Glucose as a Major Antioxidant: When, What for and Why It Fails?

 ,
,  ,
, 
 and
and 
Abstract
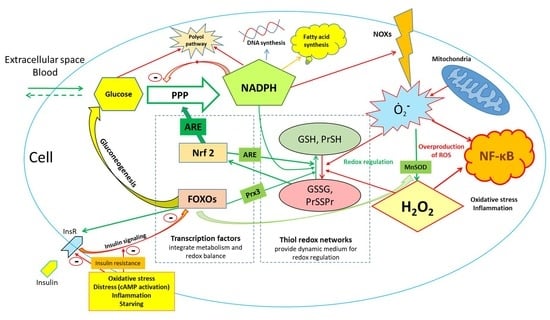
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Basic Overview of Glucose Metabolism and Its Role in Maintenance of Redox Balance
3. Oxidative PPP Is Thermodynamically More Favorable Compared to Upper Glycolysis under Conditions of Limited Glucose Supply
4. The Epidemiological Evidence of Glucose Overload in Human Population: Current vs. Historical Nutritional/Behavioral Patterns and a Growing Potential of Pharmacological Interventions
5. Glycogen Protects against (Not Only) Oxidative Stress
6. Epigenetics and Posttranslational Protein Modification Modulate Oxidative Stress Responses
7. The Evidence from Glucose-6 Phosphate Dehydrogenase Deficiency
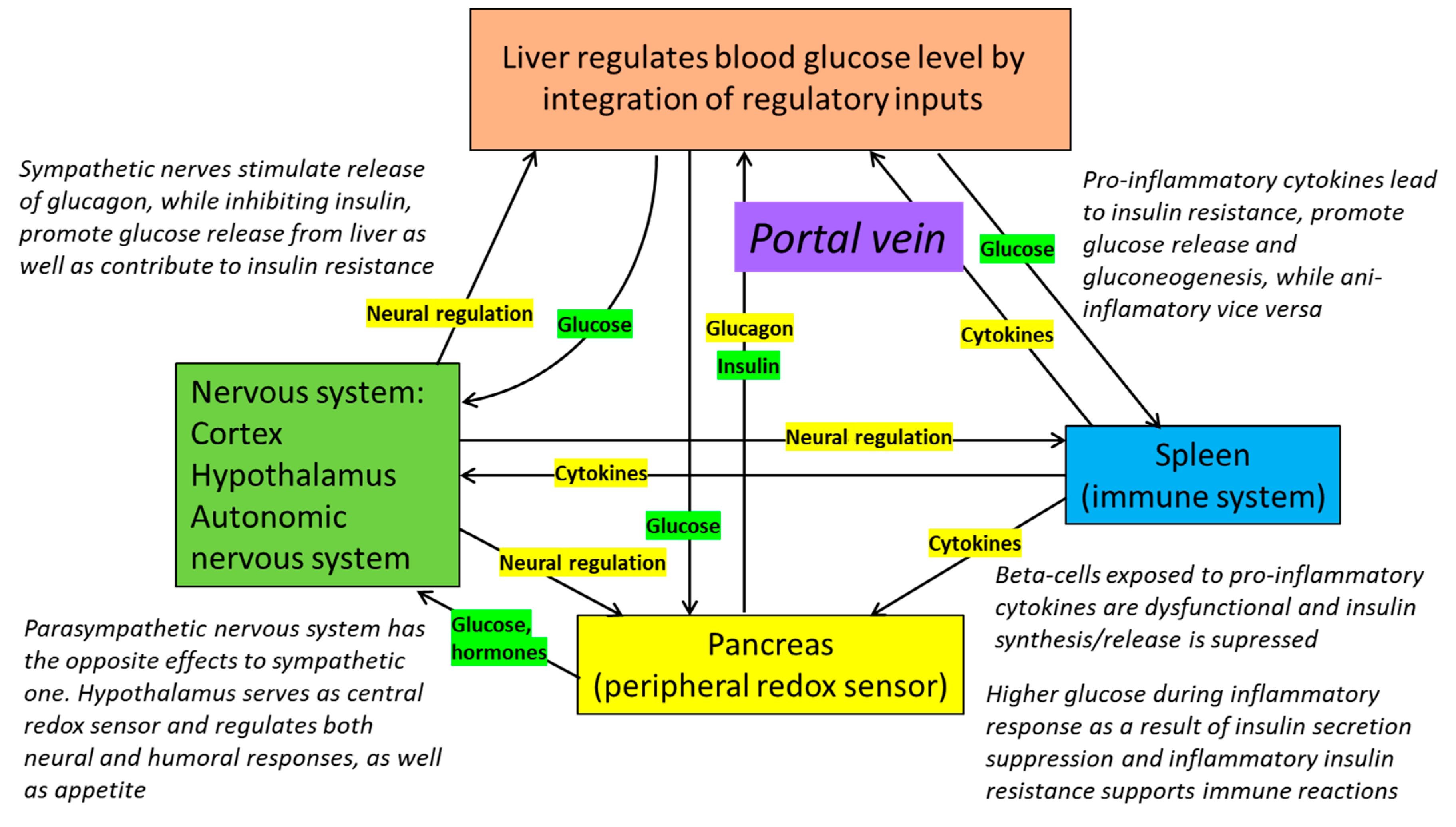
8. Redox Dependence of Pancreatic Regulation of Blood Glucose Levels
9. Inflammation, Insulin Resistance and Redox Homeostasis
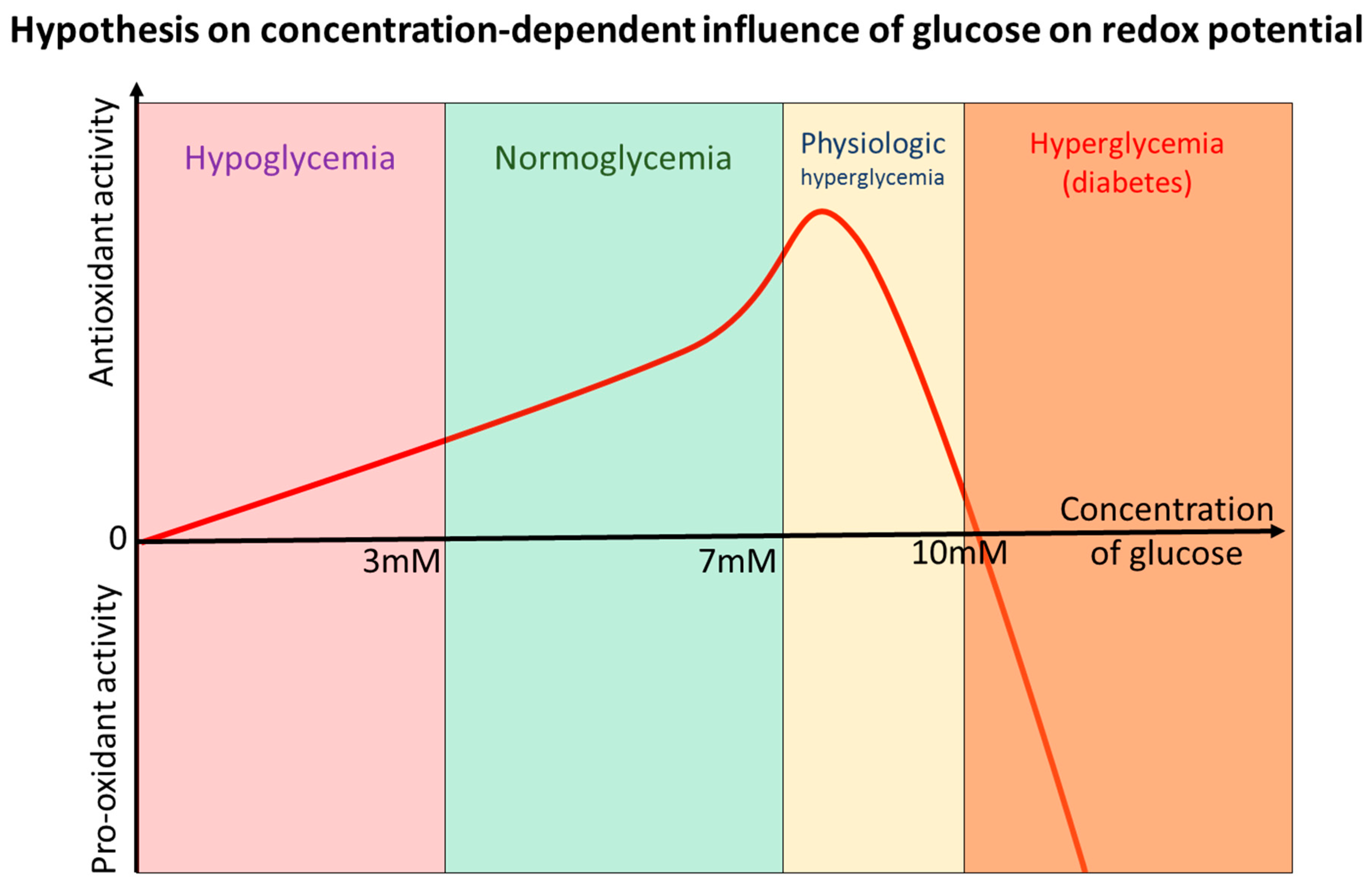
10. Glucose—“Oxidant” or “Antioxidant” after All? Sola Dosis Facit Venenum
11. Important Implications
12. Limitations of the Analysis and Directions of Further Research
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wasserman, D.H. Four grams of glucose. J. Physiol. Endocrinol. Metab. 2009, 296, E11–E21. [Google Scholar] [CrossRef]
- Aronoff, S.L.; Berkowitz, K.; Shreiner, B.; Want, L. Glucose metabolism and regulation: Beyond insulin and glucagon. Diabetes Spectr. 2004, 17, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. A fresh view of glycolysis and glucokinase regulation: History and current status. J. Biol. Chem. 2014, 289, 12189–12194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, J.; Guinovart, J.J. Brain glycogen in health and disease. Mol. Asp. Med. 2015, 46, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Soty, M.; Gautier-Stein, A.; Rajas, F.; Mithieux, G. Gut-Brain Glucose Signaling in Energy Homeostasis. Cell Metab. 2017, 25, 1231–1242. [Google Scholar] [CrossRef] [Green Version]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ralser, M.; Wamelink, M.M.; Kowald, A.; Gerisch, B.; Heeren, G.; Struys, E.A.; Klipp, E.; Jakobs, C.; Breitenbach, M.; Lehrach, H.; et al. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 2007, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehne, A.; Emmert, H.; Soehle, J.; Winnefeld, M.; Fischer, F.; Wenck, H.; Gallinat, S.; Terstegen, L.; Lucius, R.; Hildebrand, J.; et al. Acute Activation of Oxidative Pentose Phosphate Pathway as First-Line Response to Oxidative Stress in Human Skin Cells. Mol. Cell 2015, 59, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Dick, T.P.; Ralser, M. Metabolic Remodeling in Times of Stress: Who Shoots Faster than His Shadow? Mol. Cell 2015, 59, 519–521. [Google Scholar] [CrossRef] [Green Version]
- Sthijns, M.M.; Weseler, A.R.; Bast, A.; Haenen, G.R. Time in Redox Adaptation Processes: From Evolution to Hormesis. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [Green Version]
- Gebril, H.M.; Avula, B.; Wang, Y.H.; Khan, I.A.; Jekabsons, M.B. (13)C metabolic flux analysis in neurons utilizing a model that accounts for hexose phosphate recycling within the pentose phosphate pathway. Neurochem. Int. 2016, 93, 26–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, P.; Sun, H.; Xu, C.; Chen, T.; Zou, B.; Jiang, P.; Du, W. Evidence for a direct cross-talk between malic enzyme and the pentose phosphate pathway via structural interactions. J. Biol. Chem. 2017, 292, 17113–17120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luzzatto, L.; Nannelli, C.; Notaro, R. Glucose-6-Phosphate Dehydrogenase Deficiency. Hematol. Oncol. Clin. 2016, 30, 373–393. [Google Scholar] [CrossRef]
- Lai, Y.K.; Lai, N.M.; Lee, S.W. Glucose-6-phosphate dehydrogenase deficiency and risk of diabetes: A systematic review and meta-analysis. Ann. Hematol. 2017, 96, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.C.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choinière, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell 2015, 60, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J. Systems Biology of Metabolism. Annu. Rev. Biochem. 2017, 86, 245–275. [Google Scholar] [CrossRef] [PubMed]
- Pomatto, L.C.D.; Dill, T.; Carboneau, B.; Levan, S.; Kato, J.; Mercken, E.M.; Pearson, K.J.; Bernier, M; de Cabo, R. Deletion of Nrf2 shortens lifespan in C57BL6/J male mice but does not alter the health and survival benefits of caloric restriction. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Wu, X. Imbalanced insulin action in chronic over nutrition: Clinical harm, molecular mechanisms, and a way forward. Atherosclerosis 2016, 247, 225–282. [Google Scholar] [CrossRef] [Green Version]
- Dehghan, M.; Mente, A.; Zhang, X.; Swaminathan, S.; Li, W.; Mohan, V.; Iqbal, R.; Kumar, R.; Wentzel-Viljoen, E.; Rosengren, A.; et al. Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): A prospective cohort study. Lancet 2017, 390, 2050–2062. [Google Scholar] [CrossRef] [Green Version]
- Shan, Z.; Guo, Y.; Hu, F.B.; Liu, L.; Qi, Q. Association of Low-Carbohydrate and Low-Fat Diets With Mortality among US Adults. JAMA Int. Med. 2020. [Google Scholar] [CrossRef]
- Ravichandran, M.; Grandl, G.; Ristow, M. Dietary Carbohydrates Impair Healthspan and Promote Mortality. Cell Metab. 2017, 26, 585–587. [Google Scholar] [CrossRef] [Green Version]
- Muth, N.D.; Dietz, W.H.; Magge, S.N.; Johnson, R.K. Public Policies to Reduce Sugary Drink Consumption in Children and Adolescents. Pediatrics 2019, 143. [Google Scholar] [CrossRef] [Green Version]
- Hardy, K.; Brand-Miller, J.; Brown, K.D.; Thomas, M.G.; Copeland, L. The Importance of Dietary Carbohydrate in Human Evolution. Q. Rev. Biol. 2015, 90, 251–268. [Google Scholar] [CrossRef] [Green Version]
- Mosca, F.; Gianni, M.L. Human milk: Composition and health benefits. Pediatr. Med. Chir. 2017, 39, 155. [Google Scholar] [CrossRef] [Green Version]
- Andreas, N.J.; Kampmann, B.; Mehring Le-Doare, K. Human breast milk: A review on its composition and bioactivity. Early Hum. Dev. 2015, 91, 629–635. [Google Scholar] [CrossRef]
- Gromnatska, N.; Cherkas, A.; Lemishko, B.; Kulya, O. The Pattern of Metabolic Syndrome in Children with Abdominal Obesity. Georgian. Med. News 2019, 289, 68–72. [Google Scholar]
- Beauchamp, G.K. Why do we like sweet taste: A bitter tale? Physiol. Behav. 2016, 164, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiani, C.; Colombo, R.; Gaglio, D.; Mastroianni, F.; Pescini, D.; Westerhoff, H.V.; Mauri, G.; Vanoni, M.; Alberghina, L. A metabolic core model elucidates how enhanced utilization of glucose and glutamine, with enhanced glutamine-dependent lactate production, promotes cancer cell growth: The WarburQ effect. PLoS Comput. Biol. 2017, 13, e1005758. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef]
- Strongman, H.; Gadd, S.; Matthews, A.; Mansfield, K.E.; Stanway, S.; Lyon, A.R.; Dos-Santos-Silva, I.; Smeeth, L.; Bhaskaran, K. Medium and long-term risks of specific cardiovascular diseases in survivors of 20 adult cancers: A population-based cohort study using multiple linked UK electronic health records databases. Lancet 2019, 394, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Lau, E.; Paniagua, S.M.; Liu, E.; Jovani, M.; Li, S.; Takvorian, K.; Ramachandran, V.S.; Splansky, G.L.; Kreger, B.; Larson, M.; et al. American Heart Association Scientific Sessions; Circulation: Philadelphia, PA, USA, 2019; Volume 140, p. A12269. [Google Scholar]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared Risk Factors in Cardiovascular Disease and Cancer. Circulation 2016, 133, 1104–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rosa, M.J.; Veuthey, T.; Florman, J.; Grant, J.; Blanco, M.G.; Andersen, N.; Donnelly, J.; Rayes, D.; Alkema, M.J. The flight response impairs cytoprotective mechanisms by activating the insulin pathway. Nature 2019, 573, 135–138. [Google Scholar] [CrossRef]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Urban, N.; Tsitsipatis, D.; Hausig, F.; Kreuzer, K.; Erler, K.; Stein, V.; Ristow, M.; Steinbrenner, H.; Klotz, L.-O. Non-linear impact of glutathione depletion on C. elegans life span and stress resistance. Redox Biol. 2017, 11, 502–515. [Google Scholar] [CrossRef]
- Laranjeiro, R.; Harinath, G.; Burke, D.; Braeckman, B.P.; Driscoll, M. Single swim sessions in C. elegans induce key features of mammalian exercise. BMC Biol. 2017, 15, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkas, A.; Abrahamovych, O.; Golota, S.; Nersesyan, A.; Pichler, C.; Serhiyenko, V.; Knasmüller, S.; Zarkovic, N.; Eckl, P. The correlations of glycated hemoglobin and carbohydrate metabolism parameters with heart rate variability in apparently healthy sedentary young male subjects. Redox Biol. 2015, 5, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkas, A.; Golota, S. An intermittent exhaustion of the pool of glycogen in the human organism as a simple universal health promoting mechanism. Med. Hypotheses 2014, 82, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.J.; Eriksson, J.W. Emerging Role of SGLT-2 Inhibitors for the Treatment of Obesity. Drugs 2019, 79, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.C.; Cherney, D.Z.I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 2018, 61, 2098–2107. [Google Scholar] [CrossRef] [Green Version]
- Yale, J.F.; Paty, B.; Senior, P.A. Hypoglycemia. Can. J. Diabetes 2018, 42, S104–S108. [Google Scholar] [CrossRef] [Green Version]
- Ciarlone, G.E.; Hinojo, C.M.; Stavitzski, N.M.; Dean, J.B. CNS function and dysfunction during exposure to hyperbaric oxygen in operational and clinical settings. Redox Biol. 2019, 27, 101159. [Google Scholar] [CrossRef]
- Gusarov, I.; Pani, B.; Gautier, L.; Smolentseva, O.; Eremina, S.; Shamovsky, I.; Katkova-Zhukotskaya, O.; Mironov, A.; Nudler, E. Glycogen controls Caenorhabditis elegans lifespan and resistance to oxidative stress. Nat. Commun. 2017, 8, 15868. [Google Scholar] [CrossRef]
- Oliveira, J.M.; Losano, N.F.; Condessa, S.S.; de Freitas, R.; Cardoso, S.A.; Freitas, M.B.; de Oliveira, L.L. Exposure to deltamethrin induces oxidative stress and decreases of energy reserve in tissues of the Neotropical fruit-eating bat Artibeus lituratus. Ecotoxicol. Environ. Saf. 2017, 148, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.; Lopez-Martinez, G.; Fane, B.; Davidowitz, G. Hawkmoths use nectar sugar to reduce oxidative damage from flight. Science 2017, 355, 733–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczor, J.J.; Robertshaw, H.A.; Tarnopolsky, M.A. Higher oxidative stress in skeletal muscle of McArdle disease patients. Mol. Genet. Metab. Rep. 2017, 12, 69–75. [Google Scholar] [CrossRef]
- Philp, A.; Hargreaves, M.; Baar, K. More than a store: Regulatory roles for glycogen in skeletal muscle adaptation to exercise. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1343–E1351. [Google Scholar] [CrossRef] [Green Version]
- Possik, E.; Pause, A. Glycogen: A must have storage to survive stressful emergencies. Worm 2016, 5, e1156831. [Google Scholar] [CrossRef] [Green Version]
- LaMacchia, J.C.; Roth, M.B. Aquaporins-2 and -4 regulate glycogen metabolism and survival during hyposmotic-anoxic stress in Caenorhabditis elegans. Am. J. Physiol. Cell Physiol. 2015, 309, C92–C96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anton, S.D.; Moehl, K.; Donahoo, W.T.; Marosi, K.; Lee, S.A.; Mainous, A.G., 3rd; Leeuwenburgh, C.; Mattson, M.P. Flipping the Metabolic Switch: Understanding and Applying the Health Benefits of Fasting. Obesity 2017. [Google Scholar] [CrossRef]
- Perry, R.J.; Wang, Y.; Cline, G.W.; Rabin-Court, A.; Song, J.D.; Dufour, S.; Zhang, X.M.; Petersen, K.F.; Shulman, G.I. Leptin Mediates a Glucose-Fatty Acid Cycle to Maintain Glucose Homeostasis in Starvation. Cell 2018, 172, 234–248.e17. [Google Scholar] [CrossRef]
- Duerrschmid, C.; He, Y.; Wang, C.; Li, C.; Bournat, J.C.; Romere, C.; Saha, P.K.; Lee, M.E.; Phillips, K.J.; Jain, M.; et al. Asprosin is a centrally acting orexigenic hormone. Nat. Med. 2017, 23, 1444–1453. [Google Scholar] [CrossRef] [Green Version]
- Romere, C.; Duerrschmid, C.; Bournat, J.; Constable, P.; Jain, M.; Xia, F.; Saha, P.K.; Del Solar, M.; Zhu, B.; York, B.; et al. Asprosin, a Fasting-Induced Glucogenic Protein Hormone. Cell 2016, 165, 566–579. [Google Scholar] [CrossRef] [Green Version]
- Bordoni, L.; Gabbianelli, R. Primers on nutrigenetics and nutri(epi)genomics: Origins and development of precision nutrition. Biochimie 2019, 160, 156–171. [Google Scholar] [CrossRef] [PubMed]
- Gabbianelli, R. Modulation of the Epigenome by Nutrition and Xenobiotics during Early Life and across the Life Span: The Key Role of Lifestyle. Lifestyle Genom. 2018, 11, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Gabbianelli, R.; Damiani, E. Epigenetics and neurodegeneration: Role of early-life nutrition. J. Nutr. Biochem. 2018, 57, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.F.; Lehner, B. Intergenerational and transgenerational epigenetic inheritance in animals. Nat. Cell Biol. 2019, 21, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Afanas’ev, I. New nucleophilic mechanisms of ros-dependent epigenetic modifications: Comparison of aging and cancer. Aging Dis. 2014, 5, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Ni, X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef]
- Strzyz, P. A sugar rush of DNA methylation. Nat. Rev. Mol. Cell Biol. 2018, 19, 617. [Google Scholar] [CrossRef]
- Bordoni, L.; Nasuti, C.; Di Stefano, A.; Marinelli, L.; Gabbianelli, R. Epigenetic Memory of Early-Life Parental Perturbation: Dopamine Decrease and DNA Methylation Changes in Offspring. Oxidative Med. Cell. Longev. 2019, 2019, 1472623. [Google Scholar] [CrossRef]
- Liu, T.F.; McCall, C.E. Deacetylation by SIRT1 Reprograms Inflammation and Cancer. Genes Cancer 2013, 4, 135–147. [Google Scholar] [CrossRef]
- Guillaumet-Adkins, A.; Yañez, Y.; Peris-Diaz, M.D.; Calabria, I.; Palanca-Ballester, C.; Sandoval, J. Epigenetics and Oxidative Stress in Aging. Oxidative Med. Cell. Longev. 2017, 2017, 9175806. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Zarkovic, N. 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Mol. Asp. Med. 2003, 24, 281–291. [Google Scholar] [CrossRef]
- Klotz, L.O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef]
- Monsalve, M.; Prieto, I.; de Bem, A.F.; Olmos, Y. Methodological Approach for the Evaluation of FOXO as a Positive Regulator of Antioxidant Genes. Methods Mol. Biol. 2019, 1890, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Cherkas, A.; Zarkovic, N. 4-Hydroxynonenal in Redox Homeostasis of Gastrointestinal Mucosa: Implications for the Stomach in Health and Diseases. Antioxidants 2018, 7. [Google Scholar] [CrossRef]
- Jaganjac, M.; Milkovic, L.; Gegotek, A.; Cindric, M.; Zarkovic, K.; Skrzydlewska, E.; Zarkovic, N. The relevance of pathophysiological alterations in redox signaling of 4-hydroxynonenal for pharmacological therapies of major stress-associated diseases. Free Radic. Biol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Siler, U.; Romao, S.; Tejera, E.; Pastukhov, O.; Kuzmenko, E.; Valencia, R.G.; Meda Spaccamela, V.; Belohradsky, B.H.; Speer, O.; Schmugge, M.; et al. Severe glucose-6-phosphate dehydrogenase deficiency leads to susceptibility to infection and absent NETosis. J. Allergy Clin. Immunol. 2017, 139, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Ninfali, P.; Bresolin, N.; Baronciani, L.; Fortunato, F.; Comi, G.; Magnani, M.; Scarlato, G. Glucose-6-phosphate dehydrogenase Lodi844C: A study on its expression in blood cells and muscle. Enzyme 1991, 45, 180–187. [Google Scholar]
- Demir, A.Y.; van Solinge, W.W.; van Oirschot, B.; van Wesel, A.; Vergouwen, P.; Thimister, E.; Maase, K.; Rijksen, G.; Schutgens, R.; van Wijk, R. Glucose 6-phosphate dehydrogenase deficiency in an elite long-distance runner. Blood 2009, 113, 2118–2119. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, P.P.; Sonati, F.; Rivi, R.; Mason, P.; Grosveld, F.; Luzzatto, L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 1995, 14, 5209. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legan, S.K.; Rebrin, I.; Mockett, R.J.; Radyuk, S.N.; Klichko, V.I.; Sohal, R.S.; Orr, W.C. Overexpression of glucose-6-phosphate dehydrogenase extends the life span of Drosophila melanogaster. J. Biol. Chem. 2008, 283, 32492–32499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobrega-Pereira, S.; Fernandez-Marcos, P.J.; Brioche, T.; Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Flores, J.M.; Viña, J.; Serrano, M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016, 7, 10894. [Google Scholar] [CrossRef] [Green Version]
- Monte Alegre, S.; Saad, S.T.; Delatre, E.; Saad, M.J. Insulin secretion in patients deficient in glucose-6-phosphate dehydrogenase. Horm. Metab. Res. 1991, 23, 171–173. [Google Scholar] [CrossRef]
- Spegel, P.; Sharoyko, V.V.; Goehring, I.; Danielsson, A.P.; Malmgren, S.; Nagorny, C.L.; Andersson, L.E.; Koeck, T.; Sharp, G.W.; Straub, S.G.; et al. Time-resolved metabolomics analysis of beta-cells implicates the pentose phosphate pathway in the control of insulin release. Biochem. J. 2013, 450, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Kalwat, M.A.; Cobb, M.H. Mechanisms of the amplifying pathway of insulin secretion in the beta cell. Pharmacol. Ther. 2017, 179, 17–30. [Google Scholar] [CrossRef]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [Google Scholar] [CrossRef] [Green Version]
- Gylfe, E. Glucose control of glucagon secretion-’There’s a brand-new gimmick every year. Upsala J. Med. Sci. 2016, 121, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, F.M.; Rohm, M.; Clark, A.; Brereton, M.F. Is Type 2 Diabetes a Glycogen Storage Disease of Pancreatic beta Cells? Cell Metab. 2017, 26, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergari, E.; Knudsen, J.G.; Ramracheya, R.; Salehi, A.; Zhang, Q.; Adam, J.; Asterholm, I.W.; Benrick, A.; Briant, L.J.B.; Chibalina, M.V.; et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat. Commun. 2019, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Huising, M.O. The somatostatin-secreting pancreatic delta-cell in health and disease. Nat. Rev. Endocrinol. 2018, 14, 404–414. [Google Scholar] [CrossRef]
- D’Alessio, D. Is GLP-1 a hormone: Whether and When? J. Diabetes Investig. 2016, 7, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Funcke, J.B.; Scherer, P.E. Beyond adiponectin and leptin: Adipose tissue-derived mediators of inter-organ communication. J. Lipid Res. 2019, 60, 1648–1684. [Google Scholar] [CrossRef] [PubMed]
- Kalsbeek, A.; Bruinstroop, E.; Yi, C.X.; Klieverik, L.; Liu, J.; Fliers, E. Hormonal control of metabolism by the hypothalamus-autonomic nervous system-liver axis. Front. Horm. Res. 2014, 42, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. Neural regulation of pancreatic islet cell mass and function. Diabetes Obes. Metab. 2014, 16, 87–95. [Google Scholar] [CrossRef]
- Yang, H.C.; Wu, Y.H.; Liu, H.Y.; Stern, A.; Chiu, D.T. What has passed is prolog: New cellular and physiological roles of G6PD. Free Radic. Res. 2016, 50, 1047–1064. [Google Scholar] [CrossRef]
- Peiro, C.; Romacho, T.; Azcutia, V.; Villalobos, L.; Fernández, E.; Bolaños, J.P.; Moncada, S.; Sánchez-Ferrer, C.F. Inflammation, glucose, and vascular cell damage: The role of the pentose phosphate pathway. Cardiovasc. Diabetol. 2016, 15, 82. [Google Scholar] [CrossRef] [Green Version]
- Okin, D.; Medzhitov, R. The Effect of Sustained Inflammation on Hepatic Mevalonate Pathway Results in Hyperglycemia. Cell 2016, 165, 343–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Cherkas, A.; Golota, S.; Guéraud, F.; Abrahamovych, O.; Pichler, C.; Nersesyan, A.; Krupak, V.; Bugiichyk, V.; Yatskevych, O.; Pliatsko, M.; et al. A Helicobacter pylori-associated insulin resistance in asymptomatic sedentary young men does not correlate with inflammatory markers and urine levels of 8-iso-PGF2-alpha or 1,4-dihydroxynonane mercapturic acid. Arch. Physiol. Biochem. 2017, 1–11. [Google Scholar] [CrossRef]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, K.; Fujiwara, K.; Anai, M.; Okamoto, M.; Masaki, T.; Kakuma, T.; Shibata, H. Role of spleen-derived IL-10 in prevention of systemic low-grade inflammation by obesity. Endocr. J. 2017, 64, 375–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkas, A.; Eckl, P.; Gueraud, F.; Abrahamovych, O.; Serhiyenko, V.; Yatskevych, O.; Pliatsko, M.; Golota, S. Helicobacter pylori in sedentary men is linked to higher heart rate, sympathetic activity, and insulin resistance but not inflammation or oxidative stress. Croat. Med. J. 2016, 57, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Gregory, J.M.; Rivera, N.; Kraft, G.; Winnick, J.J.; Farmer, B.; Allen, E.J.; Donahue, E.P.; Smith, M.S.; Edgerton, D.S.; Williams, P.E.; et al. Glucose autoregulation is the dominant component of the hormone-independent counterregulatory response to hypoglycemia in the conscious dog. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E273–E283. [Google Scholar] [CrossRef]
- Cherkas, A.; Zarkovic, K.; Cipak Gasparovic, A.; Jaganjac, M.; Milkovic, L.; Abrahamovych, O.; Yatskevych, O.; Waeg, G.; Yelisyeyeva, O.; Zarkovic, N. Amaranth oil reduces accumulation of 4-hydroxynonenal-histidine adducts in gastric mucosa and improves heart rate variability in duodenal peptic ulcer patients undergoing Helicobacter pylori eradication. Free Radic. Res. 2017, 1–231. [Google Scholar] [CrossRef]
- Lee, Y.S.; Wollam, J.; Olefsky, J.M. An Integrated View of Immunometabolism. Cell 2018, 172, 22–40. [Google Scholar] [CrossRef] [Green Version]
- Tsatsoulis, A.; Mantzaris, M.D.; Bellou, S.; Andrikoula, M. Insulin resistance: An adaptive mechanism becomes maladaptive in the current environment—An evolutionary perspective. Metabolism 2013, 62, 622–633. [Google Scholar] [CrossRef]
- Hohn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; König, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Kingsley, S.; Walker, G.; Mondoux, M.A.; Tissenbaum, H.A. Metabolic shift from glycogen to trehalose promotes lifespan and healthspan in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibshman, J.D.; Doan, A.E.; Moore, B.T.; Kaplan, R.E.; Hung, A.; Webster, A.K.; Bhatt, D.P.; Chitrakar, R.; Hirschey, M.D.; Baugh, L.R. daf-16/FoxO promotes gluconeogenesis and trehalose synthesis during starvation to support survival. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Song, S.B.; Hwang, E.S. A Rise in ATP, ROS, and Mitochondrial Content upon Glucose Withdrawal Correlates with a Dysregulated Mitochondria Turnover Mediated by the Activation of the Protein Deacetylase SIRT1. Cells 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelmakh, A.; Abrahamovych, O.; Cherkas, A. Highly purified calf hemodialysate (Actovegin®) may improve endothelial function by activation of proteasomes: A hypothesis explaining the possible mechanisms of action. Med. Hypotheses 2016, 95, 77–81. [Google Scholar] [CrossRef] [PubMed]
- da-Silva, W.S.; Gómez-Puyou, A.; de Gómez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef] [Green Version]
- Steinhorn, B.; Sartoretto, J.L.; Sorrentino, A.; Romero, N.; Kalwa, H.; Abel, E.D.; Michel, T. Insulin-dependent metabolic and inotropic responses in the heart are modulated by hydrogen peroxide from NADPH-oxidase isoforms NOX2 and NOX4. Free Radic. Biol. Med. 2017, 113, 16–25. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2019. [Google Scholar] [CrossRef]
- Yan, L.J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Models Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef]
- Rovenko, B.M.; Kubrak, O.I.; Gospodaryov, D.V.; Perkhulyn, N.V.; Yurkevych, I.S.; Sanz, A.; Lushchak, O.V.; Lushchak, V.I. High sucrose consumption promotes obesity whereas its low consumption induces oxidative stress in Drosophila melanogaster. J. Insect Physiol. 2015, 79, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Mullen, L.; Mengozzi, M.; Hanschmann, E.M.; Alberts, B.; Ghezzi, P. How the redox state regulates immunity. Free Radic. Biol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gaitonde, M.K.; Evison, E.; Evans, G.M. The rate of utilization of glucose via hexosemonophosphate shunt in brain. J. Neurochem. 1983, 41, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yoseph, O.; Boxer, P.A.; Ross, B.D. Noninvasive assessment of the relative roles of cerebral antioxidant enzymes by quantitation of pentose phosphate pathway activity. Neurochem. Res. 1996, 21, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Bartnik, B.L.; Sutton, R.L.; Fukushima, M.; Harris, N.G.; Hovda, D.A.; Lee, S.M. Upregulation of pentose phosphate pathway and preservation of tricarboxylic acid cycle flux after experimental brain injury. J. Neurotrauma 2005, 22, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Pastor, R.; Tur, J.A. Antioxidant Supplementation and Adaptive Response to Training: A Systematic Review. Curr. Pharm. Des. 2019, 25, 1889–1912. [Google Scholar] [CrossRef]
- Kim, H.G.; Bae, J.H.; Jastrzebski, Z.; Cherkas, A.; Heo, B.G.; Gorinstein, S.; Ku, Y.G. Binding, Antioxidant and Anti-proliferative Properties of Bioactive Compounds of Sweet Paprika (Capsicum annuum L.). Plant Foods Hum. Nutr. 2016, 71, 129–136. [Google Scholar] [CrossRef]
- Cherkas, A.; Mondol, A.S.; Rüger, J.; Urban, N.; Popp, J.; Klotz, L.O.; Schie, I.W. Label-free molecular mapping and assessment of glycogen in C. elegans. Analyst 2019, 144, 2367–2374. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherkas, A.; Holota, S.; Mdzinarashvili, T.; Gabbianelli, R.; Zarkovic, N. Glucose as a Major Antioxidant: When, What for and Why It Fails? Antioxidants 2020, 9, 140. https://doi.org/10.3390/antiox9020140
Cherkas A, Holota S, Mdzinarashvili T, Gabbianelli R, Zarkovic N. Glucose as a Major Antioxidant: When, What for and Why It Fails? Antioxidants. 2020; 9(2):140. https://doi.org/10.3390/antiox9020140
Chicago/Turabian StyleCherkas, Andriy, Serhii Holota, Tamaz Mdzinarashvili, Rosita Gabbianelli, and Neven Zarkovic. 2020. "Glucose as a Major Antioxidant: When, What for and Why It Fails?" Antioxidants 9, no. 2: 140. https://doi.org/10.3390/antiox9020140
APA StyleCherkas, A., Holota, S., Mdzinarashvili, T., Gabbianelli, R., & Zarkovic, N. (2020). Glucose as a Major Antioxidant: When, What for and Why It Fails? Antioxidants, 9(2), 140. https://doi.org/10.3390/antiox9020140