Trans-Plasma Membrane Electron Transport and Ascorbate Efflux by Skeletal Muscle


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Cell Culture
2.4. DPIP Reduction Assays
2.5. WST-1 Reduction Assays Monitoring tPMET and Ascorbate Efflux
2.6. WST-1 Reduction Assays Monitoring GLUT1 Involvement in tPMET and Ascorbate Efflux
2.7. FLAG-GLUT1 Transfections and GLUT1 Involvement in tPMET
2.8. WST-1 Reduction Assays Monitoring Effects of Glucose on tPMET
2.9. WST-1 Reduction Assays Monitoring Effects of AICAR and Insulin on tPMET and Ascorbate Efflux
3. Results
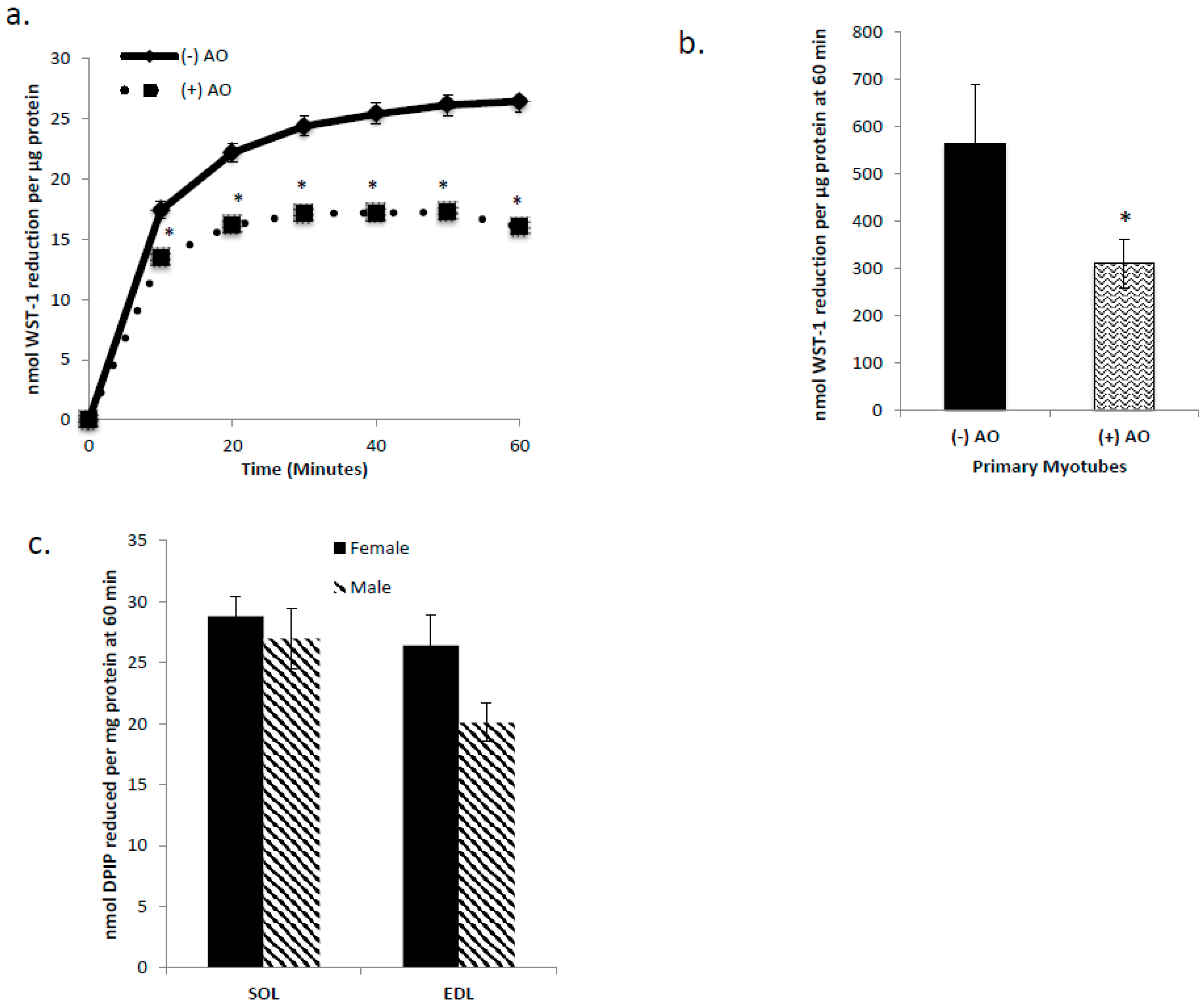
3.1. C2C12 Myotubes, Primary Myotubes, and Isolated Mouse SOL and EDL Are Capable of tPMET
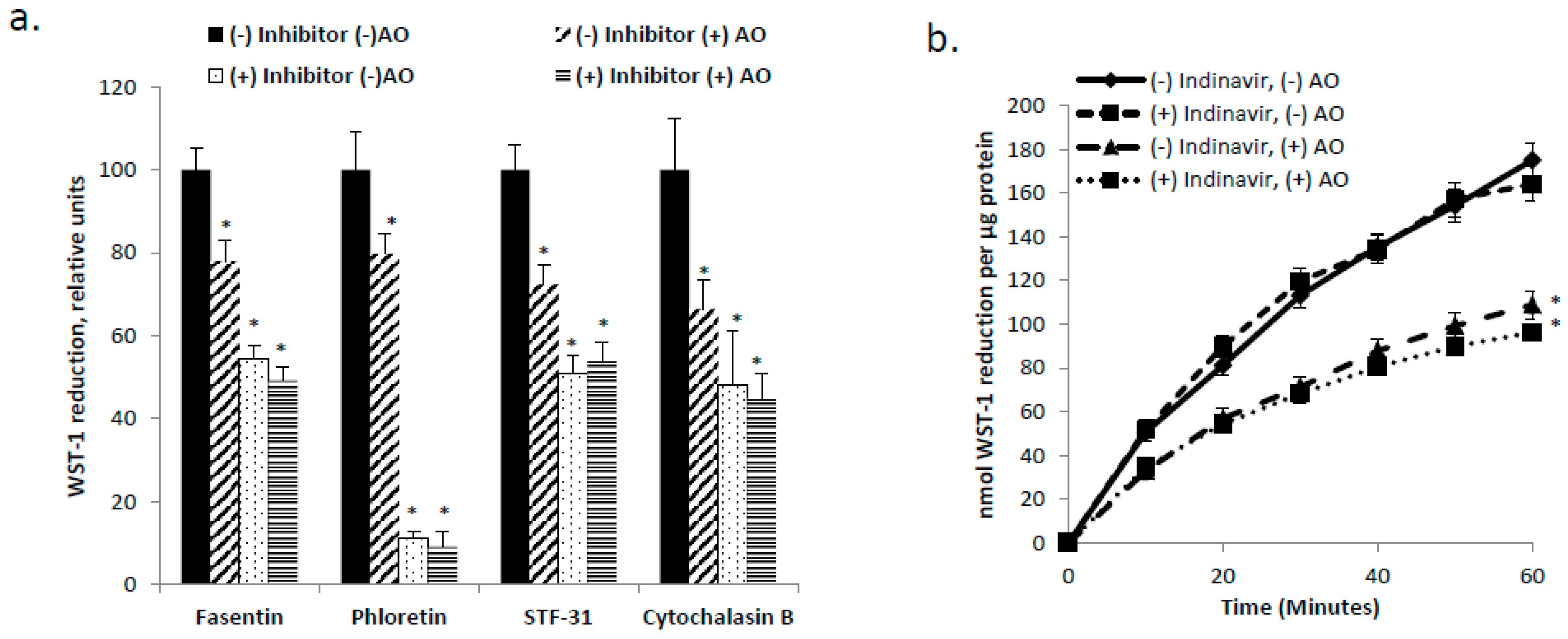
3.2. GLUT1 is the Primary Glucose Transporter Involved in tPMET
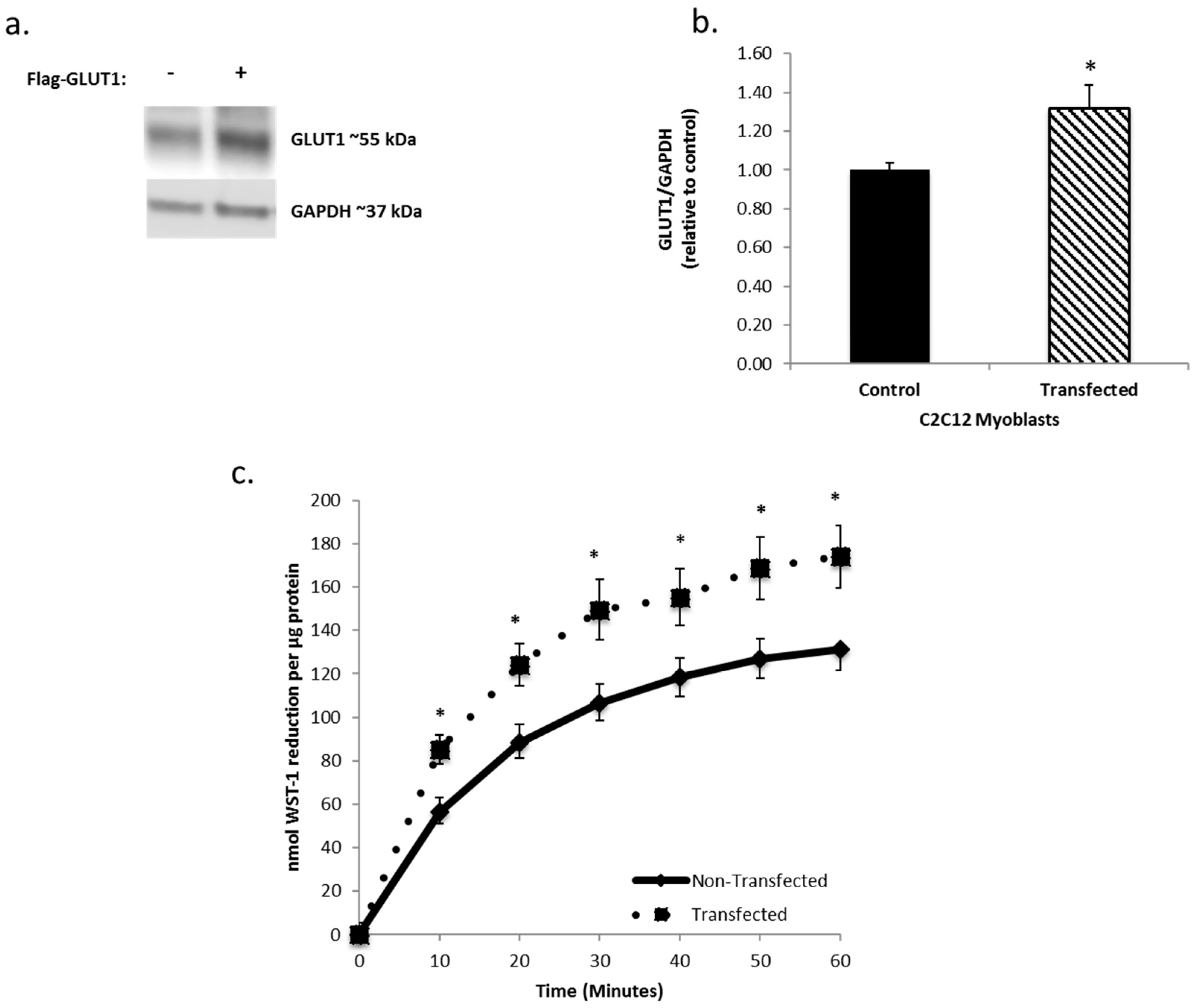
3.3. Increasing GLUT1 Expression Increases tPMET
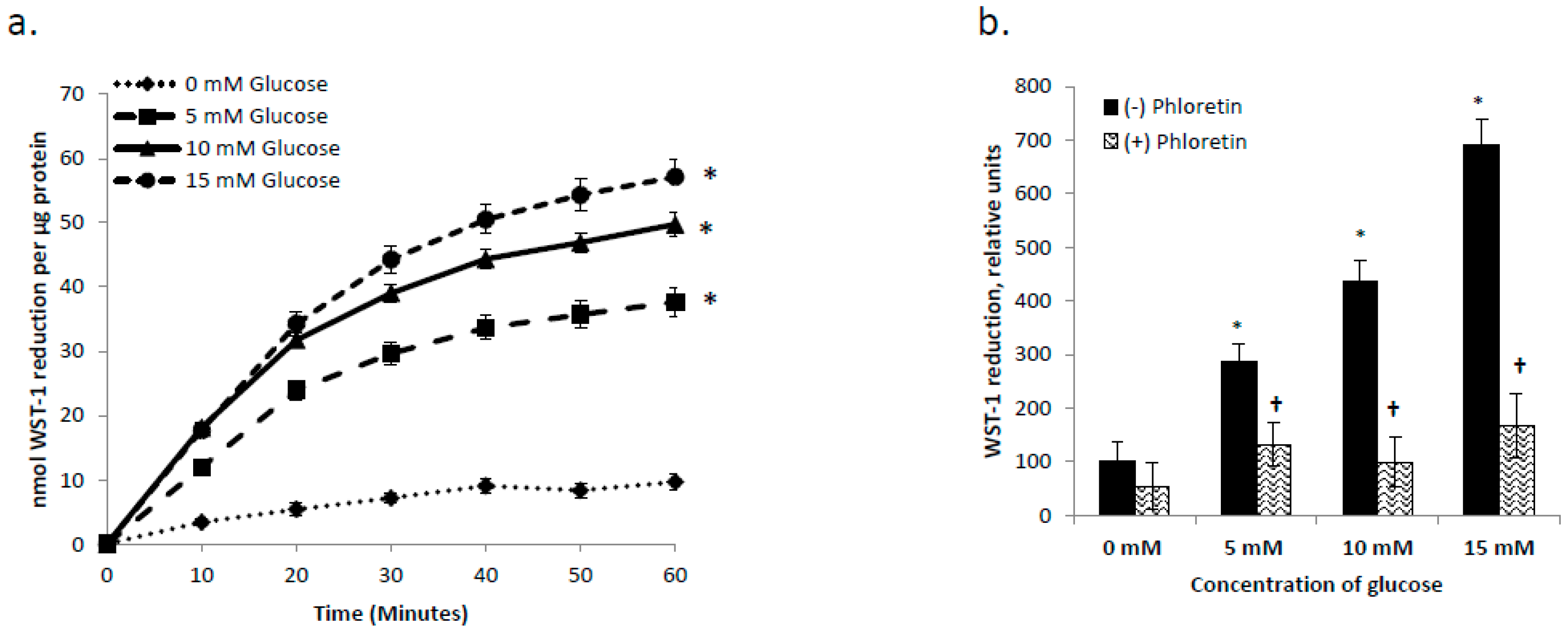
3.4. tPMET is a Glucose Dependent Process
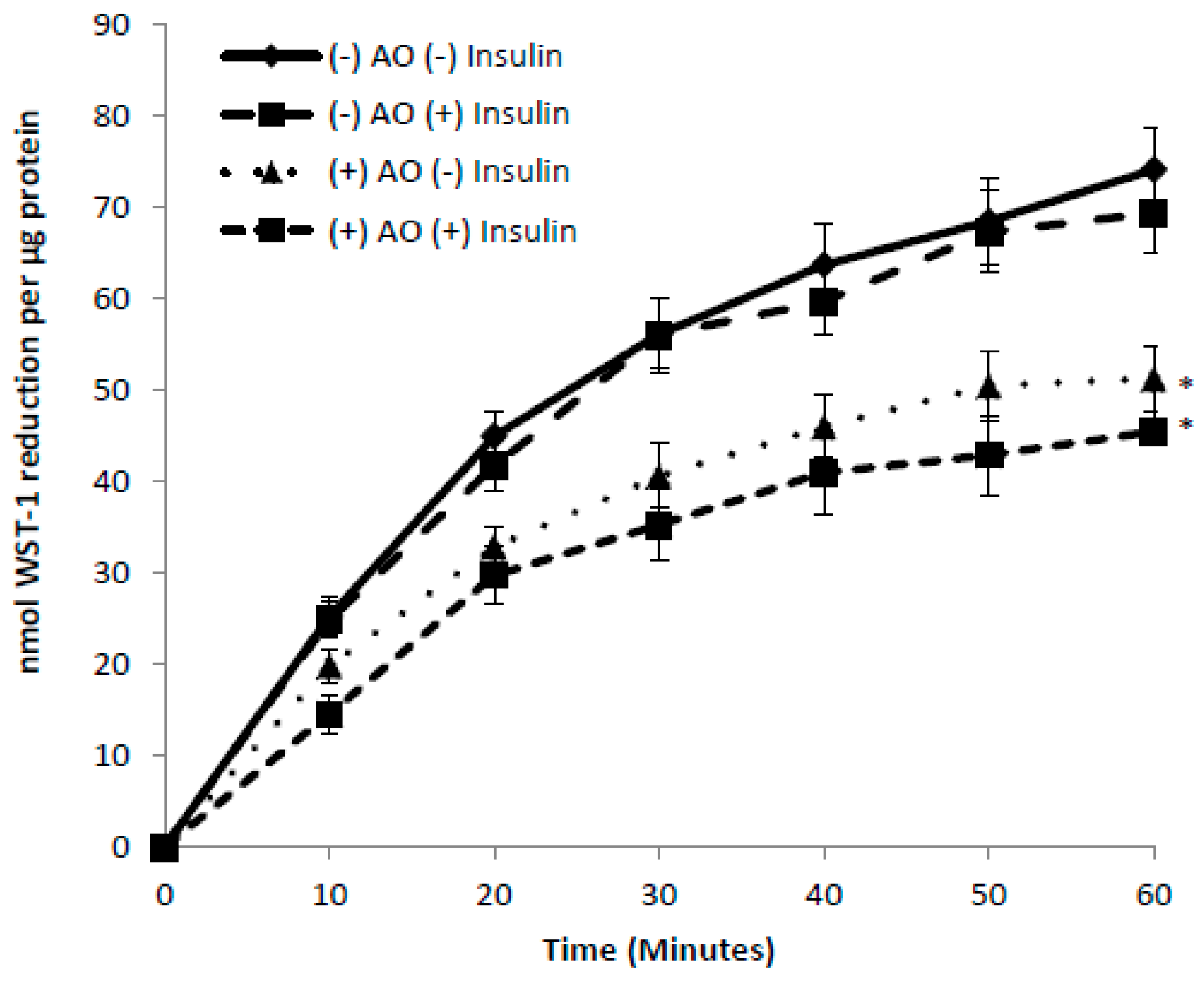
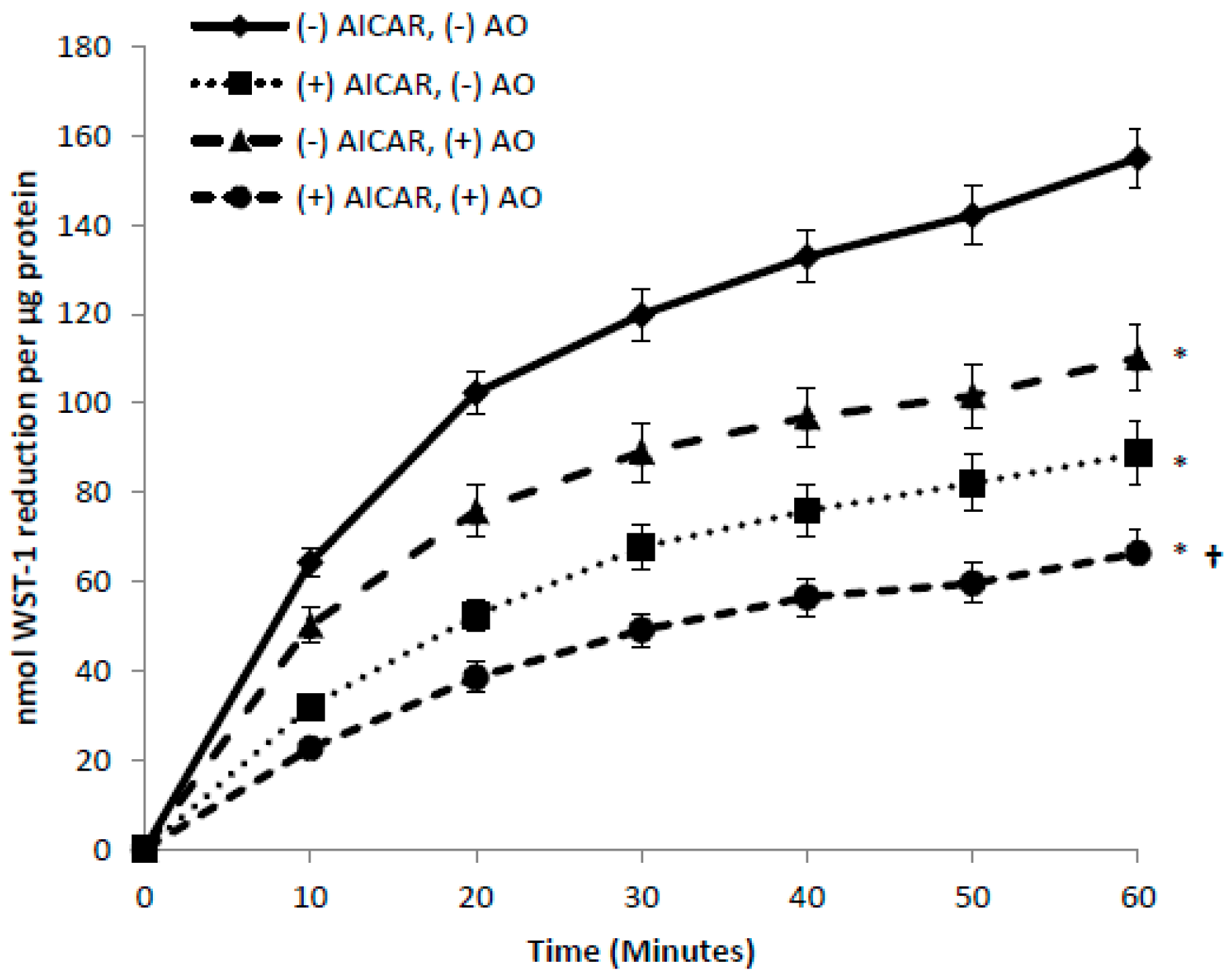
3.5. Effects of Insulin and AICAR on tPMET
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lane, D.J.; Lawen, A. Ascorbate and plasma membrane electron transport—Enzymes vs efflux. Free Radic. Biol. Med. 2009, 47, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H. Mechanisms and function of DUOX in epithelia of the lung. Antioxid. Redox Signal. 2009, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Lawen, A. Transplasma membrane electron transport comes in two flavors. Biofactors 2008, 34, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Ly, J.D.; Lawen, A. Transplasma membrane electron transport: Enzymes involved and biological function. Redox Rep. 2003, 8, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Del, P.D.; Avigliano, L.; Savini, I.; Catani, M.V. Trans-plasma membrane electron transport in mammals: Functional significance in health and disease. Antioxid. Redox Signal. 2011, 14, 2289–2318. [Google Scholar]
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. NOX family NADPH oxidases in liver and in pancreatic islets: A role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008, 36 Pt 5, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Charron, M.J.; Alper, S.L.; Lodish, H.F. A glucose transport protein expressed predominately in insulin-responsive tissues. Proc. Natl. Acad. Sci. USA 1989, 86, 2535–2539. [Google Scholar] [CrossRef] [PubMed]
- Bielli, A.; Scioli, M.G.; Mazzaglia, D.; Doldo, E.; Orlandi, A. Antioxidants and vascular health. Life Sci. 2015, 143, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Savini, I.; Catani, M.V.; Duranti, G.; Ceci, R.; Sabatini, S.; Avigliano, L. Vitamin C homeostasis in skeletal muscle cells. Free Radic. Biol. Med. 2005, 38, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Upston, J.M.; Karjalainen, A.; Bygrave, F.L.; Stocker, R. Efflux of hepatic ascorbate: A potential contributor to the maintenance of plasma vitamin C. Biochem. J. 1999, 342 Pt 1, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Casini, A.F.; Pompella, A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch. Biochem. Biophys. 2010, 500, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Flier, J.S. Regulation of glucose-transporter gene expression in vitro and in vivo. Diabetes Care 1990, 13, 548–564. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B. Dietary regulation of glucose transporter gene expression: Tissue specific effects in adipose cells and muscle. J. Nutr. 1994, 124 (Suppl. 8), 1289s–1295s. [Google Scholar] [PubMed]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [PubMed]
- Burant, C.F.; Bell, G.I. Mammalian facilitative glucose transporters: Evidence for similar substrate recognition sites in functionally monomeric proteins. Biochemistry 1992, 31, 10414–10420. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, A.; DeZutter, J.; Ganguly, A.; Devaskar, S.U. Will the original glucose transporter isoform please stand up! Am. J. Physiol. Endocrinol. Metab. 2009, 297, E836–E848. [Google Scholar] [CrossRef] [PubMed]
- Nedachi, T.; Kanzaki, M. Regulation of glucose transporters by insulin and extracellular glucose in C2C12 myotubes. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E817–E828. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.T.; Pessin, J.E. Intracellular organization of insulin signaling and GLUT4 translocation. Recent Prog. Horm. Res. 2001, 56, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Berti, L.; Kellerer, M.; Capp, E.; Häring, H.U. Leptin stimulates glucose transport and glycogen synthesis in C2C12 myotubes: Evidence for a P13-kinase mediated effect. Diabetologia 1997, 40, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J. Effects of H2O2 on insulin signaling the glucose transport system in mammalian skeletal muscle. Methods Enzymol. 2013, 528, 269–278. [Google Scholar] [PubMed]
- Rumsey, S.C.; Daruwala, R.; Al-Hasani, H.; Zarnowski, M.J.; Simpson, I.A.; Levine, M. Dehydroascorbic acid transport by GLUT4 in Xenopus oocytes and isolated rat adipocytes. J. Biol. Chem. 2000, 275, 28246–28253. [Google Scholar] [CrossRef] [PubMed]
- Andrisse, S.; Koehler, R.M.; Chen, J.E.; Patel, G.D.; Vallurupalli, V.R.; Ratliff, B.A.; Warren, D.E.; Fisher, J.S. Role of GLUT1 in regulation of reactive oxygen species. Redox Biol. 2014, 2, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Andrisse, S.; Patel, G.D.; Chen, J.E.; Webber, A.M.; Spears, L.D.; Koehler, R.M.; Robinson-Hill, R.M.; Ching, J.K.; Jeong, I.; Fisher, J.S. ATM and GLUT1-S490 Phosphorylation Regulate GLUT1 Mediated Transport in Skeletal Muscle. PLoS ONE 2013, 8, e66027. [Google Scholar] [CrossRef] [PubMed]
- Kennett, E.C.; Kuchel, P.W. Redox reactions and electron transfer across the red cell membrane. IUBMB Life 2003, 55, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Gurtoo, H.L.; Johns, D.G. On the interaction of the electron acceptor 2,6-dichlorophenolindophenol with bovine milk xanthine oxidase. J. Biol. Chem. 1971, 246, 286–293. [Google Scholar] [PubMed]
- Berridge, M.V.; Tan, A.S. Trans-plasma membrane electron transport: A cellular assay for NADH- and NADPH-oxidase based on extracellular, superoxide-mediated reduction of the sulfonated tetrazolium salt WST-1. Protoplasma 1998, 205, 74–82. [Google Scholar] [CrossRef]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine Stimulation Promotes Glucose Uptake via Phosphatidylinositol-3 Kinase/Akt Regulation of Glut1 Activity and Trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [PubMed]
- Wallach, J. Enzymatic analysis. A practical guide: By J V Passonneau and O H Lowry. pp 403. Humana Press, Totowa, NJ, USA. 1993. Biochem. Mol. Biol. Educ. 1995, 23, 90. [Google Scholar] [CrossRef]
- Patel, A.Y.; McDonald, T.M.; Spears, L.D.; Ching, J.K.; Fisher, J.S. Ataxia telangiectasia mutated influences cytochrome c oxidase activity. Biochem. Biophys. Res. Commun. 2011, 405, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar] [CrossRef] [PubMed]
- Klip, A.; Marette, A. Acute and chronic signals controlling glucose transport in skeletal muscle. J. Cell Biochem. 1992, 48, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Cura, A.J.; Carruthers, A. AMP kinase regulation of sugar transport in brain capillary endothelial cells during acute metabolic stress. Am. J. Physiol. Cell Physiol. 2012, 303, C806–C814. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zou, M.-H. Regulation of NAD(P)H oxidases by AMPK in cardiovascular systems. Free Radic. Biol. Med. 2012, 52, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Bell, C.M.; Rothbart, S.B.; Moran, R.G. AMP-activated Protein Kinase (AMPK) Control of mTORC1 Is p53- and TSC2-independent in Pemetrexed-treated Carcinoma Cells. J. Biol. Chem. 2015, 290, 27473–27486. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Kennett, E.C.; Ogawa, E.; Agar, N.S.; Godwin, I.R.; Bubb, W.A.; Kuchel, P.W. Investigation of methaemoglobin reduction by extracellular NADH in mammalian erythrocytes. Int. J. Biochem. Cell Biol. 2005, 37, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Guarente, L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 2003, 15, 241–246. [Google Scholar] [CrossRef]
- Matthew, C.K.; van Holde, K.E.; Ahern, K.G. Biochemistry, 3rd ed.; Addison-Wesley: Boston, MA, USA, 2000. [Google Scholar]
- Lenaz, G.; Paolucci, U.; Fato, R.; D’Aurelio, M.; Parenti Castelli, G.; Sgarbi, G.; Biagini, G.; Ragni, L.; Salardi, S.; Cacciari, E. Enhanced activity of the plasma membrane oxidoreductase in circulating lymphocytes from insulin-dependent diabetes mellitus patients. Biochem. Biophys. Res. Commun. 2002, 290, 1589–1592. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Luo, X.; Thangthaeng, N.; Sumien, N.; Chen, Z.; Rutledge, M.A.; Jing, S.; Forster, M.J.; Yan, L.J. Pancreatic mitochondrial complex I exhibits aberrant hyperactivity in diabetes. Biochem. Biophys. Rep. 2017, 11, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Kennett, E.C.; Kuchel, P.W. Plasma membrane oxidoreductases: Effects on erythrocyte metabolism and redox homeostasis. Antioxid. Redox Signal. 2006, 8, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Wolvetang, E.J.; Larm, J.A.; Moutsoulas, P.; Lawen, A. Apoptosis induced by inhibitors of the plasma membrane NADH-oxidase involves Bcl-2 and calcineurin. Cell Growth Differ. 1996, 7, 1315–1325. [Google Scholar] [PubMed]
- Gumaa, K.A.; McLean, P. The pentose phosphate pathway of glucose metabolism. Enzyme profiles and transient and steady-state content of intermediates of alternative pathways of glucose metabolism in Krebs ascites cells. Biochem. J. 1969, 115, 1009–1029. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Scott, J.W.; Pan, D.A.; Hudson, E.R. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003, 546, 113–120. [Google Scholar] [CrossRef]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eccardt, A.M.; Bell, T.P.; Mattathil, L.; Prasad, R.; Kelly, S.C.; Fisher, J.S. Trans-Plasma Membrane Electron Transport and Ascorbate Efflux by Skeletal Muscle. Antioxidants 2017, 6, 89. https://doi.org/10.3390/antiox6040089
Eccardt AM, Bell TP, Mattathil L, Prasad R, Kelly SC, Fisher JS. Trans-Plasma Membrane Electron Transport and Ascorbate Efflux by Skeletal Muscle. Antioxidants. 2017; 6(4):89. https://doi.org/10.3390/antiox6040089
Chicago/Turabian StyleEccardt, Amanda M., Thomas P. Bell, Lyn Mattathil, Rohan Prasad, Shannon C. Kelly, and Jonathan S. Fisher. 2017. "Trans-Plasma Membrane Electron Transport and Ascorbate Efflux by Skeletal Muscle" Antioxidants 6, no. 4: 89. https://doi.org/10.3390/antiox6040089
APA StyleEccardt, A. M., Bell, T. P., Mattathil, L., Prasad, R., Kelly, S. C., & Fisher, J. S. (2017). Trans-Plasma Membrane Electron Transport and Ascorbate Efflux by Skeletal Muscle. Antioxidants, 6(4), 89. https://doi.org/10.3390/antiox6040089