Ballottari, M.; Bassi, R.; Fleming, G.R. Zeaxanthin radical cation formation in minor light-harvesting complexes of higher plant antenna. J. Biol. Chem. 2008, 283, 3550–3558. [Google Scholar] [CrossRef] [PubMed]
- Amarie, S.; Wilk, L.; Barros, T.; Kuhlbrandt, W.; Drew, A.; Wachtveitl, J. Properties of zeaxanthin and its radical cation bound to the minor light-harvesting complexes CP24, CP26 and CP29. Biochem. Biophys. Acta. 2009, 1787, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Standfuss, J.; Scheltinga, A.C.T.; Lamborghini, M.; Kuhlbrandt, W. Mechanisms of photoprotection and nonphotochemical quenching in pea light-harvesting complex at 2.5 Å resolution. EMBO J. 2005, 24, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Barbatti, M.; Aquino, A.J.A.; Lischka, H.; Schriever, C.; Lochbrunner, S.; Riedle, E. Ultrafast internal conversion pathway and mechanism in 2-(2’-hydroxyphenyl) benzothiazole: A case study for excited-state intramolecular proton transfer systems. Phys. Chem. Chem. Phys. 2009, 11, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Formosinho, S.J.; Arnaut, L.G. Excited-state proton-transfer reactions II. Intramolecular reactions. J. Photochem. Photobiol. A 1993, 75, 21–48. [Google Scholar] [CrossRef]
- Kim, C.H.; Park, J.; Seo, J.; Park, S.Y.; Joo, T. Excited state intramolecular proton transfer and charge transfer dynamics of a 2-(2’-hydroxyphenyl) benzoxazole derivative in solution. J. Phys. Chem. A 2010, 114, 5618–5629. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Joo, T. Coherent excited state intramolecular proton transfer probed by time-resolved fluorescence. Phys. Chem. Chem. Phys. 2009, 11, 10266–10269. [Google Scholar] [CrossRef] [PubMed]
- Hasjim, P.L.; Lendzian, F.; Ponomarenko, N.; Norris, J.R. Basic molecular unit involved in charge migration in oxidized light-harvesting complex I of Rhodobacter sphaeroides. J. Phys. Chem. Lett. 2010, 1, 1687–1689. [Google Scholar] [CrossRef]
- Srivatsan, N.; Kolbasov, D.; Ponomarenko, N.; Weber, S.; Ostafin, A.E.; Norris, J.R. Cryogenic charge transport in oxidized purple bacterial light-harvesting I complexes. J. Phys. Chem. B 2003, 107, 7867–7876. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, N.; Weber, S.; Kolbasov, D.; Norris, J.R. Exploring charge migration in light-harvesting complexes using electron paramagnetic resonance line narrowing. J. Phys. Chem. B 2003, 107, 2127–2138. [Google Scholar] [CrossRef]
- Focsan, A.L.; Bowman, M.K.; Shamshina, J.; Krzyaniak, M.D.; Magyar, A.; Polyakov, N.E.; Kispert, L.D. EPR study of the astaxanthin n-octanoic acid monoester and diester radicals on silica-alumina. J. Phys. Chem. B 2012, 116, 13200–13210. [Google Scholar] [CrossRef] [PubMed]
- Focsan, A.L.; Molnar, P.; Deli, J.; Kispert, L. Structure and properties of 9’-cis-neoxanthin carotenoid radicals by electron paramagnetic resonance measurements and density functional theory calculations: Present in LHC II? J. Phys. Chem. B 2009, 113, 6087–6096. [Google Scholar] [CrossRef] [PubMed]
- Focsan, A.L.; Bowman, M.K.; Konovalova, T.A.; Molnar, P.; Deli, J.; Dixon, D.A.; Kispert, L.D. Pulsed EPR and DFT characterization of radicals produced by photo-oxidation of zeaxanthin and violaxanthin on silica-alumina. J. Phys. Chem. B 2008, 112, 1806–1819. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.; Focsan, A.L.; Konovalova, T.A.; Molnar, P.; Deli, J.; Bowman, M.K.; Kispert, L.D. Pulsed electron nuclear double resonance studies of carotenoid oxidation in Cu(II)-substituted MCM-41 molecular sieves. J. Phys. Chem. B 2008, 112, 5449–5457. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Konovalova, T.A.; Lawrence, J.N.; Smitha, M.A.; Nunley, J.; Schad, R.; Kispert, L.D. Interaction of carotenoids and Cu2+ in Cu-MCM-41: Distance-dependent reversible electron transfer. J. Phys. Chem. B 2003, 107, 2459–2465. [Google Scholar] [CrossRef]
- Konovalova, T.K.; Gao, Y.; Schad, R.; Kispert, L.D.; Saylor, C.A.; Brunel, L.-C. Photooxidation of carotenoids in mesoporous MCM-41, Ni-MCM-41 and Al-MCM-41 molecular sieves. J. Phys. Chem. B 2001, 105, 7459–7464. [Google Scholar] [CrossRef]
- Konovalova, T.K.; Gao, Y.; Kispert, L.D. Characterization of Fe-MCM-41 molecular sieves with incorporated carotenoids by multifrequency electron paramagnetic resonance. J. Phys. Chem. B 2003, 107, 1006–1011. [Google Scholar] [CrossRef]
- Augugliaro, V.; Bellardita, M.; Loddo, V.; Palmisano, G.; Palmisano, L.; Yurdakal, S. Overview on oxidation mechanisms of organic compounds by TiO2 in heterogeneous photocatalysis. J. Photochem. Photobiol. C 2012, 13, 224–245. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Chen, C.; Ji, H.; Che, Y.; Ma, W.; Zhao, J. Unraveling the photocatalytic mechanisms on TiO2 surfaces using the Oxygen-18 isotopic label technique. Molecules 2014, 19, 16291–16311. [Google Scholar] [CrossRef] [PubMed]
- Dasari, T.P.; Pathakoti, K.; Hwang, H.-M. Determination of the mechanism of photoinduced toxicity of selected metal oxide nanoparticles (ZnO, CuO, Co3O4 and TiO2) to E. coli bacteria. J. Environ. Sci. 2013, 25, 882–888. [Google Scholar] [CrossRef]
- Howe, R.F.; Grätzel, M. EPR observation of trapped electrons in colloidal titanium dioxide. J. Phys. Chem. 1985, 89, 4495–4499. [Google Scholar] [CrossRef]
- Konovalova, T.A.; Lawrence, J.; Kispert, L.D. Generation of superoxide anion and most likely singlet oxygen in irradiated TiO2 nanoparticles modified by carotenoids. J. Photochem. Photobiol. A 2004, 162, 1–8. [Google Scholar] [CrossRef]
- King, B.V.; Freund, F. Surface charges and subsurface space-charge distribution in magnesium oxides containing dissolved traces of water. Phys. Rev. B 1984, 29, 5814–5824. [Google Scholar] [CrossRef]
- Khan, A.U. Theory of electron transfer generation and quenching of singlet oxygen [1. SIGMA. g+ and 1. DELTA. g] by superoxide anion. The role of water in the dismutation of superoxide anion. J. Am. Chem. Soc. 1977, 99, 370–371. [Google Scholar] [CrossRef]
- Zhang, R.; Goldstein, S.; Samuni, A. Kinetics of superoxide-induced exchange among nitroxide antioxidants and their oxidized and reduced forms. Free Radic. Biol. Med. 1999, 26, 1245–1252. [Google Scholar] [CrossRef]
- Roberts, J.R.; Ingold, K.U. Kinetic applications or electron paramagnetic resonance spectroscopy. X. Reactions of some alkylamino radicals in solution. J. Am. Chem. Soc. 1973, 95, 3228–3255. [Google Scholar]
- Kormann, C.; Bahnemann, D.W.; Hoffmann, M.R. Preparation and characterization of quantum-size titanium dioxide. J. Phys. Chem. 1988, 92, 5196–5201. [Google Scholar] [CrossRef]
- Harbour, J.R.; Tromp, J.; Hair, M.L. Photogeneration of hydrogen peroxide in aqueous TiO2 dispersions. Can. J. Chem. 1985, 63, 204–208. [Google Scholar] [CrossRef]
- Wu, T.; Liu, G.; Zhao, J.; Hidaka, H.; Serpone, N. Evidence for H2O2 generation during the TiO2-assisted photodegradation of dyes in aqueous dispersions under visible light illumination. J. Phys. Chem. B 1999, 103, 4862–4867. [Google Scholar]
- Gao, Y.; Konovalova, T.A.; Xu, T.; Kispert, L.D. Electron transfer of carotenoids imbedded in MCM-41 and Ti-MCM-41: EPR, ENDOR, and UV-Vis studies. J. Phys. Chem. B 2002, 106, 10808–10815. [Google Scholar] [CrossRef]
- Chang, Z.; Zhu, Z.; Kevan, L. Electron spin resonance of Ni(I) in Ni-containing MCM-41 molecular sieves. J. Phys. Chem. B 1999, 103, 9442–9449. [Google Scholar] [CrossRef]
- Bal, R.; Chaudhari, K.; Srinivas, D.; Sivasanker, S.; Ratnasamy, P. Redox and catalytic chemistry of Ti in titanosilicate molecular sieves: An EPR investigation. J. Mol. Catal. 2000, 162, 199–207. [Google Scholar] [CrossRef]
- Fernando, Q. Reactions of Chelated Organic Ligands. In Advances in Inorganic Chemistry and Radiochemistry; Emeléus, H.J., Sharpe, A.G., Eds.; Academic Press: London, UK, 1965; Volume 7. [Google Scholar]
- Mujika, J.I.; Ugalde, J.M.; Lopez, X. Aluminum speciation in biological environments. The deprotonation of free and aluminum bound citrate in aqueous solution. Phys. Chem. Chem. Phys. 2012, 14, 12465–12475. [Google Scholar] [CrossRef] [PubMed]
- Coin, J.T.; Olson, J. The rate of oxygen uptake by human red blood cells. J. Biol. Chem. 1979, 254, 1178–11190. [Google Scholar] [PubMed]
- Polyakov, N.E.; Leshina, T.V.; Meteleva, E.S.; Dushkin, A.V.; Konovalova, T.A.; Kispert, L.D. Enhancement of the photocatalytic activity of TiO2 nanoparticles by water-soluble complexes of carotenoids. J. Phys. Chem. B 2010, 114, 14200–14204. [Google Scholar] [CrossRef] [PubMed]
- Kispert, L.D.; Polyakov, N.E. Carotenoid radicals: Cryptochemistry of natural colorants. Chem. Lett. 2010, 39, 148–155. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Salt | Ca(ClO4)2 | Zn(ClO4)2 | Fe(ClO4)2 |
|---|---|---|---|
| Stability constants | K1 = 23,000 M−1 K2 = 5000 M−1 | K1 = 3000 M−1 K2 = 1300 M−1 | K1 = 3000 M−1 |
| Carotenoid | vs. SCE | Kcar/Kst [20] |
|---|---|---|
| β-carotene [19] | 0.634 ± 0.001 | 0.64 |
| canthaxanthin [19] | 0.775 ± 0.001 | 1.96 |
| astaxanthin [18] | 0.768 ± 0.001 | 2.60 |
| astaxanthin monoester [18] | 0.774 ± 0.001 | 2.50 |
| astaxanthin diester [18] | 0.775 ± 0.001 | not measured |
| 8′-apo-β-carotene-8′-al [16] | 0.814 ± 0.005 | 3.22 |
| ethyl-8′-apo-β-caroten-8′-oate [16] | 0.816 ± 0.005 | 12.20 |
| 7′-apo-7′,7′-dicyano-β-carotene [16] | 0.833 ± 0.005 | 23.60 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Focsan, A.L.; Polyakov, N.E.; Kispert, L.D. Photo Protection of Haematococcus pluvialis Algae by Astaxanthin: Unique Properties of Astaxanthin Deduced by EPR, Optical and Electrochemical Studies. Antioxidants 2017, 6, 80. https://doi.org/10.3390/antiox6040080
Focsan AL, Polyakov NE, Kispert LD. Photo Protection of Haematococcus pluvialis Algae by Astaxanthin: Unique Properties of Astaxanthin Deduced by EPR, Optical and Electrochemical Studies. Antioxidants. 2017; 6(4):80. https://doi.org/10.3390/antiox6040080
Chicago/Turabian StyleFocsan, A. Ligia, Nikolay E. Polyakov, and Lowell D. Kispert. 2017. "Photo Protection of Haematococcus pluvialis Algae by Astaxanthin: Unique Properties of Astaxanthin Deduced by EPR, Optical and Electrochemical Studies" Antioxidants 6, no. 4: 80. https://doi.org/10.3390/antiox6040080
APA StyleFocsan, A. L., Polyakov, N. E., & Kispert, L. D. (2017). Photo Protection of Haematococcus pluvialis Algae by Astaxanthin: Unique Properties of Astaxanthin Deduced by EPR, Optical and Electrochemical Studies. Antioxidants, 6(4), 80. https://doi.org/10.3390/antiox6040080