The Influence of Light on Reactive Oxygen Species and NF-кB in Disease Progression

 , ,
, ,  and
and 
Abstract
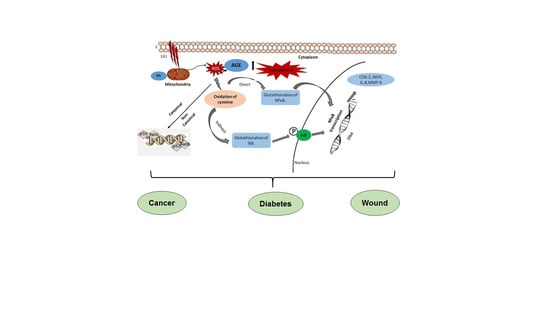
:
{kind=link}
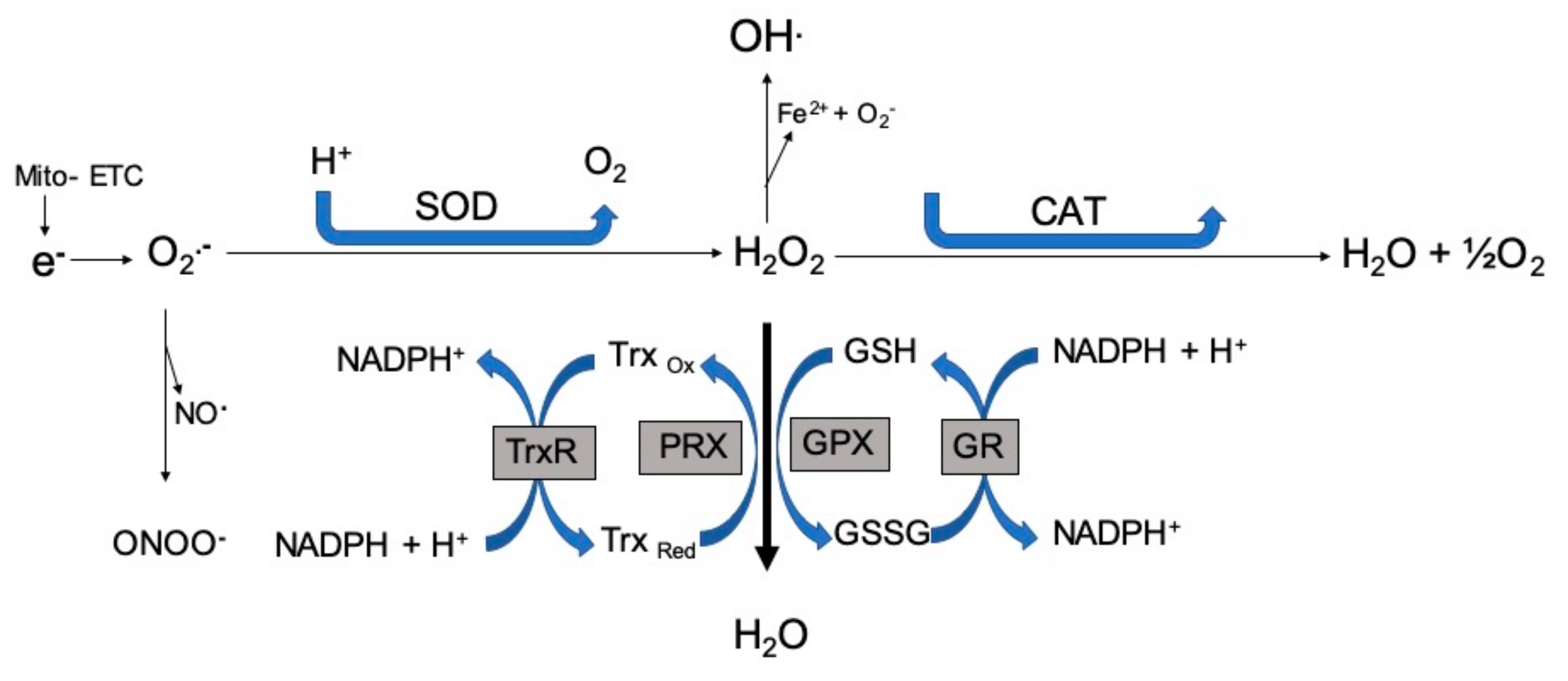{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
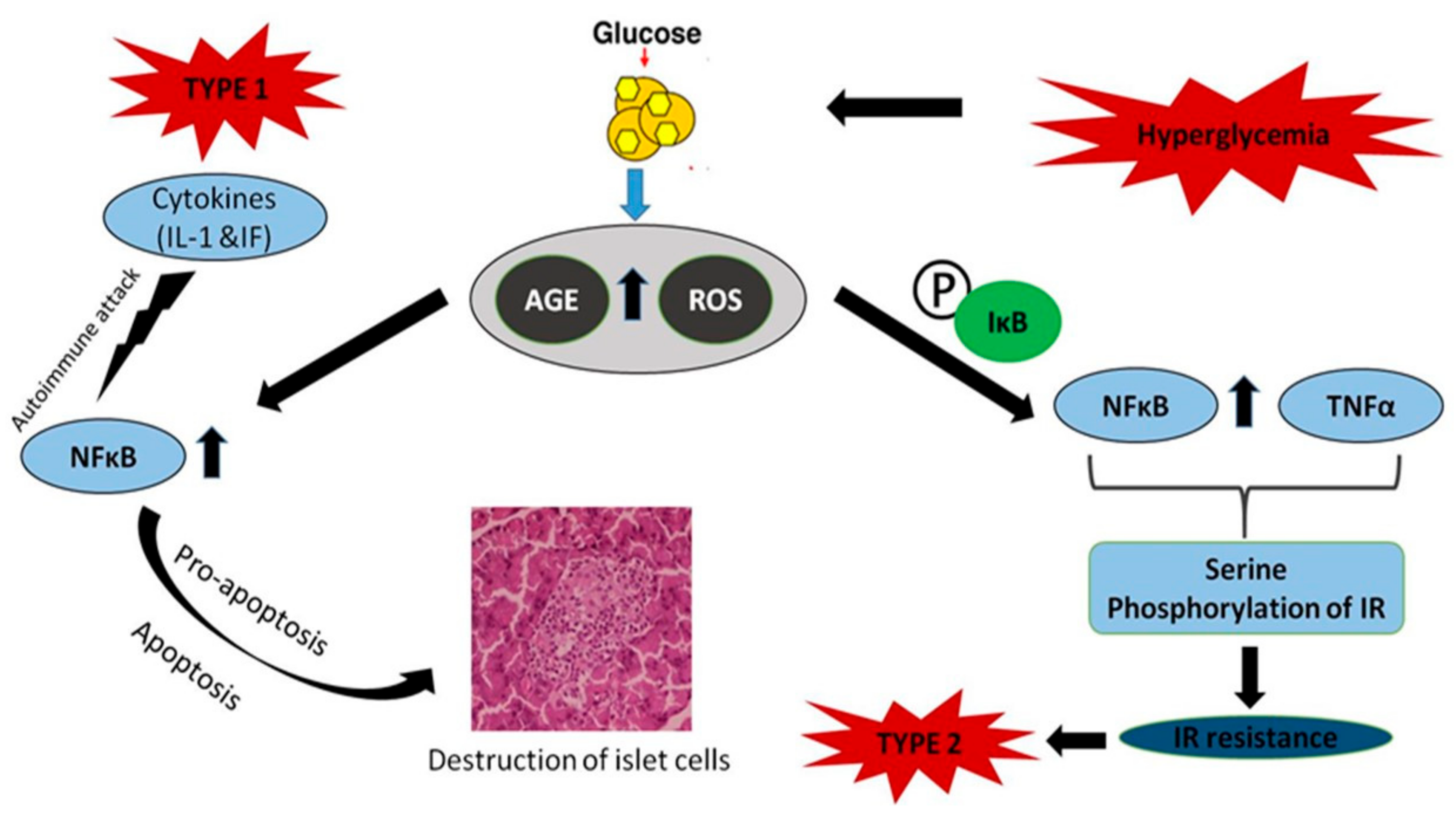{kind=link}
1. Introduction
2. Sources and Stimuli of ROS
2.1. Oxidative Stress and Cancer
2.2. Oxidative Stress and Diabetes
2.3. Oxidative Stress and Wound Healing
2.4. Influence of Light on Oxidative Stress
2.5. Effect of Light on NF-кB Activation and ROS Regulation.
2.6. Reciprocal Influence of ROS and NF-кB Activation
2.7. NF-кB in Cancer Progression
2.8. NF-кB in the Pathogenesis of Diabetes
2.9. NF-кB in Wound Healing
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liaison in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Stanicka, J.; Russell, E.G.; Woolley, J.F.; Cotter, T.G. NADPH oxidase-generated hydrogen peroxide induces DNA damage in mutant FLT3-expressing leukemia cells. J. Biol. Chem. 2015, 290, 9348–9361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, K.; Wu, Y.; Meitzler, J.L.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Antony, S.; Doroshow, J.H. NADPH oxidases and cancer. Clin. Sci. 2015, 128, 863–875. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Gonzalez, C.D.; Lee, M.S.; Marchetti, P.; Pietropaolo, M.; Towns, R.; Vaccaro, M.I.; Watada, H.; Wiley, J.W. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 2011, 7, 2–11. [Google Scholar] [CrossRef]
- Krakauer, T. Inflammasome, mTORC1 activation, and metabolic derangement contribute to the susceptibility of diabetics to infections. Med. Hypotheses 2015, 85, 997–1001. [Google Scholar] [CrossRef]
- O’Toole, E.A.; Goel, M.; Woodley, D.T. Hydrogen peroxide inhibits human keratinocyte migration. Dermatol. Surg. 1996, 22, 525–529. [Google Scholar] [CrossRef]
- Auf Dem Keller, U.; Angelika, K.; Susanne, B.; Werner, S. Reactive oxygen species and their detoxification in healing skin wounds. J. Investig. Dermatol. Symp. Proc. 2006, 11, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, L.; Olmo-Aguado, S.D.; Valenzuela, P.L.; Winge, K.; Iglesias-Soler, E.; Arguelles-Luis, J.; Alvarez-Valle, S.; Parcero-Iglesias, G.J.; Fernandez-Martinez, A.; Lucia, A. Photobiomodulation in Parkinson’s disease: A randomized controlled trial. Brain Stimul. 2019, 12, 810–812. [Google Scholar] [CrossRef]
- Chow, R.T.; Johnson, M.I.; Lopes-Martins, R.A.; Bjordal, J.M. Efficacy of low-level laser therapy in the management of neck pain: A systematic review and meta-analysis of randomised placebo or active-treatment controlled trials. Lancet 2009, 374, 1897–1908. [Google Scholar] [CrossRef]
- Lavery, L.A.; Murdoch, D.P.; Williams, J.; Lavery, D.C. Does anodyne light therapy improve peripheral neuropathy in diabetes? A double-blind, sham-controlled, randomized trial to evaluate monochromatic infrared photoenergy. Diabetes Care 2008, 31, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnall, D.A.; Nelson, A.G.; Lopez, L.; Sanz, N.; Iversen, L.; Sanz, I.; Stambaugh, L.; Arnall, S.B. The restorative effects of pulsed infrared light therapy on significant loss of peripheral protective sensation in patients with long-term type 1 and type 2 diabetes mellitus. Acta Diabetol. 2006, 43, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Brosseau, L.; Robinson, V.; Wells, G.; Debie, R.; Gam, A.; Harman, K.; Morin, M.; Shea, B.; Tugwell, P. Low level laser therapy (Classes I, II and III) for treating rheumatoid arthritis. Cochrane Database Syst. Rev. 2005, CD002049. [Google Scholar] [CrossRef]
- Huang, Z.; Ma, J.; Chen, J.; Shen, B.; Pei, F.; Kraus, V.B. The effectiveness of low-level laser therapy for nonspecific chronic low back pain: A systematic review and meta-analysis. Arthritis Res. Ther. 2015, 17, 360. [Google Scholar] [CrossRef] [Green Version]
- Yousefi-Nooraie, R.; Schonstein, E.; Heidari, K.; Rashidian, A.; Pennick, V.; Akbari-Kamrani, M.; Irani, S.; Shakiba, B.; Mortaz Hejri, S.A.; Mortaz Hejri, S.O. Low level laser therapy for nonspecific low-back pain. Cochrane Database Syst. Rev. 2008, CD005107. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.; Dai, T.; Sharma, S.K.; Huang, Y.Y.; Carroll, J.D.; Hamblin, M.R. The nuts and bolts of low-level laser (light) therapy. Ann. Biomed. Eng. 2012, 40, 516–533. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.H.; Kwok, S.J.J. Light in diagnosis, therapy and surgery. Nat. Biomed. Eng. 2017, 1, 0008. [Google Scholar] [CrossRef]
- Passarella, S.; Casamassima, E.; Molinari, S.; Pastore, D.; Quagliariello, E.; Catalano, I.M.; Cingolani, A. Increase of proton electrochemical potential and ATP synthesis in rat liver mitochondria irradiated in vitro by helium-neon laser. FEBS Lett. 1984, 175, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Lynnyk, A.; Lunova, M.; Jirsa, M.; Egorova, D.; Kulikov, A.; Kubinova, S.; Lunov, O.; Dejneka, A. Manipulating the mitochondria activity in human hepatic cell line Huh7 by low-power laser irradiation. Biomed. Opt. Express 2018, 9, 1283–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunova, M.; Smolkova, B.; Uzhytchak, M.; Janouskova, K.Z.; Jirsa, M.; Egorova, D.; Kulikov, A.; Kubinova, S.; Dejneka, A.; Lunov, O. Light-induced modulation of the mitochondrial respiratory chain activity: Possibilities and limitations. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Monro, S.; Colon, K.L.; Yin, H.; Roque, J., 3rd; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition metal complexes and photodynamic therapy from a tumor-centered approach: Challenges, opportunities, and highlights from the development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xu, H.; Meyers, A.D.; Musani, A.I.; Wang, L.; Tagg, R.; Barqawi, A.B.; Chen, Y.K. Photodynamic therapy for treatment of solid tumors—Potential and technical challenges. Technol. Cancer Res. Treat. 2008, 7, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Jayavelu, A.K.; Moloney, J.N.; Bohmer, F.D.; Cotter, T.G. NOX-driven ROS formation in cell transformation of FLT3-ITD-positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef] [Green Version]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Groeger, G.; Quiney, C.; Cotter, T.G. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid. Redox Signal. 2009, 11, 2655–2671. [Google Scholar] [CrossRef]
- Kroller-Schon, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species—Studies in white blood cells and in animal models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Terberg, J.R.; Perevoshchikova, I.V. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic. Biol. Med. 2012, 53, 1807–1817. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 11, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygenspecies. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, G.P.; Muller, A.L.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Hart, P.C.; Mao, M.; de Abreu, A.L.; Ansenberger-Fricano, K.; Ekoue, D.N.; Ganini, D.; Kajdacsy-Balla, A.; Diamond, A.M.; Minshall, R.D.; Consolaro, M.E.; et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signaling in cancer. Nat. Commun. 2015, 6, 6053. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Signal transduction by mitochondrial oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef] [Green Version]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.Y.; Woollard, A.C.; Wolff, S.P. Hydrogen peroxide production during experimental protein glycation. FEBS Lett. 1990, 268, 69–71. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, M.; Heinecke, J.W.; Chait, A. Pathophysiological concentrations of glucose promote oxidative modification of low density lipoprotein by a superoxide dependent pathway. J. Clin. Investig. 1994, 94, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creage, A.M. Advanced Glycation End Products Sparking the Development of Diabetic Vascular Injury. Basic Sci. Clin. 2006, 114, 597–605. [Google Scholar]
- McCarthy, A.D.; Etcheverry, S.B.; Cortizo, A.M. Effect of advanced glycation end products on the secretion of insulin-like growth factor-I and its binding proteins: Role in osteoblast development. Acta Diabetol. 2001, 38, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. Recent progress in advanced glycation end products and diabetic complications. Diabetes 1997, 46, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.K.; Bierhaus, A.; Schiekofer, S.; Tritschler, H.; Ziegler, R.; Nawroth, P.P. The role of oxidative stress and NF-ĸB activation in late diabetic complications. BioFactors 1999, 10, 157–167. [Google Scholar] [CrossRef]
- Ceriello, A. Cardiovascular effects of acute hyperglycemia: Pathophysiological under pinnings. Diabetes Vasc. Dis. Res. 2008, 5, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Butler, R.; Morris, A.D.; Belch, J.J.; Hill, A.; Struthers, A.D. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension 2000, 35, 746–751. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Dipietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef]
- Kauppila, J.H.; Stewart, J.B. Mitochondrial DNA: Radically free of free-radical driven mutations. Biochim. Biophys. Acta 2015, 1847, 1354–1361. [Google Scholar] [CrossRef] [Green Version]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef]
- Hornig-Do, H.T.; von Kleist-Retzow, J.C.; Lanz, K.; Wickenhauser, C.; Kudin, A.P.; Kunz, W.S.; Wiesner, R.J.; Schauen, M. Human epidermal keratinocytes accumulate superoxide due to low activity of Mn-SOD, leading to mitochondrial functional impairment. J. Investig. Dermatol. 2007, 127, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Sen, C.K.; Roy, S. Redox signals in wound healing. Biochim. Biophys. Acta 2008, 1780, 1348–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblin, M.R. Shining light on the head: Photobiomodulation for brain disorders. BBA. Clin. 2016, 6, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Freitas, L.F.; Hamblin, M.R. Proposed mechanisms of photobiomodulation or low-level light therapy. IEEE J. Sel. Top Quant. Electron 2016, 22, 4–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waypa, G.B.; Smith, K.A.; Schumacker, P.T. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Asp. Med. 2016, 47–48, 76–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, J.J.; Lanzafame, R.J.; Arany, P.R. Low-level light/laser therapy versus photobiomodulation therapy. Photomed. Laser Surg. 2015, 33, 183–184. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Aaron, C.H.; Arany, P.R.; Hamblin, M.R. Role of reactive oxygen species in low level light theraphy. In Mechanisms for Low-Light Theraphy IV; Michael, R.H., Ronald, W.W., Juanita, A., Eds.; SPIE: San Jose, CA, USA, 2009; pp. 716502–716511. [Google Scholar]
- Saini, R.; Lee, N.V.; Liu, K.Y.P.; Poh, C.F. Prospects in the Application of Photodynamic Therapy in Oral Cancer and Premalignant Lesions. Cancers 2016, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Mitton, D.; Ackroyd, R. A brief overview of photodynamic therapy in Europe. Photodiagn. Photodyn. Ther. 2008, 5, 103–111. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part two-cellular signaling, cell metabolism and modes of cell death. Photodiagn. Photodyn. Ther. 2005, 2, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Singleton, D.A.; Hang, C.; Szymanski, M.J.; Meyer, M.P.; Leach, A.G.; Kuwata, K.T.; Chen, J.S.; Greer, A.; Foote, C.S.; Houk, K.N. Mechanism of ene reactions of singlet oxygen. A two-step no-intermediate mechanism. J. Am. Chem. Soc. 2003, 125, 1319–1328. [Google Scholar] [CrossRef]
- Bansal, A.; Yang, F.; Xi, T.; Zhang, Y.; Ho, J.S. In vivo wireless photonic photodynamic therapy. Proc. Natl. Acad. Sci. USA 2018, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-kB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappa B signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercurio, F.; DiDonato, J.A.; Rosette, C.; Karin, M. p105 and p98 precursor proteins play an active role in NF-kappa B-mediated signal transduction. Genes Dev. 1993, 7, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [Green Version]
- Matthews, J.R.; Kaszubska, W.; Turcatti, G.; Wells, T.N.; Hay, R.T. Role of cysteine62 in DNA recognition by the P50 subunit of NF-kappa B. Nucleic Acids Res. 1993, 21, 1727–1734. [Google Scholar] [CrossRef]
- Klatt, P.; Lamas, S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 2000, 267, 4928–4944. [Google Scholar] [CrossRef]
- Pineda-Molinam, E.; Klatt, P.; Vazquez, J.; Mariana, A.; Garcia de Lacabo, M.; Parez-Sala, D.; Lamas, S. Glutathionylation of the p50 subunit of NF-kappaB: A mechanism for redoxinduced inhibition of DNA binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar] [CrossRef]
- Kil, I.S.; Kim, S.Y.; Park, J.W. Glutathionylation regulates IkappaB. Biochem. Biophys. Res. Commun. 2008, 373, 169–173. [Google Scholar] [CrossRef]
- Karin, M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef]
- Saccani, S.; Pantano, S.; Natoli, G. Modulation of NF-kappa-B activity by exchange of dimers. Mol. Cell 2003, 11, 1563–1574. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Marques, V.; Marinho, H.S.; Cyrne, L.; Antunes, F. Role of hydrogen peroxide in NF-κB activation: From inducer to modulator. Antioxid. Redox Signal. 2009, 11, 2223–2243. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Manabe, T.; Oka, S.; Kamata, K.; Hirata, H. Hydrogen peroxide activates IκB kinases through phosphorylation of serine residues in the activation loops. FEBS Lett. 2002, 519, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-κB through tyrosine phosphorylation of IκBα and serine phosphorylation of p65: Evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 26, 24233–24241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.S.; Matthews, D.E.; et al. Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Na, H.J.; Kim, C.K.; Kim, J.Y.; Ha, K.S.; Lee, H.; Chung, H.T.; Kwon, H.J.; Kwon, Y.G.; Kim, Y.M. The non-provitamin a carotenoid, lutein, inhibits NF-κB-dependent gene expression through redox-based regulation of the phosphatidylinositol 3-kinase/PTEN/Akt and NF-κB-inducing kinase pathways: Role of H2O2 in NF-κB activation. Free Radic. Biol. Med. 2008, 45, 885–896. [Google Scholar] [CrossRef]
- Nakano, H.; Nakajima, A.; Sakon-Komazawa, S.; Piao, J.H.; Xue, X.; Okumura, K. Reactive oxygen species mediate crosstalk between NF-κB and JNK. Cell Death Differ. 2006, 13, 730–737. [Google Scholar] [CrossRef]
- Ventura, J.J.; Cogswell, P.; Flavell, R.A.; Baldwin, A.S., Jr.; Davis, R.J. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004, 18, 2905–2915. [Google Scholar] [CrossRef] [Green Version]
- Kabe, Y.; Ando, K.; Hirao, S.; Yoshida, M.; Handa, H. Redox regulation of NF-κB activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox Signal. 2005, 7, 395–403. [Google Scholar] [CrossRef]
- Saito, Y.; Nishio, K.; Ogawa, Y.; Kimata, J.; Kinumi, T.; Yoshida, Y.; Noguchi, N.; Niki, E. Turning point in apoptosis/ necrosis induced by hydrogen peroxide. Free Radic. Res. 2006, 40, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Gilmore, T.D. Good cop, bad cop: The different faces of NF-kappa B. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ak, P.; Levine, A.J. p53 and NF-kappa B: Different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010, 24, 3643–3652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Kang, K.A.; Kim, K.C.; Na, S.Y.; Chang, W.Y.; Kim, G.Y.; Kim, H.S.; Hyun, J.W. Oxidative stress causes epigenetic alteration of CDX1 expression in colorectal cancer cells. Gene 2013, 524, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Marusawa, H.; Endo, Y.; Chiba, T. Inflammation- mediated genomic instability: Roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci. 2012, 103, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Takai, A.; Marusawa, H.; Minaki, Y.; Watanabe, T.; Nakase, H.; Kinoshita, K.; Tsujimoto, G.; Chiba, T. Targeting activation-induced cytidine deaminase prevents colon cancer development despite persistent colonic inflammation. Oncogene 2012, 31, 1733–1742. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-kappa B signaling: Balancing life and death--a new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632. [Google Scholar] [CrossRef] [Green Version]
- De Smaele, E.; Zazzeroni, F.; Papa, S.; Nguyen, D.U.; Jin, R.; Jones, J.; Cong, R.; Franzoso, G. Induction of gadd45beta by NF-kappa B downregulates pro-apoptotic JNK signaling. Nature 2001, 414, 308–313. [Google Scholar] [CrossRef]
- Chen, C.; Edelstein, L.C.; Gélinas, C. The Rel/NFkappaB family directly activates expression of the apoptosis inhibitor Bcl-x (L). Mol. Cell Biol. 2000, 20, 2687–2695. [Google Scholar] [CrossRef]
- Cardozo, A.K.; Heimberg, H.; Heremans, Y.; Leeman, R.; Kutlu, B.; Kruhøffer, M.; Ørntoft, T.; Eizirik, D.L. A comprehensive analysis of cytokine-induced and nuclear factor-B dependent genes in primary rat pancreatic beta cells. J. Biol. Chem. 2001, 276, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Heimberg, H.; Heremans, Y.; Jobin, C.; Leemans, R.; Cardozo, A.K.; Darville, M.; Eizirik, D.L. Inhibition of cytokine induced NF-B activation by adenovirus-mediated expression of a NF- B super-repressor prevents beta cell apoptosis. Diabetes 2001, 50, 2219–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoukakis, N.; Rudert, W.A.; Trucco, M.; Robbins, P.D. Protection of human islets from the effects of interleukin- by adenoviral gene transfer of an IB repressor. J. Biol. Chem. 2000, 47, 36509–36513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabley, J.G.; Haskó, G.; Liaudet, L.; Soriano, F.G.; Southan, G.J.; Salzman, A.L.; Szabó, C. NFB1 (p50)-deficient mice are not susceptible to multiple low-dose streptozotocin induced diabetes. J. Endocrinol. 2002, 173, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norlin, S.; Ahlgren, U.; Edlund, H. Nuclear factor-ĸB activity in ĸ-cells is required for glucose-stimulated insulin secretion. Diabetes 2005, 54, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK- links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef]
- Suzawa, M.; Takada, I.; Yanagisawa, J.; Ohtake, F.; Ogawa, S.; Yamauchi, T.; Kadowaki, T.; Takeuchi, Y.; Shibuya, H.; Gotoh, Y.; et al. Cytokines suppress adipogenesis and PPAR-gamma function through the TAK1/TAB1/NIK cascade. Nat. Cell Biol. 2003, 5, 224–230. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Jobin, C.; Sartor, R.B. The IκB/NF-κB system: A key determinant of mucosal inflammation and protection. Am. J. Physiol. Cell Physiol. 2000, 278, 451–462. [Google Scholar] [CrossRef]
- Na, J.; Lee, K.; Na, W.; Shin, J.Y.; Lee, M.J.; Yune, T.Y.; Lee, H.K.; Jung, H.S.; Kim, W.S.; Ju, B.G. Histone H3K27 demethylase JMJD3 in cooperation with NF-κB regulates keratinocyte wound healing. J. Investig. Dermatol. 2016, 136, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Schreml, S.; Szeimies, R.M.; Karrer, S.; Heinlin, J.; Landthaler, M.; Babilas, P. Wound healing in 21st century. J. Am. Acad. Dermatol. 2010, 63, 866–881. [Google Scholar] [CrossRef]
- Lizzul, P.F.; Aphale, A.; Malaviya, R.; Sun, Y.; Masud, S.; Dombrovskiy, V.; Gottlieb, A.B. Differential expression of phosphorylated NF-κB/ RelA in normal and psoriatic epidermis and downregulation of NF-κB in response to treatment with etanercept. J. Investig. Dermatol. 2005, 124, 1275–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrozovaa, N.; Ulrichova, J.; Galandakova, A. Models for the study of skin wound healing. The role of Nrf2 and NF-κB. Biomed. Pap. Med. Fac. Palacky Univ. Olomouc Czech Repub. 2017, 161, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar Rajendran, N.; George, B.P.; Chandran, R.; Tynga, I.M.; Houreld, N.; Abrahamse, H. The Influence of Light on Reactive Oxygen Species and NF-кB in Disease Progression. Antioxidants 2019, 8, 640. https://doi.org/10.3390/antiox8120640
Kumar Rajendran N, George BP, Chandran R, Tynga IM, Houreld N, Abrahamse H. The Influence of Light on Reactive Oxygen Species and NF-кB in Disease Progression. Antioxidants. 2019; 8(12):640. https://doi.org/10.3390/antiox8120640
Chicago/Turabian StyleKumar Rajendran, Naresh, Blassan P. George, Rahul Chandran, Ivan Mfouo Tynga, Nicolette Houreld, and Heidi Abrahamse. 2019. "The Influence of Light on Reactive Oxygen Species and NF-кB in Disease Progression" Antioxidants 8, no. 12: 640. https://doi.org/10.3390/antiox8120640
APA StyleKumar Rajendran, N., George, B. P., Chandran, R., Tynga, I. M., Houreld, N., & Abrahamse, H. (2019). The Influence of Light on Reactive Oxygen Species and NF-кB in Disease Progression. Antioxidants, 8(12), 640. https://doi.org/10.3390/antiox8120640