Insights into the Dichotomous Regulation of SOD2 in Cancer


Abstract
:1. Introduction
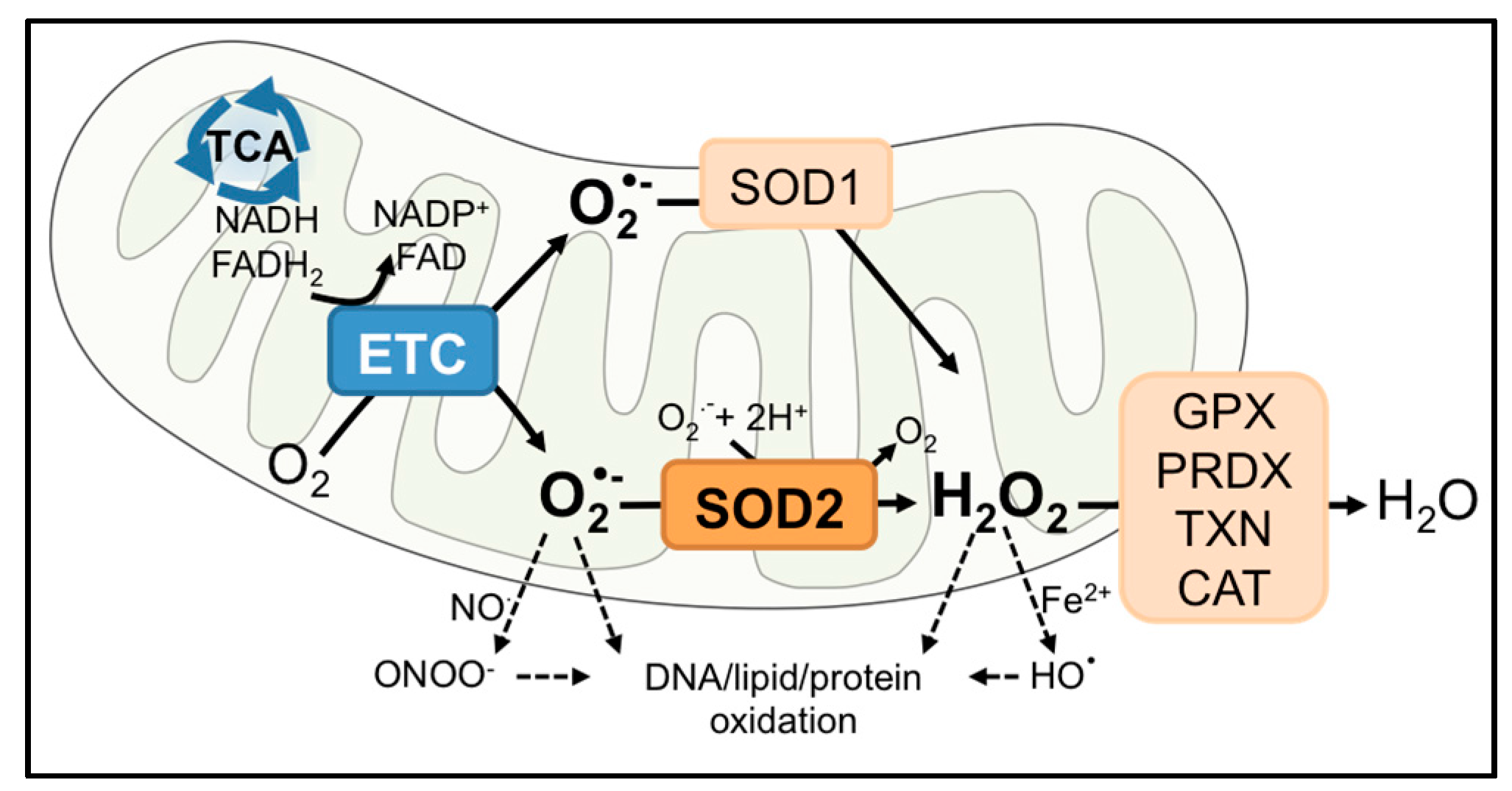
2. SOD2
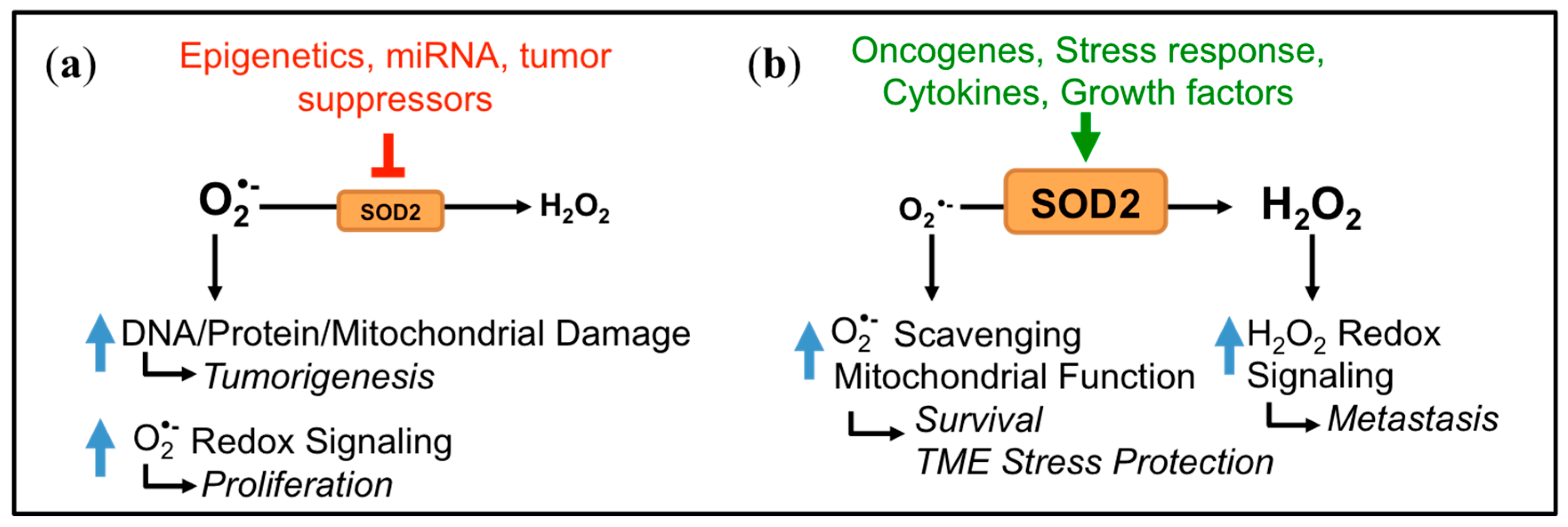
3. Dichotomous Role of SOD2 in Cancer
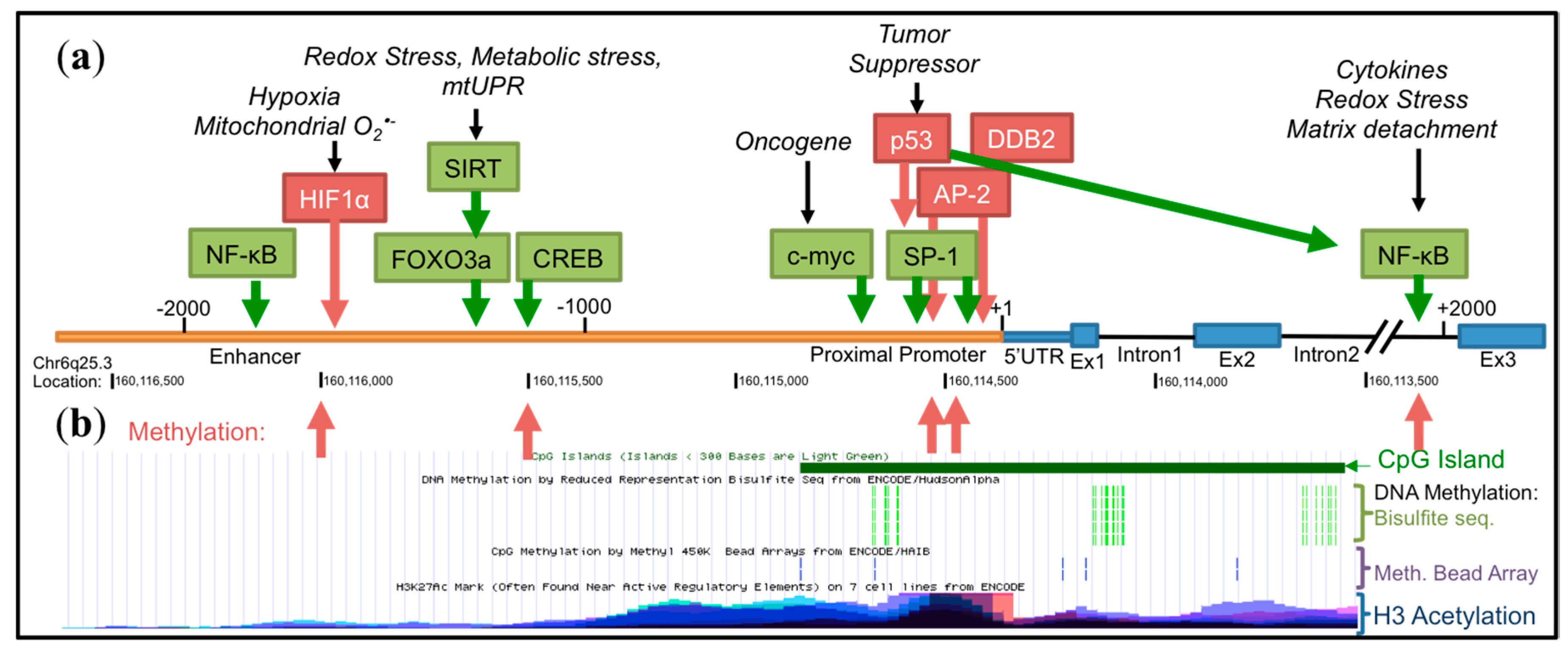
4. Transcriptional Regulation of SOD2
4.1. Basal Transcription
4.2. Epigenetic Regulation
4.3. Influence of Oncogenes and Tumor Suppressors
4.4. Inducible Transcription
4.5. Other Transcriptional Regulators
5. Post-Transcriptional Regulation of SOD2
6. Post-Translational Regulation of SOD2
6.1. Localization and Protein Interaction
6.2. Transition Metal Incorporation
6.3. Post-Translational Modifications
6.3.1. Acetylation
6.3.2. Phosphorylation
6.3.3. Oxidation, Nitration and S-Glutathionylation
6.3.4. Ubiquitination
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001, 509, 435–438. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Hao, P.; Xue, L.; Wei, S.; Zhang, Y.; Chen, Y. Inhibition of aldehyde dehydrogenase 2 by oxidative stress is associated with cardiac dysfunction in diabetic rats. Mol. Med. 2011, 17, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Lundberg, K.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc. Natl. Acad. Sci. USA 2005, 102, 5987–5991. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Kindy, M.S.; Holtsberg, F.W.; St. Clair, D.K.; Yen, H.C.; Germeyer, A.; Steiner, S.M.; Bruce-Keller, A.J.; Hutchins, J.B.; Mattson, M.P. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: Suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci. 1998, 18, 687–697. [Google Scholar] [PubMed]
- Aguirre, J.D.; Culotta, V.C. Battles with iron: Manganese in oxidative stress protection. J. Biol. Chem. 2012, 287, 13541–13548. [Google Scholar] [CrossRef] [PubMed]
- Yamakura, F.; Kawasaki, H. Post-translational modifications of superoxide dismutase. Biochim. Biophys. Acta 2010, 1804, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Copin, J.C.; Gasche, Y.; Chan, P.H. Overexpression of copper/zinc superoxide dismutase does not prevent neonatal lethality in mutant mice that lack manganese superoxide dismutase. Free Radic. Biol. Med. 2000, 28, 1571–1576. [Google Scholar] [CrossRef]
- Zhong, W.; Oberley, L.W.; Oberley, T.D.; St. Clair, D.K. Suppression of the malignant phenotype of human glioma cells by overexpression of manganese superoxide dismutase. Oncogene 1997, 14, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Weydert, C.; Roling, B.; Liu, J.; Hinkhouse, M.M.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Suppression of the malignant phenotype in human pancreatic cancer cells by the overexpression of manganese superoxide dismutase. Mol. Cancer Ther. 2003, 2, 361–369. [Google Scholar] [PubMed]
- Bravard, A.; Sabatier, L.; Hoffschir, F.; Ricoul, M.; Luccioni, C.; Dutrillaux, B. SOD2: A new type of tumor-suppressor gene? Int. J. Cancer 1992, 51, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, T.; Domann, F.E. Decreased expression of manganese superoxide dismutase in transformed cells is associated with increased cytosine methylation of the SOD2 gene. DNA Cell Biol. 1999, 18, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W.; Buettner, G.R. Role of superoxide dismutase in cancer: A review. Cancer Res. 1979, 39, 1141–1149. [Google Scholar] [PubMed]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef] [PubMed]
- Sarsour, E.H.; Kalen, A.L.; Goswami, P.C. Manganese superoxide dismutase regulates a redox cycle within the cell cycle. Antioxid. Redox Signal. 2014, 20, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W.; Oberley, T.D.; Buettner, G.R. Cell differentiation, aging and cancer: The possible roles of superoxide and superoxide dismutases. Med. Hypotheses 1980, 6, 249–268. [Google Scholar] [CrossRef]
- Oberley, L.W.; Oberley, T.D.; Buettner, G.R. Cell division in normal and transformed cells: The possible role of superoxide and hydrogen peroxide. Med. Hypotheses 1981, 7, 21–42. [Google Scholar] [CrossRef]
- Chung-man Ho, J.; Zheng, S.; Comhair, S.A.; Farver, C.; Erzurum, S.C. Differential expression of manganese superoxide dismutase and catalase in lung cancer. Cancer Res. 2001, 61, 8578–8585. [Google Scholar] [PubMed]
- Hempel, N.; Ye, H.; Abessi, B.; Mian, B.; Melendez, J.A. Altered redox status accompanies progression to metastatic human bladder cancer. Free Radic. Biol. Med. 2009, 46, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Miar, A.; Hevia, D.; Muñoz-Cimadevilla, H.; Astudillo, A.; Velasco, J.; Sainz, R.M.; Mayo, J.C. Manganese superoxide dismutase (SOD2/MnSOD)/catalase and SOD2/GPx1 ratios as biomarkers for tumor progression and metastasis in prostate, colon, and lung cancer. Free Radic. Biol. Med. 2015, 85, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.; Carrico, P.M.; Melendez, J.A. Manganese superoxide dismutase (SOD2) and redox-control of signaling events that drive metastasis. Anticancer Agents Med. Chem. 2011, 11, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; Tangpong, J.; Chaiswing, L.; Oberley, T.D.; St. Clair, D.K. Manganese superoxide dismutase is a p53-regulated gene that switches cancers between early and advanced stages. Cancer Res. 2011, 71, 6684–6695. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Kamarajugadda, S.; Cai, Q.; Chen, H.; Nayak, S.; Zhu, J.; He, M.; Jin, Y.; Zhang, Y.; Ai, L.; Martin, S.S.; et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013, 4, e504. [Google Scholar] [CrossRef] [PubMed]
- Buchheit, C.L.; Weigel, K.J.; Schafer, Z.T. Cancer cell survival during detachment from the ECM: Multiple barriers to tumour progression. Nat. Rev. Cancer 2014, 14, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.M.; Hempel, N.; Nelson, K.K.; Dabiri, G.; Gamarra, A.; Belarmino, J.; Van De Water, L.; Mian, B.M.; Melendez, J.A. Manganese superoxide dismutase enhances the invasive and migratory activity of tumor cells. Cancer Res. 2007, 67, 10260–10267. [Google Scholar] [CrossRef] [PubMed]
- Hemachandra, L.P.; Shin, D.H.; Dier, U.; Iuliano, J.N.; Engelberth, S.A.; Uusitalo, L.M.; Murphy, S.K.; Hempel, N. Mitochondrial Superoxide Dismutase Has a Protumorigenic Role in Ovarian Clear Cell Carcinoma. Cancer Res. 2015, 75, 4973–4984. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.; Melendez, J.A. Intracellular redox status controls membrane localization of pro- and anti-migratory signaling molecules. Redox Biol. 2014, 2, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, S.; Cai, Y.; Wang, A.; He, Q.; Zheng, C.; Zhao, T.; Ding, X.; Zhou, X. Manganese superoxide dismutase induces migration and invasion of tongue squamous cell carcinoma via H2O2-dependent Snail signaling. Free Radic. Biol. Med. 2012, 53, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.; Bartling, T.R.; Mian, B.; Melendez, J.A. Acquisition of the metastatic phenotype is accompanied by H2O2-dependent activation of the p130Cas signaling complex. Mol. Cancer Res. 2013, 11, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Miriyala, S.; Holley, A.K.; St. Clair, D.K. Mitochondrial superoxide dismutase—Signals of distinction. Anticancer Agents Med. Chem. 2011, 11, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Idelchik, M.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Li, X.; Wang, H.Y.; Zheng, X.F.S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Van Driel, B.E.; Lyon, H.; Hoogenraad, D.C.; Anten, S.; Hansen, U.; Van Noorden, C.J. Expression of CuZn- and Mn-superoxide dismutase in human colorectal neoplasms. Free Radic. Biol. Med. 1997, 23, 435–444. [Google Scholar] [CrossRef]
- Czeczot, H.; Skrzycki, M.; Podsiad, M.; Gawryszewska, E.; Nyckowski, P.; Porembska, Z. Antioxidant status of patients with primary colorectal cancer and liver metastases of colorectal cancer. Polski Merkuriusz Lekarski Organ Polskiego Towarzystwa Lekarskiego 2005, 18, 58–61. [Google Scholar] [PubMed]
- Cobbs, C.S.; Levi, D.S.; Aldape, K.; Israel, M.A. Manganese superoxide dismutase expression in human central nervous system tumors. Cancer Res. 1996, 56, 3192–3195. [Google Scholar] [PubMed]
- Landriscina, M.; Remiddi, F.; Ria, F.; Palazzotti, B.; De Leo, M.E.; Iacoangeli, M.; Rosselli, R.; Scerrati, M.; Galeotti, T. The level of MnSOD is directly correlated with grade of brain tumours of neuroepithelial origin. Br. J. Cancer 1996, 74, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Jung, J.H.; Moon, M.J.; Kim, Y.Y.; Kim, J.H.; Park, S.H.; Kim, C.Y.; Paek, S.H.; Kim, D.G.; Jung, H.W.; et al. Tissue expression of manganese superoxide dismutase is a candidate prognostic marker for glioblastoma. Oncology 2009, 77, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Cholia, R.P.; Kumari, S.; Kumar, S.; Kaur, M.; Kumar, R.; Dhiman, M.; Mantha, A.K. An in vitro study ascertaining the role of H2O2 and glucose oxidase in modulation of antioxidant potential and cancer cell survival mechanisms in glioblastoma U-87 MG cells. Metab. Brain Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tews, D.S. Cell death and oxidative stress in gliomas. Neuropathol. Appl. Neurobiol. 1999, 25, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Yan, T.; Lim, R.; Oberley, L.W. Expression of superoxide dismutases, catalase, and glutathione peroxidase in glioma cells. Free Radic. Biol. Med. 1999, 27, 1334–1345. [Google Scholar] [CrossRef]
- Haapasalo, H.; Kyläniemi, M.; Paunul, N.; Kinnula, V.L.; Soini, Y. Expression of antioxidant enzymes in astrocytic brain tumors. Brain Pathol. 2003, 13, 155–164. [Google Scholar] [CrossRef] [PubMed]
- French, P.J.; Swagemakers, S.M.; Nagel, J.H.; Kouwenhoven, M.C.; Brouwer, E.; van der Spek, P.; Luider, T.M.; Kros, J.M.; van den Bent, M.J.; Sillevis Smitt, P.A. Gene expression profiles associated with treatment response in oligodendrogliomas. Cancer Res. 2005, 65, 11335–11344. [Google Scholar] [CrossRef] [PubMed]
- Järvelä, S.; Sally, J.; Bragge, H.; Helena, B.; Paunu, N.; Niina, P.; Järvelä, T.; Timo, J.; Paljärvi, L.; Leo, P.; et al. Antioxidant enzymes in oligodendroglial brain tumors: Association with proliferation, apoptotic activity and survival. J. Neurooncol. 2006, 77, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dai, C.; Zhang, J. SIRT3-SOD2-ROS pathway is involved in linalool-induced glioma cell apoptotic death. Acta Biochim. Pol. 2017, 64, 343–350. [Google Scholar] [CrossRef] [PubMed]
- John, J.P.; Pollak, A.; Lubec, G. Complete sequencing and oxidative modification of manganese superoxide dismutase in medulloblastoma cells. Electrophoresis 2009, 30, 3006–3016. [Google Scholar] [CrossRef] [PubMed]
- Minig, V.; Kattan, Z.; van Beeumen, J.; Brunner, E.; Becuwe, P. Identification of DDB2 protein as a transcriptional regulator of constitutive SOD2 gene expression in human breast cancer cells. J. Biol. Chem. 2009, 284, 14165–14176. [Google Scholar] [CrossRef] [PubMed]
- Ennen, M.; Minig, V.; Grandemange, S.; Touche, N.; Merlin, J.L.; Besancenot, V.; Brunner, E.; Domenjoud, L.; Becuwe, P. Regulation of the high basal expression of the manganese superoxide dismutase gene in aggressive breast cancer cells. Free Radic. Biol. Med. 2011, 50, 1771–1779. [Google Scholar] [CrossRef] [PubMed]
- Hitchler, M.J.; Oberley, L.W.; Domann, F.E. Epigenetic silencing of SOD2 by histone modifications in human breast cancer cells. Free Radic. Biol. Med. 2008, 45, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Hitchler, M.J.; Wikainapakul, K.; Yu, L.; Powers, K.; Attatippaholkun, W.; Domann, F.E. Epigenetic regulation of manganese superoxide dismutase expression in human breast cancer cells. Epigenetics 2006, 1, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.C.; Ratti, B.A.; Mao, M.; Ansenberger-Fricano, K.; Shajahan-Haq, A.N.; Tyner, A.L.; Minshall, R.D.; Bonini, M.G. Caveolin-1 regulates cancer cell metabolism via scavenging Nrf2 and suppressing MnSOD-driven glycolysis. Oncotarget 2016, 7, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Santa-Maria, C.A.; O’Brien, J.; Gius, D.; Zhu, Y. Manganese Superoxide Dismutase Acetylation and Dysregulation, Due to Loss of SIRT3 Activity, Promote a Luminal B-Like Breast Carcinogenic-Permissive Phenotype. Antioxid. Redox Signal. 2016, 25, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Portakal, O.; Ozkaya, O.; Erden Inal, M.; Bozan, B.; Koşan, M.; Sayek, I. Coenzyme Q10 concentrations and antioxidant status in tissues of breast cancer patients. Clin. Biochem. 2000, 33, 279–284. [Google Scholar] [CrossRef]
- Xu, Y.; Krishnan, A.; Wan, X.S.; Majima, H.; Yeh, C.C.; Ludewig, G.; Kasarskis, E.J.; St. Clair, D.K. Mutations in the promoter reveal a cause for the reduced expression of the human manganese superoxide dismutase gene in cancer cells. Oncogene 1999, 18, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Govatati, S.; Malempati, S.; Saradamma, B.; Divyamaanasa, D.; Naidu, B.P.; Bramhachari, P.V.; Narayana, N.; Shivaji, S.; Bhanoori, M.; Tamanam, R.R.; et al. Manganese-superoxide dismutase (Mn-SOD) overexpression is a common event in colorectal cancers with mitochondrial microsatellite instability. Tumour Biol. 2016, 37, 10357–10364. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.G.; Wang, Y.D.; Chen, L.Q.; Wang, S.J.; Liu, G.L.; Yu, X.R.; Cheng, Y.J.; Liu, Q. Novel cancer suppressor gene for esophageal cancer: Manganese superoxide dismutase. Dis. Esophagus 2011, 24, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W.; Bize, I.B.; Sahu, S.K.; Leuthauser, S.W.; Gruber, H.E. Superoxide dismutase activity of normal murine liver, regenerating liver, and H6 hepatoma. J. Natl. Cancer Inst. 1978, 61, 375–379. [Google Scholar] [PubMed]
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Yang, H.; et al. MCU-dependent mitochondrial Ca2+ inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 2017, 36, 5897–5909. [Google Scholar] [CrossRef] [PubMed]
- Coursin, D.B.; Cihla, H.P.; Sempf, J.; Oberley, T.D.; Oberley, L.W. An immunohistochemical analysis of antioxidant and glutathione S-transferase enzyme levels in normal and neoplastic human lung. Histol. Histopathol. 1996, 11, 851–860. [Google Scholar] [PubMed]
- Liu, G.; Zhou, W.; Park, S.; Wang, L.I.; Miller, D.P.; Wain, J.C.; Lynch, T.J.; Su, L.; Christiani, D.C. The SOD2 Val/Val genotype enhances the risk of nonsmall cell lung carcinoma by p53 and XRCC1 polymorphisms. Cancer 2004, 101, 2802–2808. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Denu, R.A.; Krautkramer, K.A.; Grindle, K.M.; Yang, D.T.; Asimakopoulos, F.; Hematti, P.; Denu, J.M. Loss of SIRT3 Provides Growth Advantage for B Cell Malignancies. J. Biol. Chem. 2016, 291, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.J.; Palmer, J.M.; Walters, M.K.; Nancarrow, D.J.; Parsons, P.G.; Hayward, N.K. Simple tandem repeat allelic deletions confirm the preferential loss of distal chromosome 6q in melanoma. Int. J. Cancer 1994, 58, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.R.; Peng, B.; Pompeia, C.; Thomas, S.; Cho, E.; Clausen, P.A.; Marquez, V.E.; Farrar, W.L. Epigenetic silencing of manganese superoxide dismutase (SOD-2) in KAS 6/1 human multiple myeloma cells increases cell proliferation. Cancer Biol. Ther. 2005, 4, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Integrated molecular profiling of SOD2 expression in multiple myeloma. Blood 2007, 109, 3953–3962. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Rosen, D.G.; Zhou, Y.; Feng, L.; Yang, G.; Liu, J.; Huang, P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: Role in cell proliferation and response to oxidative stress. J. Biol. Chem. 2005, 280, 39485–39492. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines. Br. J. Cancer 2007, 97, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Pandit, H.; Zhang, W.; Li, Y.; Agle, S.; Li, X.; Li, S.P.; Cui, G.; Martin, R.C. Manganese superoxide dismutase expression is negatively associated with microRNA-301a in human pancreatic ductal adenocarcinoma. Cancer Gene Ther. 2015, 22, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Du, J.; Liu, J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Metastatic progression of pancreatic cancer: Changes in antioxidant enzymes and cell growth. Clin. Exp. Metastasis 2005, 22, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Wheatley-Price, P.; Asomaning, K.; Reid, A.; Zhai, R.; Su, L.; Zhou, W.; Zhu, A.; Ryan, D.P.; Christiani, D.C.; Liu, G. Myeloperoxidase and superoxide dismutase polymorphisms are associated with an increased risk of developing pancreatic adenocarcinoma. Cancer 2008, 112, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fang, F.; Zhang, J.; Josson, S.; St. Clair, W.H.; St. Clair, D.K. miR-17* suppresses tumorigenicity of prostate cancer by inhibiting mitochondrial antioxidant enzymes. PLoS ONE 2010, 5, e14356. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.H.; Li, C.X.; Shen, S.M.; Li, H.; Chen, G.Q.; Wei, Q.; Wang, L.S. Hypoxia-inducible factor 1α mediates the down-regulation of superoxide dismutase 2 in von Hippel-Lindau deficient renal clear cell carcinoma. Biochem. Biophys. Res. Commun. 2013, 435, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Azadzoi, K.M.; Choi, H.P.; Jing, R.; Lu, X.; Li, C.; Wang, F.; Lu, J.; Yang, J.H. LC-MS/MS Analysis Unravels Deep Oxidation of Manganese Superoxide Dismutase in Kidney Cancer. Int. J. Mol. Sci. 2017, 18, 319. [Google Scholar] [CrossRef] [PubMed]
- Mallery, S.R.; Pei, P.; Landwehr, D.J.; Clark, C.M.; Bradburn, J.E.; Ness, G.M.; Robertson, F.M. Implications for oxidative and nitrative stress in the pathogenesis of AIDS-related Kaposi’s sarcoma. Carcinogenesis 2004, 25, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, J.; Jiang, L.; Wang, A.; Shi, F.; Ye, H.; Zhou, X. MicroRNA-222 regulates cell invasion by targeting matrix metalloproteinase 1 (MMP1) and manganese superoxide dismutase 2 (SOD2) in tongue squamous cell carcinoma cell lines. Cancer Genom. Proteom. 2009, 6, 131–139. [Google Scholar]
- Liu, Z.; He, Q.; Ding, X.; Zhao, T.; Zhao, L.; Wang, A. SOD2 is a C-myc target gene that promotes the migration and invasion of tongue squamous cell carcinoma involving cancer stem-like cells. Int. J. Biochem. Cell Biol. 2015, 60, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Oncomine Research Edition: 715 Datasets and 86733 Samples. Available online: https://www.oncomine.org/resource/login.html (accessed on 1 October 2017).
- Bravard, A.; Hoffschir, F.; Sabatier, L.; Ricoul, M.; Pinton, A.; Cassingena, R.; Estrade, S.; Luccioni, C.; Dutrillaux, B. Early superoxide dismutase alterations during SV40-transformation of human fibroblasts. Int. J. Cancer 1992, 52, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Porntadavity, S.; St. Clair, D.K. Transcriptional regulation of the human manganese superoxide dismutase gene: The role of specificity protein 1 (Sp1) and activating protein-2 (AP-2). Biochem. J. 2002, 362, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.H.; Huang, Y.; Oberley, L.W.; Domann, F.E. A family of AP-2 proteins down-regulate manganese superoxide dismutase expression. J. Biol. Chem. 2001, 276, 14407–14413. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fang, F.; Dhar, S.K.; Bosch, A.; St. Clair, W.H.; Kasarskis, E.J.; St. Clair, D.K. Mutations in the SOD2 promoter reveal a molecular basis for an activating protein 2-dependent dysregulation of manganese superoxide dismutase expression in cancer cells. Mol. Cancer Res. 2008, 6, 1881–1893. [Google Scholar] [CrossRef] [PubMed]
- Douglas, D.B.; Akiyama, Y.; Carraway, H.; Belinsky, S.A.; Esteller, M.; Gabrielson, E.; Weitzman, S.; Williams, T.; Herman, J.G.; Baylin, S.B. Hypermethylation of a small CpGuanine-rich region correlates with loss of activator protein-2alpha expression during progression of breast cancer. Cancer Res. 2004, 64, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Peng, J.; Oberley, L.W.; Domann, F.E. Transcriptional inhibition of manganese superoxide dismutase (SOD2) gene expression by DNA methylation of the 5′ CpG island. Free Radic. Biol. Med. 1997, 23, 314–320. [Google Scholar] [CrossRef]
- Cyr, A.R.; Hitchler, M.J.; Domann, F.E. Regulation of SOD2 in cancer by histone modifications and CpG methylation: Closing the loop between redox biology and epigenetics. Antioxid. Redox Signal. 2013, 18, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Drane, P.; Bravard, A.; Bouvard, V.; May, E. Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis. Oncogene 2001, 20, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Bedogni, B.; Anzevino, R.; Colavitti, R.; Palazzotti, B.; Borrello, S.; Galeotti, T. Deregulated manganese superoxide dismutase expression and resistance to oxidative injury in p53-deficient cells. Cancer Res. 2000, 60, 4654–4660. [Google Scholar] [PubMed]
- Hussain, S.P.; Amstad, P.; He, P.; Robles, A.; Lupold, S.; Kaneko, I.; Ichimiya, M.; Sengupta, S.; Mechanic, L.; Okamura, S.; et al. p53-Induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004, 64, 2350–2356. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; Xu, Y.; Chen, Y.; St. Clair, D.K. Specificity protein 1-dependent p53-mediated suppression of human manganese superoxide dismutase gene expression. J. Biol. Chem. 2006, 281, 21698–21709. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; Xu, Y.; St. Clair, D.K. Nuclear factor kappaB- and specificity protein 1-dependent p53-mediated bi-directional regulation of the human manganese superoxide dismutase gene. J. Biol. Chem. 2010, 285, 9835–9846. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chaiswing, L.; Velez, J.M.; Batinic-Haberle, I.; Colburn, N.H.; Oberley, T.D.; St. Clair, D.K. p53 Translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005, 65, 3745–3750. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Oberley, T.D.; Chaiswing, L.; Lin, S.M.; Epstein, C.J.; Huang, T.T.; St. Clair, D. Manganese superoxide dismutase deficiency enhances cell turnover via tumor promoter-induced alterations in AP-1 and p53-mediated pathways in a skin cancer model. Oncogene 2002, 21, 3836–3846. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.P.; Roe, J.H.; Chock, P.B.; Yim, M.B. Transcriptional activation of the human manganese superoxide dismutase gene mediated by tetradecanoylphorbol acetate. J. Biol. Chem. 1999, 274, 37455–37460. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Acquaah-Mensah, G.; Singhal, M.; Malhotra, D.; Biswal, S. Network inference algorithms elucidate Nrf2 regulation of mouse lung oxidative stress. PLoS Comput. Biol. 2008, 4, e1000166. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kiningham, K.K.; Devalaraja, M.N.; Yeh, C.C.; Majima, H.; Kasarskis, E.J.; St. Clair, D.K. An intronic NF-kappaB element is essential for induction of the human manganese superoxide dismutase gene by tumor necrosis factor-alpha and interleukin-1beta. DNA Cell Biol. 1999, 18, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Ping, D.; Boss, J.M. Tumor necrosis factor alpha and interleukin-1beta regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-beta and NF-kappaB. Mol. Cell. Biol. 1997, 17, 6970–6981. [Google Scholar] [CrossRef] [PubMed]
- Kiningham, K.K.; Cardozo, Z.A.; Cook, C.; Cole, M.P.; Stewart, J.C.; Tassone, M.; Coleman, M.C.; Spitz, D.R. All-trans-retinoic acid induces manganese superoxide dismutase in human neuroblastoma through NF-kappaB. Free Radic. Biol. Med. 2008, 44, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Josson, S.; Xu, Y.; Fang, F.; Dhar, S.K.; St. Clair, D.K.; St. Clair, W.H. RelB regulates manganese superoxide dismutase gene and resistance to ionizing radiation of prostate cancer cells. Oncogene 2006, 25, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; St. Clair, D.K.; Fang, F.; Warren, G.W.; Rangnekar, V.M.; Crooks, P.A.; St. Clair, W.H. The radiosensitization effect of parthenolide in prostate cancer cells is mediated by nuclear factor-kappaB inhibition and enhanced by the presence of PTEN. Mol. Cancer Ther. 2007, 6, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Delhalle, S.; Deregowski, V.; Benoit, V.; Merville, M.P.; Bours, V. NF-kappaB-dependent MnSOD expression protects adenocarcinoma cells from TNF-alpha-induced apoptosis. Oncogene 2002, 21, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap ‘n’ Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Solis, L.M.; Behrens, C.; Dong, W.; Suraokar, M.; Ozburn, N.C.; Moran, C.A.; Corvalan, A.H.; Biswal, S.; Swisher, S.G.; Bekele, B.N.; et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010, 16, 3743–3753. [Google Scholar] [CrossRef] [PubMed]
- Hanada, N.; Takahata, T.; Zhou, Q.; Ye, X.; Sun, R.; Itoh, J.; Ishiguro, A.; Kijima, H.; Mimura, J.; Itoh, K.; et al. Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 2012, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, H.; Zhou, J.S.; Cao, J.F.; Zhou, X.B.; Choi, A.M.; Chen, Z.H.; Shen, H.H. Caveolin-1 inhibits expression of antioxidant enzymes through direct interaction with nuclear erythroid 2 p45-related factor-2 (Nrf2). J. Biol. Chem. 2012, 287, 20922–20930. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Sutter, T.R.; Kensler, T.W. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1,2-dimethiole-3-thione. Mol. Med. 2001, 7, 135–145. [Google Scholar] [PubMed]
- Li, M.; Chiu, J.F.; Mossman, B.T.; Fukagawa, N.K. Down-regulation of manganese-superoxide dismutase through phosphorylation of FOXO3a by Akt in explanted vascular smooth muscle cells from old rats. J. Biol. Chem. 2006, 281, 40429–40439. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Yuan, H.; Xu, Z.G.; Lanting, L.; Li, S.L.; Wang, M.; Hu, M.C.; Reddy, M.A.; Natarajan, R. Role of the Akt/FoxO3a pathway in TGF-beta1-mediated mesangial cell dysfunction: A novel mechanism related to diabetic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 3325–3335. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Nguyen, M.; Qin, F.X.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Pennington, J.D.; Bisht, K.S.; Aykin-Burns, N.; Kim, H.S.; Mishra, M.; Sun, L.; Nguyen, P.; Ahn, B.H.; Leclerc, J.; et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int. J. Biol. Sci. 2008, 4, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Kenny, T.C.; Hart, P.; Ragazzi, M.; Sersinghe, M.; Chipuk, J.; Sagar, M.A.K.; Eliceiri, K.W.; LaFramboise, T.; Grandhi, S.; Santos, J.; et al. Selected mitochondrial DNA landscapes activate the SIRT3 axis of the UPRmt to promote metastasis. Oncogene 2017, 36, 4393–4404. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Gonzalez, J.; Zhang, T.; Kamel-Reid, S.; Wells, R.A. The aryl hydrocarbon receptor nuclear translocator (ARNT) modulates the antioxidant response in AML cells. Leuk. Res. 2013, 37, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Solari, C.; Vázquez Echegaray, C.; Cosentino, M.S.; Petrone, M.V.; Waisman, A.; Luzzani, C.; Francia, M.; Villodre, E.; Lenz, G.; Miriuka, S.; et al. Manganese Superoxide Dismutase Gene Expression Is Induced by Nanog and Oct4, Essential Pluripotent Stem Cells’ Transcription Factors. PLoS ONE 2015, 10, e0144336. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bartolome, R.; Patel, V.; Cotrim, A.; Leelahavanichkul, K.; Molinolo, A.A.; Mitchell, J.B.; Gutkind, J.S. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell 2012, 11, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Epperly, M.W.; Bray, J.A.; Esocobar, P.; Bigbee, W.L.; Watkins, S.; Greenberger, J.S. Overexpression of the human manganese superoxide dismutase (MnSOD) transgene in subclones of murine hematopoietic progenitor cell line 32D cl 3 decreases irradiation-induced apoptosis but does not alter G2/M or G1/S phase cell cycle arrest. Radiat. Oncol. Investig. 1999, 7, 331–342. [Google Scholar] [CrossRef]
- Deng, X.; Ewton, D.Z.; Friedman, E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 2009, 69, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.J.; Egry, L.A.; Wong, G.H.; Kaspar, R.L. The 3′ UTR of human MnSOD mRNA hybridizes to a small cytoplasmic RNA and inhibits gene expression. Biochem. Biophys. Res. Commun. 2000, 274, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, A.J.; Fang, Y.; Liu, Y.; Tian, Z.; Mladinov, D.; Matus, I.R.; Ding, X.; Greene, A.S.; Liang, M. MicroRNA-target pairs in human renal epithelial cells treated with transforming growth factor beta 1: A novel role of miR-382. Nucleic Acids Res. 2010, 38, 8338–8347. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; She, K.; Tian, D.; Zhang, P.; Xin, X. miR-146a Inhibits Proliferation and Enhances Chemosensitivity in Epithelial Ovarian Cancer via Reduction of SOD2. Oncol. Res. 2016, 23, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Omar, R.A.; Chyan, Y.J.; Andorn, A.C.; Poeggeler, B.; Robakis, N.K.; Pappolla, M.A. Increased Expression but Reduced Activity of Antioxidant Enzymes in Alzheimer’s Disease. J. Alzheimers Dis. 1999, 1, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Zhong, W.; Zhao, R.; Oberley, T.D. Thioredoxin 1 as a subcellular biomarker of redox imbalance in human prostate cancer progression. Free Radic. Biol. Med. 2010, 49, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Becuwe, P.; Ennen, M.; Klotz, R.; Barbieux, C.; Grandemange, S. Manganese superoxide dismutase in breast cancer: From molecular mechanisms of gene regulation to biological and clinical significance. Free Radic. Biol. Med. 2014, 77, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Barra, D.; Schinina, M.E.; Simmaco, M.; Bannister, J.V.; Bannister, W.H.; Rotilio, G.; Bossa, F. The primary structure of human liver manganese superoxide dismutase. J. Biol. Chem. 1984, 259, 12595–12601. [Google Scholar] [PubMed]
- Miao, L.; St. Clair, D.K. Regulation of superoxide dismutase genes: Implications in disease. Free Radic. Biol. Med. 2009, 47, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Borgstahl, G.E.; Parge, H.E.; Hickey, M.J.; Johnson, M.J.; Boissinot, M.; Hallewell, R.A.; Lepock, J.R.; Cabelli, D.E.; Tainer, J.A. Human mitochondrial manganese superoxide dismutase polymorphic variant Ile58Thr reduces activity by destabilizing the tetrameric interface. Biochemistry 1996, 35, 4287–4297. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Saavedra, D.; McCord, J.M. Paradoxical effects of thiol reagents on Jurkat cells and a new thiol-sensitive mutant form of human mitochondrial superoxide dismutase. Cancer Res. 2003, 63, 159–163. [Google Scholar] [PubMed]
- Zhang, H.J.; Yan, T.; Oberley, T.D.; Oberley, L.W. Comparison of effects of two polymorphic variants of manganese superoxide dismutase on human breast MCF-7 cancer cell phenotype. Cancer Res. 1999, 59, 6276–6283. [Google Scholar] [PubMed]
- Shimoda-Matsubayashi, S.; Matsumine, H.; Kobayashi, T.; Nakagawa-Hattori, Y.; Shimizu, Y.; Mizuno, Y. Structural dimorphism in the mitochondrial targeting sequence in the human manganese superoxide dismutase gene. A predictive evidence for conformational change to influence mitochondrial transport and a study of allelic association in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1996, 226, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.; Khoury, H.; Prip-Buus, C.; Cepanec, C.; Pessayre, D.; Degoul, F. The Ala16Val genetic dimorphism modulates the import of human manganese superoxide dismutase into rat liver mitochondria. Pharmacogenetics 2003, 13, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, J.S.; Gilula, N.B.; Lerner, R.A. On signal sequence polymorphisms and diseases of distribution. Proc. Natl. Acad. Sci. USA 1996, 93, 4471–4473. [Google Scholar] [CrossRef] [PubMed]
- Bag, A.; Bag, N. Target sequence polymorphism of human manganese superoxide dismutase gene and its association with cancer risk: A review. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3298–3305. [Google Scholar] [CrossRef] [PubMed]
- Woodson, K.; Tangrea, J.A.; Lehman, T.A.; Modali, R.; Taylor, K.M.; Snyder, K.; Taylor, P.R.; Virtamo, J.; Albanes, D. Manganese superoxide dismutase (MnSOD) polymorphism, alpha-tocopherol supplementation and prostate cancer risk in the alpha-tocopherol, beta-carotene cancer prevention study (Finland). Cancer Causes Control 2003, 14, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.H.; Carlson, M.D.; Ostrer, H.; Harlap, S.; Stone, A.; Winters, M.; Ambrosone, C.B. Genetic variants in SOD2, MPO, and NQO1, and risk of ovarian cancer. Gynecol. Oncol. 2004, 93, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Berto, M.D.; Bica, C.G.; de Sá, G.P.; Barbisan, F.; Azzolin, V.F.; Rogalski, F.; Duarte, M.M.; da Cruz, I.B. The effect of superoxide anion and hydrogen peroxide imbalance on prostate cancer: An integrative in vivo and in vitro analysis. Med. Oncol. 2015, 32, 251. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Terada, K.; Yano, M.; Sergeev, I.; Mori, M. Oxidative stress inhibits the mitochondrial import of preproteins and leads to their degradation. Exp. Cell Res. 2001, 263, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.; Lewis, V.; Collins, S.J.; Haigh, C.L. Cytosolic caspases mediate mislocalised SOD2 depletion in an in vitro model of chronic prion infection. Dis. Models Mech. 2013, 6, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Luk, E.; Yang, M.; Jensen, L.T.; Bourbonnais, Y.; Culotta, V.C. Manganese activation of superoxide dismutase 2 in the mitochondria of Saccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 22715–22720. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Whittaker, M.M.; Bächinger, H.P.; Whittaker, J.W. Calorimetric studies on the tight binding metal interactions of Escherichia coli manganese superoxide dismutase. J. Biol. Chem. 2004, 279, 27339–27344. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, O. Manganese complexes displaying superoxide dismutase activity: A balance between different factors. Bioorg. Chem. 2011, 39, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; He, Y.X.; Zhao, M.X.; Li, W.F. Structures of native and Fe-substituted SOD2 from Saccharomyces cerevisiae. Acta Crystallogr. 2011, 67, 1173–1178. [Google Scholar] [CrossRef]
- Yamakura, F.; Kobayashi, K.; Furukawa, S.; Suzuki, Y. In vitro preparation of iron-substituted human manganese superoxide dismutase: Possible toxic properties for mitochondria. Free Radic. Biol. Med. 2007, 43, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Beyer, W.F.; Fridovich, I. In vivo competition between iron and manganese for occupancy of the active site region of the manganese-superoxide dismutase of Escherichia coli. J. Biol. Chem. 1991, 266, 303–308. [Google Scholar] [PubMed]
- Vance, C.K.; Miller, A.F. Spectroscopic comparisons of the pH dependencies of Fe-substituted (Mn)superoxide dismutase and Fe-superoxide dismutase. Biochemistry 1998, 37, 5518–5527. [Google Scholar] [CrossRef] [PubMed]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Cavadini, P.; Biasiotto, G.; Poli, M.; Levi, S.; Verardi, R.; Zanella, I.; Derosas, M.; Ingrassia, R.; Corrado, M.; Arosio, P. RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood 2007, 109, 3552–3559. [Google Scholar] [CrossRef] [PubMed]
- Ganini, D.; Petrovich, R.M.; Edwards, L.L.; Mason, R.P. Iron incorporation into MnSOD A (bacterial Mn-dependent superoxide dismutase) leads to the formation of a peroxidase/catalase implicated in oxidative damage to bacteria. Biochim. Biophys. Acta 2015, 1850, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Ansenberger-Fricano, K.; Ganini, D.; Mao, M.; Chatterjee, S.; Dallas, S.; Mason, R.P.; Stadler, K.; Santos, J.H.; Bonini, M.G. The peroxidase activity of mitochondrial superoxide dismutase. Free Radic. Biol. Med. 2013, 54, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Cheng, K.; Zhang, B.; Xu, H.; Cao, Y.; Guo, F.; Feng, X.; Xia, Q. Novel mechanisms for superoxide-scavenging activity of human manganese superoxide dismutase determined by the K68 key acetylation site. Free Radic. Biol. Med. 2015, 85, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Deng, C.X.; Finley, L.W.; Kim, H.S.; Gius, D. SIRT3 is a mitochondrial tumor suppressor: A scientific tale that connects aberrant cellular ROS, the Warburg effect, and carcinogenesis. Cancer Res. 2012, 72, 2468–2472. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.; Guarente, L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhou, Y.; Qiao, C.; Ni, T.; Li, Z.; You, Q.; Guo, Q.; Lu, N. Oroxylin A inhibits glycolysis-dependent proliferation of human breast cancer via promoting SIRT3-mediated SOD2 transcription and HIF1α destabilization. Cell Death Dis. 2015, 6, e1714. [Google Scholar] [CrossRef] [PubMed]
- Alhazzazi, T.Y.; Kamarajan, P.; Verdin, E.; Kapila, Y.L. SIRT3 and cancer: Tumor promoter or suppressor? Biochim. Biophys. Acta 2011, 1816, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Alhazzazi, T.Y.; Kamarajan, P.; Joo, N.; Huang, J.Y.; Verdin, E.; D’Silva, N.J.; Kapila, Y.L. Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer 2011, 117, 1670–1678. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Nihal, M.; Singh, C.K.; Zhong, W.; Liu, X.; Ahmad, N. Pro-Proliferative Function of Mitochondrial Sirtuin Deacetylase SIRT3 in Human Melanoma. J. Investig. Dermatol. 2016, 136, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Swindell, W.R.; Laurent, G.; Vyas, S.; Bulyk, M.L.; Haigis, M.C. Nuclear respiratory factor 2 induces SIRT3 expression. Aging Cell 2015, 14, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Qin, L.; Shi, Y.; Candas, D.; Fan, M.; Lu, C.L.; Vaughan, A.T.; Shen, R.; Wu, L.S.; Liu, R.; et al. CDK4-mediated MnSOD activation and mitochondrial homeostasis in radioadaptive protection. Free Radic. Biol. Med. 2015, 81, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Candas, D.; Fan, M.; Nantajit, D.; Vaughan, A.T.; Murley, J.S.; Woloschak, G.E.; Grdina, D.J.; Li, J.J. CyclinB1/Cdk1 phosphorylates mitochondrial antioxidant MnSOD in cell adaptive response to radiation stress. J. Mol. Cell Biol. 2013, 5, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Castellano, I.; Cecere, F.; De Vendittis, A.; Cotugno, R.; Chambery, A.; Di Maro, A.; Michniewicz, A.; Parlato, G.; Masullo, M.; Avvedimento, E.V.; et al. Rat mitochondrial manganese superoxide dismutase: Amino acid positions involved in covalent modifications, activity, and heat stability. Biopolymers 2009, 91, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Archambaud, C.; Nahori, M.A.; Pizarro-Cerda, J.; Cossart, P.; Dussurget, O. Control of Listeria superoxide dismutase by phosphorylation. J. Biol. Chem. 2006, 281, 31812–31822. [Google Scholar] [CrossRef] [PubMed]
- Bykova, N.V.; Egsgaard, H.; Møller, I.M. Identification of 14 new phosphoproteins involved in important plant mitochondrial processes. FEBS Lett. 2003, 540, 141–146. [Google Scholar] [CrossRef]
- Cai, Z.; Yan, L.J. Protein Oxidative Modifications: Beneficial Roles in Disease and Health. J. Biochem. Pharmacol. Res. 2013, 1, 15–26. [Google Scholar] [PubMed]
- MacMillan-Crow, L.A.; Crow, J.P.; Kerby, J.D.; Beckman, J.S.; Thompson, J.A. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc. Natl. Acad. Sci. USA 1996, 93, 11853–11858. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Thompson, J.A. Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34F) by peroxynitrite. Arch. Biochem. Biophys. 1999, 366, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.J.; Hearn, A.S.; Cabelli, D.E.; Nick, H.S.; Tainer, J.A.; Silverman, D.N. Contribution of human manganese superoxide dismutase tyrosine 34 to structure and catalysis. Biochemistry 2009, 48, 3417–3424. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ying, J.; Jiang, B.; Guo, W.; Adachi, T.; Sharov, V.; Lazar, H.; Menzoian, J.; Knyushko, T.V.; Bigelow, D.; et al. Detection of sequence-specific tyrosine nitration of manganese SOD and SERCA in cardiovascular disease and aging. Am. J. Physiol. 2006, 290, H2220–H2227. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R. Analysis of thiols. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3271–3273. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.K.; Saba, H.; MacMillan-Crow, L.A. Effect of S-nitrosoglutathione on renal mitochondrial function: A new mechanism for reversible regulation of manganese superoxide dismutase activity? Free Radic. Biol. Med. 2013, 56, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Yamakura, F.; Taka, H.; Fujimura, T.; Murayama, K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J. Biol. Chem. 1998, 273, 14085–14089. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Basu, A.; Dai, A.; Heldak, M.; Makino, A. Coronary endothelial dysfunction and mitochondrial reactive oxygen species in type 2 diabetic mice. Am. J. Physiol. Cell Physiol. 2013, 305, C1033–C1040. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Hong, S.S.; Zhang, M.; Cai, Q.Q.; Zhang, M.X.; Xu, C.J. Proteomic alterations of fibroblasts induced by ovarian cancer cells reveal potential cancer targets. Neoplasma 2017. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Decrease | Mechanism | Increase | Mechanism |
|---|---|---|---|---|
| Bladder | ↓Expression (Oncomine) 1 | N/D 2 | ||
| Brain/CNS | ↓Expression [36,37] | N/D | ↑Expression [38,39,40,41,42,43,44,45,46] (Oncomine) | N/D |
| ↓Activity [47] | ↓SIRT3 3: SOD2 hyperacetylation [47] | |||
| oxidation [48] | ||||
| Breast | ↓Expression (Oncomine) | p53 transcriptional inhibition (tumor initiation) [22]; | ↑Expression [22] (Oncomine) | Loss of p53 (tumor progression) [22]; |
| DDB2 4 [49] | NF-κB 8 [50]; | |||
| Epigenetics [51,52] | Nrf2 9 [53] | |||
| ↓Activity | ↓SIRT3: SOD2 hyperacetylation [54] | ↑Activity [55] | N/D | |
| Colorectal | ↓Expression [56] | Increased AP-1 5 occupancy at promoter SNP 6 [56] | ↑Expression [20,57] (Oncomine) | N/D |
| Esophageal | ↓Expression [58] (Oncomine) | N/D | ↑Expression (Oncomine) | N/D |
| Leukemia | ↓Expression (Oncomine) | ↑Expression | ARNT 10 | |
| Liver | ↑Expression (Oncomine) | N/D | ||
| ↓Activity [59] | Ca2+ inhibition of SIRT3 [60] | |||
| Lung | ↑Expression [20,61] | N/D | ||
| ↓Activity | Ala16Val [62] | |||
| Lymphoma | ↑Expression (Oncomine) | N/D | ||
| ↓Activity | ↓SIRT3: SOD2 hyperacetylation [63] | |||
| Melanoma | ↓Expression | LOH 7 [64] | ||
| Multiple Myeloma | ↓Expression | Epigenetic silencing [65,66] | ||
| Ovarian | ↑Expression [27,67] (Oncomine) | Keap1 mutation/Nrf2 activation [68] | ||
| Pancreatic | ↓Expression | Epigenetic silencing [69]; miR-301a [70] | ↑Expression [71] (Oncomine) | N/D |
| ↓Activity | Ala16Val [72] | |||
| Prostate | ↑Expression [20] | Low miR-17* expression [73] | ||
| Renal Clear Cell | ↓Expression | HIF-1α [74] | ||
| ↓Activity | Oxidation [75] | |||
| Sarcoma | ↓Expression (Oncomine) | N/D | ↑Expression (Oncomine) | N/D |
| ↓Activity | Nitration [76] | |||
| Tongue Squamous Cell | ↓Expression | miR-222 [77] | ↑Expression | c-myc 16 [78] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.S.; Gupta Vallur, P.; Phaëton, R.; Mythreye, K.; Hempel, N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants 2017, 6, 86. https://doi.org/10.3390/antiox6040086
Kim YS, Gupta Vallur P, Phaëton R, Mythreye K, Hempel N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants. 2017; 6(4):86. https://doi.org/10.3390/antiox6040086
Chicago/Turabian StyleKim, Yeon Soo, Piyushi Gupta Vallur, Rébécca Phaëton, Karthikeyan Mythreye, and Nadine Hempel. 2017. "Insights into the Dichotomous Regulation of SOD2 in Cancer" Antioxidants 6, no. 4: 86. https://doi.org/10.3390/antiox6040086
APA StyleKim, Y. S., Gupta Vallur, P., Phaëton, R., Mythreye, K., & Hempel, N. (2017). Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants, 6(4), 86. https://doi.org/10.3390/antiox6040086