Treatment with a Catalytic Superoxide Dismutase (SOD) Mimetic Improves Liver Steatosis, Insulin Sensitivity, and Inflammation in Obesity-Induced Type 2 Diabetes

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Palmitic Acid (PA) Solution
2.3. Measurement of Superoxide by Dihydroethidium (DHE)
2.4. Mice and Treatment
2.5. Body Weight and Liver Weight
2.6. Comprehensive Diagnostic Panel
2.7. Quantification of Hepatic Lipid Accumulation
2.8. Liver Histology
2.9. 8-Hzydroxy-2’-deoxyguanosine (8-OHdG)
2.10. Serum Adipokine Measurement
2.11. Intraperitoneal Glucose Tolerance Test (IPGTT)
2.12. Insulin Tolerance Test (ITT)
2.13. Homeostasic Model Assessment-Estimated Insulin Resistance (HOMA-IR)
2.14. Nuclear Protein Extractions and Electrophoretic Mobility Shift Assay (EMSA)
2.15. Quantitative Real-Time PCR (qRT-PCR)
2.16. Enzyme-Linked Immunosorbent Assay (ELISA)
2.17. Statistical Analysis
3. Results
3.1. MnP Diminishes Superoxide Production In Vitro
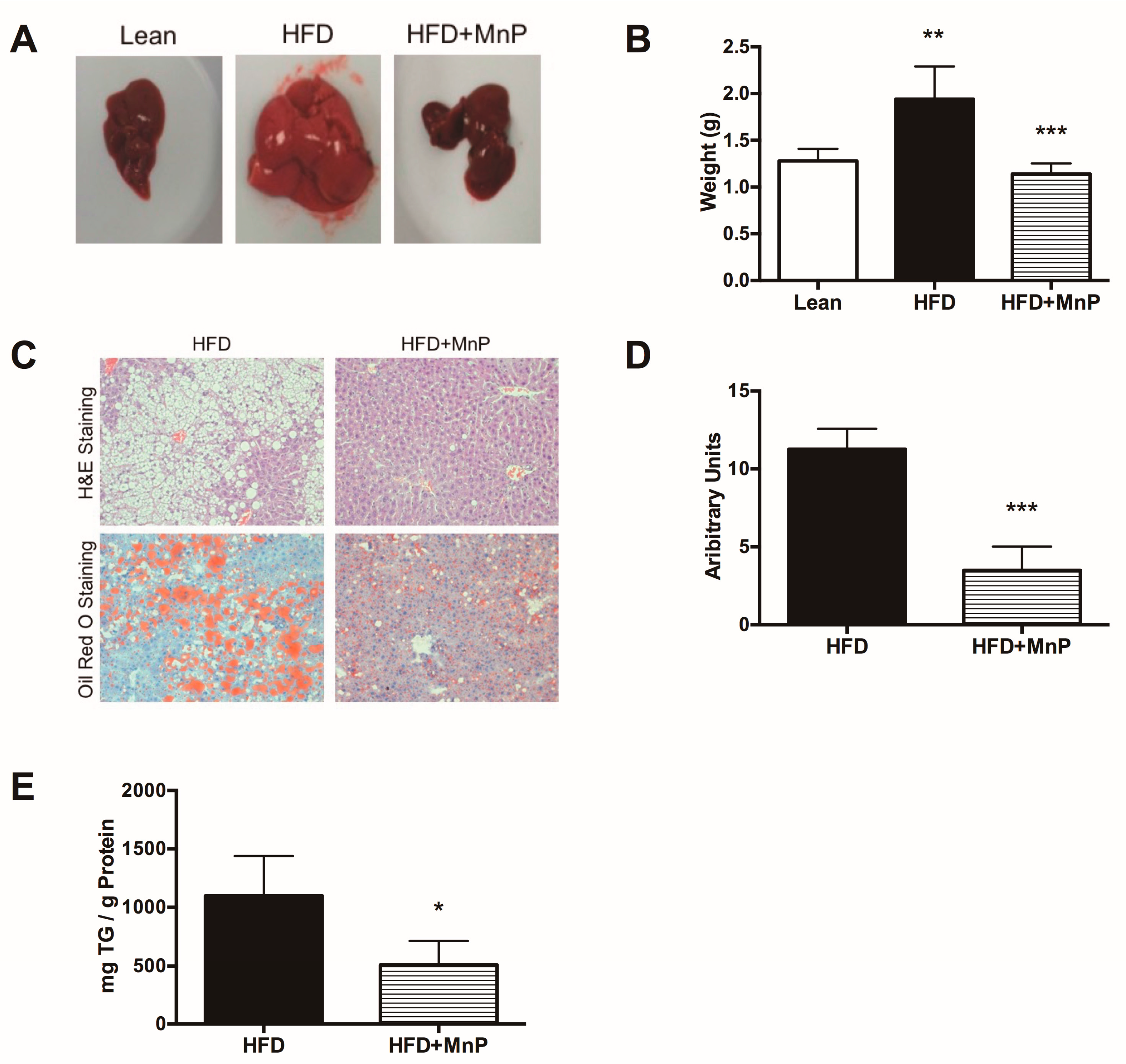
3.2. MnP Treatment Reduces Weight Gain and Imposes Minimal Toxicity
3.3. Redox Modulation Reduces Liver Steatosis and Hepatic Lipid Accumulation
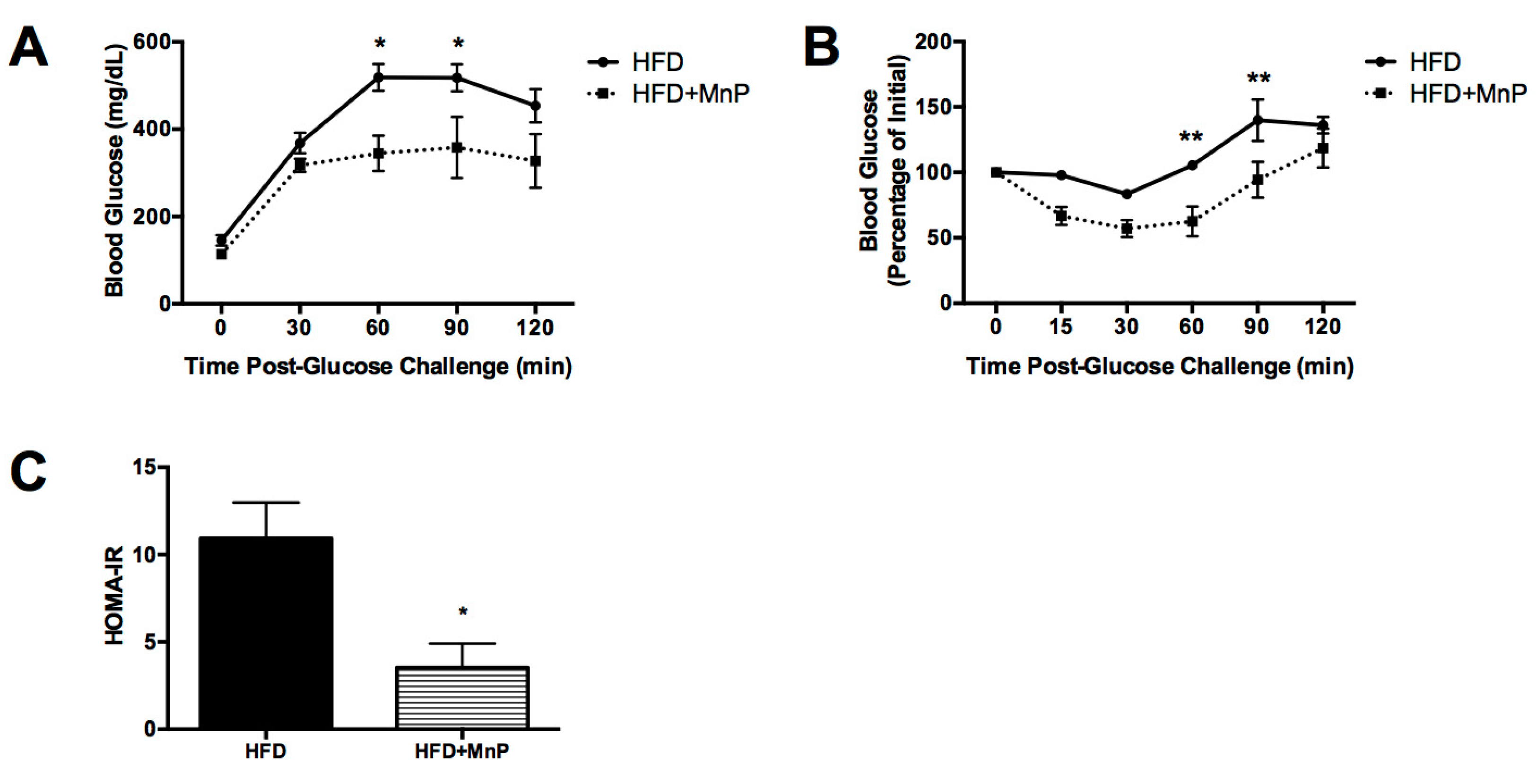
3.4. Hepatic Insulin Resistance is Improved with MnP Treatment
3.5. Insulin, Leptin, and Plasminogen Activator Inhibitor Type-1 (PAI-1) Levels are Diminished Following MnP Treatment
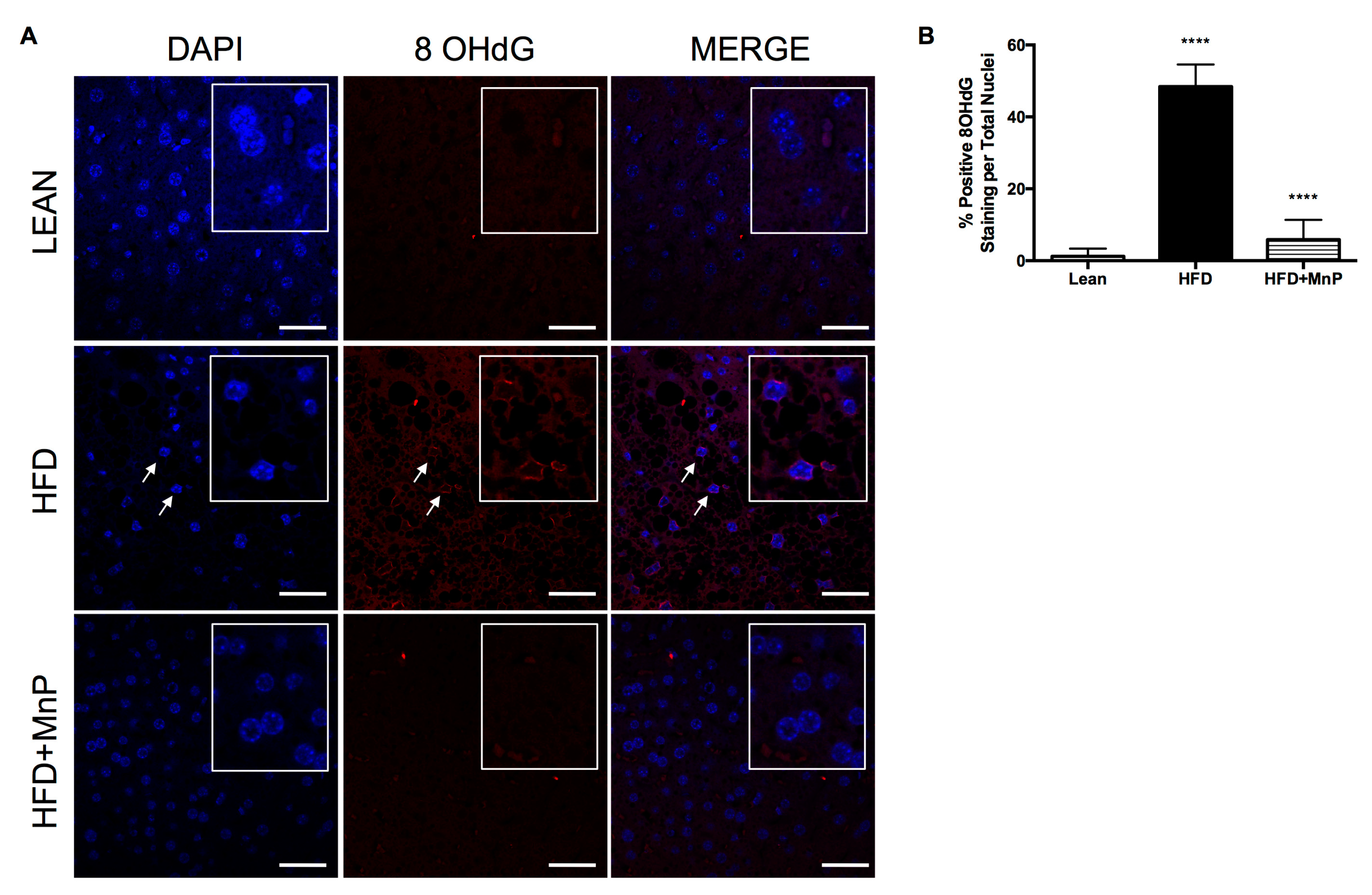
3.6. Redox Modulation Minimizes Oxidative Stress-Induced DNA Damage and Pro-Inflammatory Cytokine Production During HFD
3.7. MnP Treatment Inhibits Hepatic Nuclear NF-κB Binding
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flegal, K.M.; Kruszon-Moran, D.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Trends in obesity among adults in the united states, 2005 to 2014. JAMA 2016, 315, 2284–2291. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, P.A.; Ortega, E. A burning question: Does an adipokine-induced activation of the immune system mediate the effect of overnutrition on type 2 diabetes? Diabetes 2005, 54, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B. Globalization of diabetes: The role of diet, lifestyle, and genes. Diabetes Care 2011, 34, 1249–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism 2016, 65, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and prevention of hepatic steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Ahmed, M. Non-alcoholic fatty liver disease in 2015. World J. Hepatol. 2015, 7, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, H.B.; Smith, R.J. Fatty liver disease in diabetes mellitus. Hepatobiliary Surg. Nutr. 2015, 4, 101–108. [Google Scholar] [PubMed]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y. Inflammation as a sensor of metabolic stress in obesity and type 2 diabetes. Endocrinology 2011, 152, 4005–4006. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care 2004, 27, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. Ikk-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappab. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C.; Crook, M.A. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 1998, 41, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Roden, M. Mechanisms of disease: Hepatic steatosis in type 2 diabetes—Pathogenesis and clinical relevance. Nat. Rev. Endocrinol. 2006, 2, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammatory pathways and insulin action. Int. J. Obes. Relat. Metab. Disord. 2003, 27, S53–S55. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Spasojevic, I.; Tse, H.M.; Tovmasyan, A.; Rajic, Z.; Clair, D.K.; Vujaskovic, Z.; Dewhirst, M.W.; Piganelli, J.D. Design of mn porphyrins for treating oxidative stress injuries and their redox-based regulation of cellular transcriptional activities. Amino Acids 2012, 42, 95–113. [Google Scholar] [CrossRef] [PubMed]
- Gad, S.C.; Sullivan, D.W., Jr.; Crapo, J.D.; Spainhour, C.B. A nonclinical safety assessment of MnTE-2-PyP, a manganese porphyrin. Int. J. Toxicol. 2013, 32, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.M.; Milton, M.J.; Schreiner, S.; Profozich, J.L.; Trucco, M.; Piganelli, J.D. Disruption of innate-mediated proinflammatory cytokine and reactive oxygen species third signal leads to antigen-specific hyporesponsiveness. J. Immunol. 2007, 178, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.M.; Milton, M.J.; Piganelli, J.D. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen-presenting cells: Implication for their use in targeting oxidation-reduction reactions in innate immunity. Free Radic. Biol. Med. 2004, 36, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Sklavos, M.M.; Tse, H.M.; Piganelli, J.D. Redox modulation inhibits CD8 T cell effector function. Free Radic. Biol. Med. 2008, 45, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Sklavos, M.M.; Bertera, S.; Tse, H.M.; Bottino, R.; He, J.; Beilke, J.N.; Coulombe, M.G.; Gill, R.G.; Crapo, J.D.; Trucco, M.; et al. Redox modulation protects islets from transplant-related injury. Diabetes 2010, 59, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Piganelli, J.D.; Flores, S.C.; Cruz, C.; Koepp, J.; Batinic-Haberle, I.; Crapo, J.; Day, B.; Kachadourian, R.; Young, R.; Bradley, B.; et al. A metalloporphyrin-based superoxide dismutase mimic inhibits adoptive transfer of autoimmune diabetes by a diabetogenic T-cell clone. Diabetes 2002, 51, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Delmastro-Greenwood, M.M.; Votyakova, T.; Goetzman, E.; Marre, M.L.; Previte, D.M.; Tovmasyan, A.; Batinic-Haberle, I.; Trucco, M.M.; Piganelli, J.D. Mn porphyrin regulation of aerobic glycolysis: Implications on the activation of diabetogenic immune cells. Antioxid. Redox. Signal. 2013, 19, 1902–1915. [Google Scholar] [CrossRef] [PubMed]
- Delmastro, M.M.; Styche, A.J.; Trucco, M.M.; Workman, C.J.; Vignali, D.A.; Piganelli, J.D. Modulation of redox balance leaves murine diabetogenic TH1 T cells “LAG-3-ing” behind. Diabetes 2012, 61, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Previte, D.M.; O’Connor, E.C.; Novak, E.A.; Martins, C.P.; Mollen, K.P.; Piganelli, J.D. Reactive oxygen species are required for driving efficient and sustained aerobic glycolysis during CD4+ T cell activation. PLoS ONE 2017, 12, e0175549. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. An educational overview of the chemistry, biochemistry and therapeutic aspects of mn porphyrins—From superoxide dismutation to H2O2-driven pathways. Redox Biol. 2015, 5, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Perdomo, G.; Brown, N.F.; O’Doherty, R.M. Fatty acid-induced insulin resistance in L6 myotubes is prevented by inhibition of activation and nuclear localization of nuclear factor kappa B. J. Biol. Chem. 2004, 279, 41294–41301. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Bikineyeva, A.; Dikalov, S.I. Rapid and specific measurements of superoxide using fluorescence spectroscopy. J. Biomol. Screen. 2013, 18, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Calabuig-Navarro, V.; Yamauchi, J.; Lee, S.; Zhang, T.; Liu, Y.Z.; Sadlek, K.; Coudriet, G.M.; Piganelli, J.D.; Jiang, C.L.; Miller, R.; et al. Forkhead Box O6 (FoxO6) Depletion Attenuates Hepatic Gluconeogenesis and Protects against Fat-Induced Glucose Disorder in Mice. J. Biol. Chem. 2015, 290, 15581–15594. [Google Scholar] [CrossRef] [PubMed]
- Blough, E.; Dineen, B.; Esser, K. Extraction of nuclear proteins from striated muscle tissue. Biotechniques 1999, 26, 202–204, 206. [Google Scholar] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.A.; King, G.L. Oxidative stress and antioxidant treatment in diabetes. Ann. N. Y. Acad. Sci. 2004, 1031, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Bottino, R.; Balamurugan, A.N.; Tse, H.; Thirunavukkarasu, C.; Ge, X.; Profozich, J.; Milton, M.; Ziegenfuss, A.; Trucco, M.; Piganelli, J.D. Response of human islets to isolation stress and the effect of antioxidant treatment. Diabetes 2004, 53, 2559–2568. [Google Scholar] [CrossRef] [PubMed]
- Bottino, R.; Balamurugan, A.N.; Bertera, S.; Pietropaolo, M.; Trucco, M.; Piganelli, J.D. Preservation of human islet cell functional mass by anti-oxidative action of a novel SOD mimic compound. Diabetes 2002, 51, 2561–2567. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lechon, M.J.; Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Nong, S.; Huang, X.; Lu, Y.; Zhao, H.; Lin, Y.; Man, Y.; Wang, S.; Yang, J.; Li, J. The effects of palmitate on hepatic insulin resistance are mediated by NADPH Oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J. Biol. Chem. 2010, 285, 29965–29973. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Ahren, B. The high-fat diet-fed mouse: A model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004, 53 (Suppl. 3), S215–S219. [Google Scholar] [CrossRef] [PubMed]
- Physiological Data Summary—C57BL/6J (000664). Available online: http://cheval.pratique.free.fr/STAGE%20ENVL/m%E9moire/C57BL6J/B6data000664%5B1%5D.pdf (accessed on 22 September 2017).
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and abuse of homa modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, M. Insulin resistance and pancreatic beta cell failure. J. Clin. Investig. 2006, 116, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Ren, J. Leptin and hyperleptinemia-from friend to foe for cardiovascular function. J. Endocrinol. 2004, 181, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Hannan, K.S.; Greenberg, M.L.; Friedman, J.M. Hyperleptinemia is required for the development of leptin resistance. PLoS ONE 2010, 5, e11376. [Google Scholar] [CrossRef] [PubMed]
- Poirier, P.; Giles, T.D.; Bray, G.A.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Binder, B.R.; Christ, G.; Gruber, F.; Grubic, N.; Hufnagl, P.; Krebs, M.; Mihaly, J.; Prager, G.W. Plasminogen activator inhibitor 1: Physiological and pathophysiological roles. News Physiol. Sci. 2002, 17, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.J.; Sobel, B.E. PAI-1 and diabetes: A journey from the bench to the bedside. Diabetes Care 2012, 35, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Alessi, M.C.; Bastelica, D.; Mavri, A.; Morange, P.; Berthet, B.; Grino, M.; Juhan-Vague, I. Plasma PAI-1 levels are more strongly related to liver steatosis than to adipose tissue accumulation. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Alessi, M.C.; Juhan-Vague, I. PAI-1 and the metabolic syndrome: Links, causes, and consequences. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Loskutoff, D.J. Tissue distribution and regulation of plasminogen activator inhibitor-1 in obese mice. Mol. Med. 1996, 2, 568–582. [Google Scholar] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C 2009, 27, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Spiegelman, B.M. Tumor necrosis factor alpha: A key component of the obesity-diabetes link. Diabetes 1994, 43, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Uysal, K.T.; Wiesbrock, S.M.; Pandey, M.; Hotamisligil, G.S.; Loskutoff, D.J. Tumor necrosis factor alpha is a key component in the obesity-linked elevation of plasminogen activator inhibitor 1. Proc. Natl. Acad. Sci. USA 1999, 96, 6902–6907. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Stanton, M.C.; Chen, S.C.; Jackson, J.V.; Rojas-Triana, A.; Kinsley, D.; Cui, L.; Fine, J.S.; Greenfeder, S.; Bober, L.A.; Jenh, C.H. Inflammatory signals shift from adipose to liver during high fat feeding and influence the development of steatohepatitis in mice. J. Inflamm. 2011, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Sugita, H.; Kaneki, M.; Tokunaga, E.; Sugita, M.; Koike, C.; Yasuhara, S.; Tompkins, R.G.; Martyn, J.A. Inducible nitric oxide synthase plays a role in LPS-induced hyperglycemia and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E386–E394. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Shimizu, N.; Kunii, K.; Martyn, J.A.; Ueki, K.; Kaneki, M. A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes 2005, 54, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, S.; Choi, C.S.; Shimizu, N.; Yamada, M.; Kim, M.; Zhang, T.; Shiota, G.; Dong, H.H.; Kim, Y.B.; Kaneki, M. Liver-specific inducible nitric-oxide synthase expression is sufficient to cause hepatic insulin resistance and mild hyperglycemia in mice. J. Biol. Chem. 2011, 286, 34959–34975. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Weaver, M.R.; Kosmacek, E.A.; O’Connor, B.P.; Harmacek, L.; Venkataraman, S.; Oberley-Deegan, R.E. MnTE-2-PyP reduces prostate cancer growth and metastasis by suppressing p300 activity and p300/HIF-1/CREB binding to the promoter region of the PAI-1 gene. Free Radic. Biol. Med. 2016, 94, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed]
- Campfield, L.A.; Smith, F.J.; Guisez, Y.; Devos, R.; Burn, P. Recombinant mouse OB protein: Evidence for a peripheral signal linking adiposity and central neural networks. Science 1995, 269, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Caro, J.F.; Sinha, M.K.; Kolaczynski, J.W.; Zhang, P.L.; Considine, R.V. Leptin: The tale of an obesity gene. Diabetes 1996, 45, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V. Increased serum leptin indicates leptin resistance in obesity. Clin. Chem. 2011, 57, 1461–1462. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.E. PAI-1 and atherothrombosis. J. Thromb. Haemost. 2005, 3, 1879–1883. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. NASH is an inflammatory disorder: Pathogenic, prognostic and therapeutic implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Brodsky, T.; Sosinsky, A.Z.; McLoughlin, R.; Stansky, E.; Fussell, L.; Sheppard, A.; DiSanto-Rose, M.; Kershaw, E.E.; Reynolds, T.H., IV. Manganese [III] tetrakis [5,10,15,20]-benzoic acid porphyrin reduces adiposity and improves insulin action in mice with pre-existing obesity. PLoS ONE 2015, 10, e0137388. [Google Scholar] [CrossRef] [PubMed]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Pires, K.M.; Ilkun, O.; Valente, M.; Boudina, S. Treatment with a SOD mimetic reduces visceral adiposity, adipocyte death, and adipose tissue inflammation in high fat-fed mice. Obesity 2014, 22, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Hatano, E.; Bennett, B.L.; Manning, A.M.; Qian, T.; Lemasters, J.J.; Brenner, D.A. NF-kappab stimulates inducible nitric oxide synthase to protect mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis. Gastroenterology 2001, 120, 1251–1262. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Lean | HFD | HFD + MnP | One-Way ANOVA |
|---|---|---|---|---|
| Albumin (g/dL) | 2.617 | 3.433 | 4.233 | * Lean vs. HFD + MnP |
| Alanine Aminotransferase (U/L) | 63 | 58.33 | 79 | ns |
| Alkaline Phosphatase (U/L) | 55.17 | 54.5 | 54 | ns |
| Amylase (U/L) | 921.5 | 1042 | 1092 | ns |
| Blood Urea Nitrogen (mg/dL) | 23.17 | 19.67 | 22.5 | ns |
| Calcium (mg/dL) | 10.5 | 10.18 | 10.32 | ns |
| Creatinine (mg/dL) | 0.1667 | 0.2 | 0.2 | ns |
| Globulin (g/dL) | 2.167 | 2.317 | 1.85 | ns |
| Glucose (mg/dL) | 121.5 | 141.5 | 142.2 | ns |
| Phosphorus (mg/dL) | 8.75 | 8.083 | 7.833 | ns |
| Potassium (mmol/L) | 5.583 | 5.083 | 4.633 | ns |
| Sodium (mmol/L) | 151.5 | 150.8 | 152.5 | ns |
| Total Bilirubin (mg/dL) | 0.1833 | 0.2333 | 0.2333 | ns |
| Total Serum Protein (g/dL) | 5.817 | 5.733 | 6.067 | ns |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coudriet, G.M.; Delmastro-Greenwood, M.M.; Previte, D.M.; Marré, M.L.; O’Connor, E.C.; Novak, E.A.; Vincent, G.; Mollen, K.P.; Lee, S.; Dong, H.H.; et al. Treatment with a Catalytic Superoxide Dismutase (SOD) Mimetic Improves Liver Steatosis, Insulin Sensitivity, and Inflammation in Obesity-Induced Type 2 Diabetes. Antioxidants 2017, 6, 85. https://doi.org/10.3390/antiox6040085
Coudriet GM, Delmastro-Greenwood MM, Previte DM, Marré ML, O’Connor EC, Novak EA, Vincent G, Mollen KP, Lee S, Dong HH, et al. Treatment with a Catalytic Superoxide Dismutase (SOD) Mimetic Improves Liver Steatosis, Insulin Sensitivity, and Inflammation in Obesity-Induced Type 2 Diabetes. Antioxidants. 2017; 6(4):85. https://doi.org/10.3390/antiox6040085
Chicago/Turabian StyleCoudriet, Gina M., Meghan M. Delmastro-Greenwood, Dana M. Previte, Meghan L. Marré, Erin C. O’Connor, Elizabeth A. Novak, Garret Vincent, Kevin P. Mollen, Sojin Lee, H. Henry Dong, and et al. 2017. "Treatment with a Catalytic Superoxide Dismutase (SOD) Mimetic Improves Liver Steatosis, Insulin Sensitivity, and Inflammation in Obesity-Induced Type 2 Diabetes" Antioxidants 6, no. 4: 85. https://doi.org/10.3390/antiox6040085
APA StyleCoudriet, G. M., Delmastro-Greenwood, M. M., Previte, D. M., Marré, M. L., O’Connor, E. C., Novak, E. A., Vincent, G., Mollen, K. P., Lee, S., Dong, H. H., & Piganelli, J. D. (2017). Treatment with a Catalytic Superoxide Dismutase (SOD) Mimetic Improves Liver Steatosis, Insulin Sensitivity, and Inflammation in Obesity-Induced Type 2 Diabetes. Antioxidants, 6(4), 85. https://doi.org/10.3390/antiox6040085