Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA)



Abstract
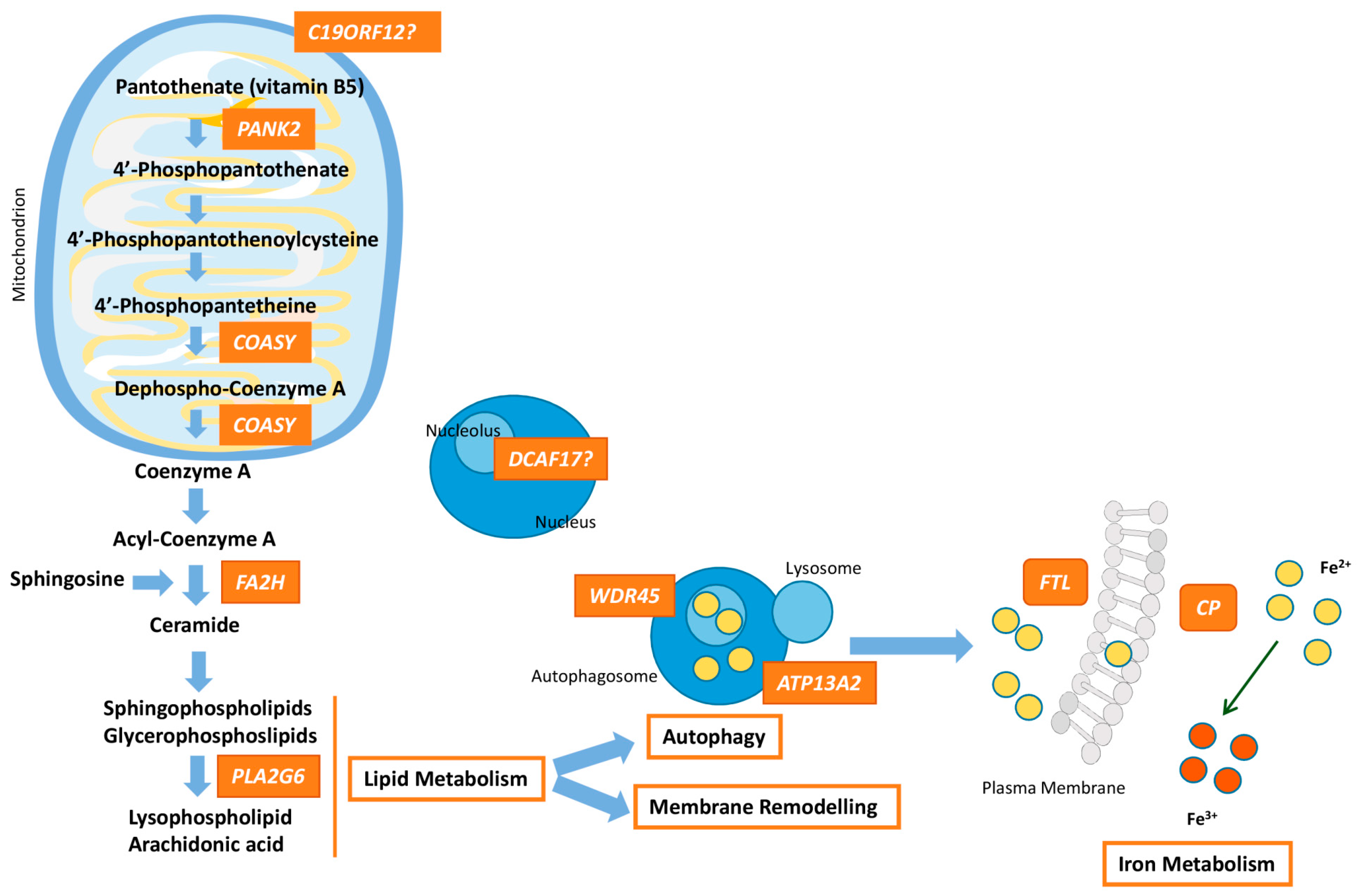
:1. Overview of the NBIA Syndromes
2. NBIA Errors of Coenzyme a Biosynthesis
2.1. Pantothenate Kinase-Associated Neurodegeneration (PKAN)
2.2. COASY Protein-Associated Neurodegeneration (CoPAN)
3. NBIA Types Related to Lipid Metabolism and Membrane Remodeling
3.1. PLA2G6-Associated Neurodegeneration (PLAN)
3.2. Mitochondrial Membrane Protein-Associated Neurodegeneration (MPAN)
3.3. Fatty Acid Hydroxylase-Associated Neurodegeneration (FAHN)
4. Autophagosome/Lysosome Regulation
4.1. β-propeller-Associated Neurodegeneration (BPAN)
4.2. Kufor–Rakeb Syndrome
5. NBIA Forms Caused by Mutations in Iron-Related Genes
5.1. Aceruloplasminemia
5.2. Neuropherritinopathy (NF)
6. Another NBIA Subtype: Woodhouse-Sakati Syndrome
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Espinós, C.; Galindo, M.I.; Garcia-Gimeno, M.A.; Ibañez-Cabellos, J.S.; Martinez-Rubio, D.; Millan, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative Stress, a Crossroad between Rare Diseases and Neurodegeneration. Antioxidants 2020, 9, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa-Vela, M.; Lupo, V.; Montpeyo, M.; Sancho, P.; Marce-Grau, A.; Hernandez-Vara, J.; Darling, A.; Jenkins, A.; Fernandez-Rodriguez, S.; Tello, C.; et al. Impaired proteasome activity and neurodegeneration with brain iron accumulation in FBXO7 defect. Ann. Clin. Transl. Neurol. 2020, 7, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Gautschi, M.; Merlini, L.; Calza, A.M.; Hayflick, S.; Nuoffer, J.M.; Fluss, J. Late diagnosis of fucosidosis in a child with progressive fixed dystonia, bilateral pallidal lesions and red spots on the skin. Eur. J. Paediatr. Neurol. 2014, 18, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Kosaki, R.; Nakabayashi, K.; Hata, K.; Takahashi, T.; Kosaki, K. Paramagnetic signals in the globus pallidus as late radiographic sign of juvenile-onset GM1 gangliosidosis. Pediatr. Neurol. 2015, 52, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.P.; Conceicao, C.; Scortenschi, E. GM1 gangliosidosis, late infantile onset dystonia, and T2 Hypointensity in the globus pallidus and substantia Nigra. Pediatr. Neurol. 2013, 49, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Jaberi, E.; Rohani, M.; Shahidi, G.A.; Nafissi, S.; Arefian, E.; Soleimani, M.; Rasooli, P.; Ahmadieh, H.; Daftarian, N.; KaramiNejadRanjbar, M.; et al. Identification of mutation in GTPBP2 in patients of a family with neurodegeneration accompanied by iron deposition in the brain. Neurobiol. Aging 2016, 38, 216.e11–216.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, R.; Lewis-Smith, D.; Douroudis, K.; Duff, J.; Keogh, M.; Pyle, A.; Fletcher, N.; Chinnery, P.F. SCP2 mutations and neurodegeneration with brain iron accumulation. Neurology 2015, 85, 1909–1911. [Google Scholar] [CrossRef] [Green Version]
- Sferra, A.; Baillat, G.; Rizza, T.; Barresi, S.; Flex, E.; Tasca, G.; D’Amico, A.; Bellacchio, E.; Ciolfi, A.; Caputo, V.; et al. TBCE Mutations Cause Early-Onset Progressive Encephalopathy with Distal Spinal Muscular Atrophy. Am. J. Hum. Genet. 2016, 99, 974–983. [Google Scholar] [CrossRef] [Green Version]
- Meyer, E.; Carss, K.J.; Rankin, J.; Nichols, J.M.; Grozeva, D.; Joseph, A.P.; Mencacci, N.E.; Papandreou, A.; Ng, J.; Barral, S.; et al. Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat. Genet. 2017, 49, 223–237. [Google Scholar] [CrossRef]
- Dard, R.; Meyniel, C.; Touitou, V.; Stevanin, G.; Lamari, F.; Durr, A.; Ewenczyk, C.; Mochel, F. Mutations in DDHD1, encoding a phospholipase A1, is a novel cause of retinopathy and neurodegeneration with brain iron accumulation. Eur. J. Med. Genet. 2017, 60, 639–642. [Google Scholar] [CrossRef]
- Roubertie, A.; Hieu, N.; Roux, C.J.; Leboucq, N.; Manes, G.; Charif, M.; Echenne, B.; Goizet, C.; Guissart, C.; Meyer, P.; et al. AP4 deficiency: A novel form of neurodegeneration with brain iron accumulation? Neurol. Genet. 2018, 4, e217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vill, K.; Muller-Felber, W.; Alhaddad, B.; Strom, T.M.; Teusch, V.; Weigand, H.; Blaschek, A.; Meitinger, T.; Haack, T.B. A homozygous splice variant in AP4S1 mimicking neurodegeneration with brain iron accumulation. Mov. Disord. 2017, 32, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Svingen, L.; Goheen, M.; Godfrey, R.; Wahl, C.; Baker, E.H.; Gahl, W.A.; Malicdan, M.C.V.; Toro, C. Late diagnosis and atypical brain imaging of Aicardi-Goutieres syndrome: Are we failing to diagnose Aicardi-Goutieres syndrome-2? Dev. Med. Child Neurol. 2017, 59, 1307–1311. [Google Scholar] [CrossRef] [Green Version]
- Zoons, E.; de Koning, T.J.; Abeling, N.G.; Tijssen, M.A. Neurodegeneration with Brain Iron Accumulation on MRI: An Adult Case of alpha-Mannosidosis. JIMD Rep. 2012, 4, 99–102. [Google Scholar]
- Drecourt, A.; Babdor, J.; Dussiot, M.; Petit, F.; Goudin, N.; Garfa-Traore, M.; Habarou, F.; Bole-Feysot, C.; Nitschke, P.; Ottolenghi, C.; et al. Impaired Transferrin Receptor Palmitoylation and Recycling in Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2018, 102, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herebian, D.; Alhaddad, B.; Seibt, A.; Schwarzmayr, T.; Danhauser, K.; Klee, D.; Harmsen, S.; Meitinger, T.; Strom, T.M.; Schulz, A.; et al. Coexisting variants in OSTM1 and MANEAL cause a complex neurodegenerative disorder with NBIA-like brain abnormalities. Eur. J. Hum. Genet. 2017, 25, 1092–1095. [Google Scholar] [CrossRef] [Green Version]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.A.; et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Lyon, G.J.; Marchi, E.; Ekstein, J.; Meiner, V.; Hirsch, Y.; Scher, S.; Yang, E.; De Vivo, D.C.; Madrid, R.; Li, Q.; et al. VAC14 syndrome in two siblings with retinitis pigmentosa and neurodegeneration with brain iron accumulation. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid Med. Cell Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [Green Version]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Dolgacheva, L.P. Interaction of misfolded proteins and mitochondria in neurodegenerative disorders. Biochem. Soc. Trans. 2017, 45, 1025–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef]
- Hayflick, S.J. Unraveling the Hallervorden-Spatz syndrome: Pantothenate kinase-associated neurodegeneration is the name. Curr. Opin. Pediatr. 2003, 15, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.T.; Valente, E.M.; Cif, L.; Salvi, S.; Albanese, A.; Scarano, V.; Bonuccelli, U.; Bentivoglio, A.R.; D’Amico, A.; Marelli, C.; et al. The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration. Neurology 2005, 64, 1810–1812. [Google Scholar] [CrossRef]
- Hartig, M.B.; Hortnagel, K.; Garavaglia, B.; Zorzi, G.; Kmiec, T.; Klopstock, T.; Rostasy, K.; Svetel, M.; Kostic, V.S.; Schuelke, M.; et al. Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann. Neurol. 2006, 59, 248–256. [Google Scholar] [CrossRef]
- Gordon, N. Pantothenate kinase-associated neurodegeneration (Hallervorden-Spatz syndrome). Eur. J. Paediatr. Neurol. 2002, 6, 243–247. [Google Scholar] [CrossRef]
- Egan, R.A.; Weleber, R.G.; Hogarth, P.; Gregory, A.; Coryell, J.; Westaway, S.K.; Gitschier, J.; Das, S.; Hayflick, S.J. Neuro-ophthalmologic and electroretinographic findings in pantothenate kinase-associated neurodegeneration (formerly Hallervorden-Spatz syndrome). Am. J. Ophthalmol. 2005, 140, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Yoon, W.T.; Lee, W.Y.; Shin, H.Y.; Lee, S.T.; Ki, C.S. Novel PANK2 gene mutations in korean patient with pantothenate kinase-associated neurodegeneration presenting unilateral dystonic tremor. Mov. Disord. 2010, 25, 245–247. [Google Scholar] [CrossRef]
- Antonini, A.; Goldwurm, S.; Benti, R.; Prokisch, H.; Ebhardt, M.; Cilia, R.; Zini, M.; Righini, A.; Cossu, G.; Pezzoli, G. Genetic, clinical, and imaging characterization of one patient with late-onset, slowly progressive, pantothenate kinase-associated neurodegeneration. Mov. Disord. 2006, 21, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Song, S.K.; Lee, P.H. A Novel PANK2 Mutation in a Patient with Atypical Pantothenate-Kinase-Associated Neurodegeneration Presenting with Adult-Onset Parkinsonism. J. Clin. Neurol. 2009, 5, 192–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, N. Late onset atypical pantothenate-kinase-associated neurodegeneration. Case Rep. Neurol. Med. 2013, 2013, 860201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Hayflick, S.J.; Jankovic, J. Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) and pantothenate kinase-associated neurodegeneration. Mov. Disord. 2004, 19, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J. Neurodegeneration with brain iron accumulation: From genes to pathogenesis. Semin. Pediatr. Neurol. 2006, 13, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Delgado, R.F.; Sanchez, P.R.; Speckter, H.; Then, E.P.; Jimenez, R.; Oviedo, J.; Dellani, P.R.; Foerster, B.; Stoeter, P. Missense PANK2 mutation without “eye of the tiger” sign: MR findings in a large group of patients with pantothenate kinase-associated neurodegeneration (PKAN). J. Magn. Reson. Imaging 2012, 35, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.L.; Gaddikeri, S.; Chapman, P.R.; Roy, R.; Gaddikeri, R.S.; Marussi, V.H.; Bag, A.K. Neurodegeneration with Brain Iron Accumulation: Clinicoradiological Approach to Diagnosis. J. Neuroimaging 2015, 25, 539–551. [Google Scholar] [CrossRef]
- Stankiewicz, J.; Panter, S.S.; Neema, M.; Arora, A.; Batt, C.E.; Bakshi, R. Iron in chronic brain disorders: Imaging and neurotherapeutic implications. Neurotherapeutics 2007, 4, 371–386. [Google Scholar] [CrossRef] [Green Version]
- Malandrini, A.; Fabrizi, G.M.; Bartalucci, P.; Salvadori, C.; Berti, G.; Sabo, C.; Guazzi, G.C. Clinicopathological study of familial late infantile Hallervorden-Spatz disease: A particular form of neuroacanthocytosis. Childs Nerv. Syst. 1996, 12, 155–160. [Google Scholar] [CrossRef]
- Johnson, M.A.; Kuo, Y.M.; Westaway, S.K.; Parker, S.M.; Ching, K.H.; Gitschier, J.; Hayflick, S.J. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann. N. Y. Acad. Sci. 2004, 1012, 282–298. [Google Scholar] [CrossRef]
- Kruer, M.C.; Hiken, M.; Gregory, A.; Malandrini, A.; Clark, D.; Hogarth, P.; Grafe, M.; Hayflick, S.J.; Woltjer, R.L. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 2011, 134, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Woltjer, R.L.; Reese, L.C.; Richardson, B.E.; Tran, H.; Green, S.; Pham, T.; Chalupsky, M.; Gabriel, I.; Light, T.; Sanford, L.; et al. Pallidal neuronal apolipoprotein E in pantothenate kinase-associated neurodegeneration recapitulates ischemic injury to the globus pallidus. Mol. Genet. Metab. 2015, 116, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hortnagel, K.; Prokisch, H.; Meitinger, T. An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum. Mol. Genet. 2003, 12, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.S.; Senisterra, G.; Rabeh, W.M.; Vedadi, M.; Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S.; Park, H.W. Crystal structures of human pantothenate kinases. Insights into allosteric regulation and mutations linked to a neurodegeneration disorder. J. Biol. Chem. 2007, 282, 27984–27993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.M.; Rock, C.O.; Jackowski, S. Biochemical properties of human pantothenate kinase 2 isoforms and mutations linked to pantothenate kinase-associated neurodegeneration. J. Biol. Chem. 2006, 281, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Kotzbauer, P.T.; Truax, A.C.; Trojanowski, J.Q.; Lee, V.M. Altered neuronal mitochondrial coenzyme A synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J. Neurosci. 2005, 25, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, R.; Rock, C.O.; Jackowski, S.; Zhang, Y.M. Activation of human mitochondrial pantothenate kinase 2 by palmitoylcarnitine. Proc. Natl. Acad. Sci. USA 2007, 104, 1494–1499. [Google Scholar] [CrossRef] [Green Version]
- Vallari, D.S.; Jackowski, S.; Rock, C.O. Regulation of pantothenate kinase by coenzyme A and its thioesters. J. Biol. Chem. 1987, 262, 2468–2471. [Google Scholar]
- Kurian, M.A.; Hayflick, S.J. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): Review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int. Rev. Neurobiol. 2013, 110, 49–71. [Google Scholar]
- Gregory, A.; Hayflick, S.J. Neurodegeneration with brain iron accumulation. Folia Neuropathol. 2005, 43, 286–296. [Google Scholar]
- Leoni, V.; Strittmatter, L.; Zorzi, G.; Zibordi, F.; Dusi, S.; Garavaglia, B.; Venco, P.; Caccia, C.; Souza, A.L.; Deik, A.; et al. Metabolic consequences of mitochondrial coenzyme A deficiency in patients with PANK2 mutations. Mol. Genet. Metab. 2012, 105, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A corrects pathological defects in human neurons of PANK2-associated neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Angelova, P.R.; Wiethoff, S.; Tsuchiya, Y.; Mazzacuva, F.; Preza, E.; Bhatia, K.P.; Mills, K.; Gout, I.; Abramov, A.Y.; et al. iPSC-derived neuronal models of PANK2-associated neurodegeneration reveal mitochondrial dysfunction contributing to early disease. PLoS ONE 2017, 12, e0184104. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.; Derosas, M.; Luscieti, S.; Cavadini, P.; Campanella, A.; Verardi, R.; Finazzi, D.; Arosio, P. Pantothenate kinase-2 (Pank2) silencing causes cell growth reduction, cell-specific ferroportin upregulation and iron deregulation. Neurobiol. Dis. 2010, 39, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clardy, S.L.; Wang, X.; Boyer, P.J.; Earley, C.J.; Allen, R.P.; Connor, J.R. Is ferroportin-hepcidin signaling altered in restless legs syndrome? J. Neurol. Sci. 2006, 247, 173–179. [Google Scholar] [CrossRef]
- Zizioli, D.; Tiso, N.; Guglielmi, A.; Saraceno, C.; Busolin, G.; Giuliani, R.; Khatri, D.; Monti, E.; Borsani, G.; Argenton, F.; et al. Knock-down of pantothenate kinase 2 severely affects the development of the nervous and vascular system in zebrafish, providing new insights into PKAN disease. Neurobiol. Dis. 2016, 85, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, Z.; Kuo, Y.M.; Zhou, B. Dietary rescue of fumble—A Drosophila model for pantothenate-kinase-associated neurodegeneration. J. Inherit. Metab. Dis. 2005, 28, 1055–1064. [Google Scholar] [CrossRef]
- Wu, Z.; Li, C.; Lv, S.; Zhou, B. Pantothenate kinase-associated neurodegeneration: Insights from a Drosophila model. Hum. Mol. Genet. 2009, 18, 3659–3672. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, D.; Dusi, S.; Giordano, C.; Lamperti, C.; Morbin, M.; Fugnanesi, V.; Marchet, S.; Fagiolari, G.; Sibon, O.; Moggio, M.; et al. Pantethine treatment is effective in recovering the disease phenotype induced by ketogenic diet in a pantothenate kinase-associated neurodegeneration mouse model. Brain 2014, 137, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Kuo, Y.M.; Duncan, J.L.; Westaway, S.K.; Yang, H.; Nune, G.; Xu, E.Y.; Hayflick, S.J.; Gitschier, J. Deficiency of pantothenate kinase 2 (Pank2) in mice leads to retinal degeneration and azoospermia. Hum. Mol. Genet. 2005, 14, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, D.; Dusi, S.; Morbin, M.; Uggetti, A.; Moda, F.; D’Amato, I.; Giordano, C.; d’Amati, G.; Cozzi, A.; Levi, S.; et al. Pantothenate kinase-associated neurodegeneration: Altered mitochondria membrane potential and defective respiration in Pank2 knock-out mouse model. Hum. Mol. Genet. 2012, 21, 5294–5305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.Y.; Hogarth, P.; Placzek, A.; Gregory, A.M.; Fox, R.; Zhen, D.; Hamada, J.; van der Zwaag, M.; Lambrechts, R.; Jin, H.; et al. 4’-Phosphopantetheine corrects CoA, iron, and dopamine metabolic defects in mammalian models of PKAN. EMBO Mol. Med. 2019, 11, e10489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Chohnan, S.; Virga, K.G.; Stevens, R.D.; Ilkayeva, O.R.; Wenner, B.R.; Bain, J.R.; Newgard, C.B.; Lee, R.E.; Rock, C.O.; et al. Chemical knockout of pantothenate kinase reveals the metabolic and genetic program responsible for hepatic coenzyme A homeostasis. Chem. Biol. 2007, 14, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, Y.M.; Hayflick, S.J.; Gitschier, J. Deprivation of pantothenic acid elicits a movement disorder and azoospermia in a mouse model of pantothenate kinase-associated neurodegeneration. J. Inherit. Metab. Dis. 2007, 30, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, C.; Yao, J.; Frank, M.W.; Rock, C.O.; Jackowski, S. A pantothenate kinase-deficient mouse model reveals a gene expression program associated with brain coenzyme a reduction. Biochim Biophys Acta Mol. Basis Dis. 2020, 1866, 165663. [Google Scholar] [CrossRef]
- Zorzi, G.; Zibordi, F.; Chiapparini, L.; Bertini, E.; Russo, L.; Piga, A.; Longo, F.; Garavaglia, B.; Aquino, D.; Savoiardo, M.; et al. Iron-related MRI images in patients with pantothenate kinase-associated neurodegeneration (PKAN) treated with deferiprone: Results of a phase II pilot trial. Mov. Disord. 2011, 26, 1756–1759. [Google Scholar] [CrossRef]
- Pratini, N.R.; Sweeters, N.; Vichinsky, E.; Neufeld, J.A. Treatment of classic pantothenate kinase-associated neurodegeneration with deferiprone and intrathecal baclofen. Am. J. Phys. Med. Rehabil. 2013, 92, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Cossu, G.; Abbruzzese, G.; Matta, G.; Murgia, D.; Melis, M.; Ricchi, V.; Galanello, R.; Barella, S.; Origa, R.; Balocco, M.; et al. Efficacy and safety of deferiprone for the treatment of pantothenate kinase-associated neurodegeneration (PKAN) and neurodegeneration with brain iron accumulation (NBIA): Results from a four years follow-up. Parkinsonism Relat. Disord. 2014, 20, 651–654. [Google Scholar] [CrossRef]
- Rohani, M.; Razmeh, S.; Shahidi, G.A.; Alizadeh, E.; Orooji, M. A pilot trial of deferiprone in pantothenate kinase-associated neurodegeneration patients. Neurol. Int. 2017, 9, 7279. [Google Scholar] [CrossRef] [PubMed]
- Klopstock, T.; Escolar, M.L.; Marshall, R.D.; Perez-Duenas, B.; Tuller, S.; Videnovic, A.; Greblikas, F. The FOsmetpantotenate Replacement Therapy (FORT) randomized, double-blind, Placebo-controlled pivotal trial: Study design and development methodology of a novel primary efficacy outcome in patients with pantothenate kinase-associated neurodegeneration. Clin. Trials 2019, 16, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Sharma, L.K.; Subramanian, C.; Yun, M.K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat. Commun. 2018, 9, 4399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Cordoba, M.; Villanueva-Paz, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Talaveron-Rey, M.; Abril-Jaramillo, J.; Vintimilla-Tosi, A.B.; Sanchez-Alcazar, J.A. Precision medicine in pantothenate kinase-associated neurodegeneration. Neural Regen. Res. 2019, 14, 1177–1185. [Google Scholar] [PubMed]
- Rana, A.; Seinen, E.; Siudeja, K.; Muntendam, R.; Srinivasan, B.; van der Want, J.J.; Hayflick, S.; Reijngoud, D.J.; Kayser, O.; Sibon, O.C. Pantethine rescues a Drosophila model for pantothenate kinase-associated neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 6988–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogarth, P.; Kurian, M.A.; Gregory, A.; Csanyi, B.; Zagustin, T.; Kmiec, T.; Wood, P.; Klucken, A.; Scalise, N.; Sofia, F.; et al. Consensus clinical management guideline for pantothenate kinase-associated neurodegeneration (PKAN). Mol. Genet. Metab. 2017, 120, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Dusi, S.; Valletta, L.; Haack, T.B.; Tsuchiya, Y.; Venco, P.; Pasqualato, S.; Goffrini, P.; Tigano, M.; Demchenko, N.; Wieland, T.; et al. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2014, 94, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Annesi, G.; Gagliardi, M.; Iannello, G.; Quattrone, A. Mutational analysis of COASY in an Italian patient with NBIA. Parkinsonism Relat. Disord. 2016, 28, 150–151. [Google Scholar] [CrossRef]
- Evers, C.; Seitz, A.; Assmann, B.; Opladen, T.; Karch, S.; Hinderhofer, K.; Granzow, M.; Paramasivam, N.; Eils, R.; Diessl, N.; et al. Diagnosis of CoPAN by whole exome sequencing: Waking up a sleeping tiger’s eye. Am. J. Med. Genet. A 2017, 173, 1878–1886. [Google Scholar] [CrossRef]
- Ceccatelli Berti, C.; Dallabona, C.; Lazzaretti, M.; Dusi, S.; Tosi, E.; Tiranti, V.; Goffrini, P. Modeling human Coenzyme A synthase mutation in yeast reveals altered mitochondrial function, lipid content and iron metabolism. Microb. Cell. 2015, 2, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Bosveld, F.; Rana, A.; van der Wouden, P.E.; Lemstra, W.; Ritsema, M.; Kampinga, H.H.; Sibon, O.C. De novo CoA biosynthesis is required to maintain DNA integrity during development of the Drosophila nervous system. Hum. Mol. Genet. 2008, 17, 2058–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatri, D.; Zizioli, D.; Tiso, N.; Facchinello, N.; Vezzoli, S.; Gianoncelli, A.; Memo, M.; Monti, E.; Borsani, G.; Finazzi, D. Down-regulation of coasy, the gene associated with NBIA-VI, reduces Bmp signaling, perturbs dorso-ventral patterning and alters neuronal development in zebrafish. Sci. Rep. 2016, 6, 37660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrechts, R.A.; Schepers, H.; Yu, Y.; van der Zwaag, M.; Autio, K.J.; Vieira-Lara, M.A.; Bakker, B.M.; Tijssen, M.A.; Hayflick, S.J.; Grzeschik, N.A.; et al. CoA-dependent activation of mitochondrial acyl carrier protein links four neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10488. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, K.J.; Castillo-Quan, J.I. Mitochondrial dysfunction and defects in lipid homeostasis as therapeutic targets in neurodegeneration with brain iron accumulation. Rare Dis. 2016, 4, e1128616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, A.; Kurian, M.A.; Maher, E.R.; Hogarth, P.; Hayflick, S. PLA2G6-Associated Neurodegeneration. In Genereviews®; Pagon, R.A., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Fong, C.T., Mefford, H.C., Smith, R.J.H., Stephens, K., Eds.; University of Washington: Seattle, WA, USA, 2008; pp. 1993–2016. [Google Scholar]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar] [PubMed]
- Illingworth, M.A.; Meyer, E.; Chong, W.K.; Manzur, A.Y.; Carr, L.J.; Younis, R.; Hardy, C.; McDonald, F.; Childs, A.M.; Stewart, B.; et al. PLA2G6-associated neurodegeneration (PLAN): Further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Mol. Genet. Metab. 2014, 112, 183–189. [Google Scholar] [CrossRef]
- Mascalchi, M.; Mari, F.; Berti, B.; Bartolini, E.; Lenge, M.; Bianchi, A.; Antonucci, L.; Santorelli, F.M.; Garavaglia, B.; Guerrini, R. Fast Progression of Cerebellar Atrophy in PLA2G6-Associated Infantile Neuronal Axonal Dystrophy. Cerebellum 2017, 16, 742–745. [Google Scholar] [CrossRef]
- Mubaidin, A.; Roberts, E.; Hampshire, D.; Dehyyat, M.; Shurbaji, A.; Mubaidien, M.; Jamil, A.; Al-Din, A.; Kurdi, A.; Woods, C.G. Karak syndrome: A novel degenerative disorder of the basal ganglia and cerebellum. J. Med. Genet. 2003, 40, 543–546. [Google Scholar] [CrossRef] [Green Version]
- Crompton, D.; Rehal, P.K.; MacPherson, L.; Foster, K.; Lunt, P.; Hughes, I.; Brady, A.F.; Pike, M.G.; De Gressi, S.; Morgan, N.V.; et al. Multiplex ligation-dependent probe amplification (MLPA) analysis is an effective tool for the detection of novel intragenic PLA2G6 mutations: Implications for molecular diagnosis. Mol. Genet. Metab. 2010, 100, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, A.; Romaniello, R.; Grasso, R.; Cavallini, A.; Righini, A.; Bresolin, N.; Borgatti, R.; Bassi, M.T. Novel splice-site mutations and a large intragenic deletion in PLA2G6 associated with a severe and rapidly progressive form of infantile neuroaxonal dystrophy. Clin. Genet. 2010, 78, 432–440. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shimojima, K.; Shibata, T.; Akiyama, M.; Oka, M.; Akiyama, T.; Yoshinaga, H.; Kobayashi, K. Novel PLA2G6 mutations associated with an exonic deletion due to non-allelic homologous recombination in a patient with infantile neuroaxonal dystrophy. Hum. Genome Var. 2015, 2, 15048. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Bhatia, K.P.; Li, A.; Hernandez, D.; Davis, M.; Wood, N.W.; Hardy, J.; Houlden, H.; Singleton, A.; Schneider, S.A. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann. Neurol. 2009, 65, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Li, A.; Schneider, S.A.; Holton, J.L.; Johnson, R.; Kidd, D.; Chataway, J.; Bhatia, K.P.; Lees, A.J.; Hardy, J.; et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol. Aging 2012, 33, 814–823. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S. Terminal axon pathology in infantile neuroaxonal dystrophy. Pediatr. Neurol. 1991, 7, 116–120. [Google Scholar] [CrossRef]
- Sumi-Akamaru, H.; Beck, G.; Kato, S.; Mochizuki, H. Neuroaxonal dystrophy in PLA2G6 knockout mice. Neuropathology 2015, 35, 289–302. [Google Scholar] [CrossRef]
- Malik, I.; Turk, J.; Mancuso, D.J.; Montier, L.; Wohltmann, M.; Wozniak, D.F.; Schmidt, R.E.; Gross, R.W.; Kotzbauer, P.T. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am. J. Pathol. 2008, 172, 406–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinzawa, K.; Sumi, H.; Ikawa, M.; Matsuoka, Y.; Okabe, M.; Sakoda, S.; Tsujimoto, Y. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: A model of human neurodegenerative disease. J. Neurosci. 2008, 28, 2212–2220. [Google Scholar] [CrossRef] [Green Version]
- Malley, K.R.; Koroleva, O.; Miller, I.; Sanishvili, R.; Jenkins, C.M.; Gross, R.W.; Korolev, S. The structure of iPLA2beta reveals dimeric active sites and suggests mechanisms of regulation and localization. Nat. Commun. 2018, 9, 765. [Google Scholar] [CrossRef] [Green Version]
- Winstead, M.V.; Balsinde, J.; Dennis, E.A. Calcium-independent phospholipase A(2): Structure and function. Biochim. Biophys. Acta 2000, 1488, 28–39. [Google Scholar] [CrossRef]
- Tang, J.; Kriz, R.W.; Wolfman, N.; Shaffer, M.; Seehra, J.; Jones, S.S. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J. Biol. Chem. 1997, 272, 8567–8575. [Google Scholar] [CrossRef] [Green Version]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, J.; White, T.D.; Nelson, A.J.; Lei, X.; Ramanadham, S. iPLA2beta and its role in male fertility, neurological disorders, metabolic disorders, and inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Mosior, M.; Johnson, C.A.; Chen, Y.; Dennis, E.A. Group-specific assays that distinguish between the four major types of mammalian phospholipase A2. Anal. Biochem. 1999, 269, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.Y.; Yeo, J.F.; Ling, S.F.; Farooqui, A.A. Distribution of calcium-independent phospholipase A2 (iPLA 2) in monkey brain. J. Neurocytol. 2005, 34, 447–458. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, X.; Zhao, C.; Choi, J.; Shi, J.; Song, K.; Turk, J.; Ma, Z.A. Protection of pancreatic beta-cells by group VIA phospholipase A(2)-mediated repair of mitochondrial membrane peroxidation. Endocrinology 2010, 151, 3038–3048. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.D.; Gottlieb, R.A. Inhibition of mitochondrial calcium-independent phospholipase A2 (iPLA2) attenuates mitochondrial phospholipid loss and is cardioprotective. Biochem. J. 2002, 362, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Seleznev, K.; Zhao, C.; Zhang, X.H.; Song, K.; Ma, Z.A. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J. Biol. Chem. 2006, 281, 22275–22288. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Bao, S.; Lei, X.; Jin, C.; Zhang, S.; Turk, J.; Ramanadham, S. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA(2)beta. Biochim. Biophys. Acta 2010, 1801, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Davids, M.; Kane, M.S.; He, M.; Wolfe, L.A.; Li, X.; Raihan, M.A.; Chao, K.R.; Bone, W.P.; Boerkoel, C.F.; Gahl, W.A.; et al. Disruption of Golgi morphology and altered protein glycosylation in PLA2G6-associated neurodegeneration. J. Med. Genet. 2016, 53, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Lee, P.T.; Chen, K.; Mao, D.; Tan, K.L.; Zuo, Z.; Lin, W.W.; Wang, L.; Bellen, H.J. Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to alpha-Synuclein Gain. Cell Metab. 2018, 28, 605–618.e6. [Google Scholar] [CrossRef] [Green Version]
- Beck, G.; Sugiura, Y.; Shinzawa, K.; Kato, S.; Setou, M.; Tsujimoto, Y.; Sakoda, S. Sumi-Akamaru, H. Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 2011, 31, 11411–11420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, Y.; Kim, H.W.; Igarashi, M.; Modi, H.R.; Chang, L.; Ma, K.; Greenstein, D.; Wohltmann, M.; Turk, J.; Rapoport, S.I.; et al. Disturbed brain phospholipid and docosahexaenoic acid metabolism in calcium-independent phospholipase A(2)-VIA (iPLA(2)beta)-knockout mice. Biochim. Biophys. Acta 2012, 1821, 1278–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strokin, M.; Seburn, K.L.; Cox, G.A.; Martens, K.A.; Reiser, G. Severe disturbance in the Ca2+ signaling in astrocytes from mouse models of human infantile neuroaxonal dystrophy with mutated Pla2g6. Hum. Mol. Genet. 2012, 21, 2807–2814. [Google Scholar] [CrossRef] [PubMed]
- Strokin, M.; Reiser, G. Mitochondria from a mouse model of the human infantile neuroaxonal dystrophy (INAD) with genetic defects in VIA iPLA2 have disturbed Ca(2+) regulation with reduction in Ca(2+) capacity. Neurochem. Int. 2016, 99, 187–193. [Google Scholar] [CrossRef]
- Strokin, M.; Reiser, G. Neurons and astrocytes in an infantile neuroaxonal dystrophy (INAD) mouse model show characteristic alterations in glutamate-induced Ca(2+) signaling. Neurochem. Int. 2017, 108, 121–132. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cocheme, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Porter, N.A.; Schneider, C.; Brash, A.R.; Yin, H. Formation of 4-hydroxynonenal from cardiolipin oxidation: Intramolecular peroxyl radical addition and decomposition. Free Radic. Biol. Med. 2011, 50, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.C.; Lu, C.S.; Weng, Y.H.; Chen, Y.L.; Huang, Y.Z.; Chen, R.S.; Cheng, Y.C.; Huang, Y.C.; Liu, Y.C.; Lai, S.C.; et al. PARK14 (D331Y) PLA2G6 Causes Early-Onset Degeneration of Substantia Nigra Dopaminergic Neurons by Inducing Mitochondrial Dysfunction, ER Stress, Mitophagy Impairment and Transcriptional Dysregulation in a Knockin Mouse Model. Mol. Neurobiol. 2019, 56, 3835–3853. [Google Scholar] [CrossRef]
- Iliadi, K.G.; Gluscencova, O.B.; Iliadi, N.; Boulianne, G.L. Mutations in the Drosophila homolog of human PLA2G6 give rise to age-dependent loss of psychomotor activity and neurodegeneration. Sci. Rep. 2018, 8, 2939. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.C.; Yeh, T.H.; Lu, C.S.; Huang, Y.C.; Cheng, Y.C.; Huang, Y.Z.; Weng, Y.H.; Liu, Y.C.; Lai, S.C.; Chen, Y.L.; et al. PARK14 PLA2G6 mutants are defective in preventing rotenone-induced mitochondrial dysfunction, ROS generation and activation of mitochondrial apoptotic pathway. Oncotarget 2017, 8, 79046–79060. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wang, J.; Zhao, C.; Bi, W.; Yue, Z.; Ma, Z.A. Genetic ablation of PLA2G6 in mice leads to cerebellar atrophy characterized by Purkinje cell loss and glial cell activation. PLoS ONE 2011, 6, e26991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, H.; Taha, A.Y.; Cheon, Y.; Kim, H.W.; Turk, J.; Rapoport, S.I. iPLA2beta knockout mouse, a genetic model for progressive human motor disorders, develops age-related neuropathology. Neurochem. Res. 2014, 39, 1522–1532. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.P.; Su, K.P.; Mondelli, V.; Pariante, C.M. Omega-3 Polyunsaturated Fatty Acids in Youths with Attention Deficit Hyperactivity Disorder: A Systematic Review and Meta-Analysis of Clinical Trials and Biological Studies. Neuropsychopharmacology 2018, 43, 534–545. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.; Midei, M.; Dastgir, J.; Flora, C.; Molinari, R.J.; Heerinckx, F.; Endemann, S.; Atwal, P.; Milner, P.; Shchepinov, M.S. Treatment of infantile neuroaxonal dystrophy with RT001: A di-deuterated ethyl ester of linoleic acid: Report of two cases. JIMD Rep. 2020, 54, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, J.; Wang, H.; Fu, Z.; Yu, Y. Identification and expression analysis of ERF transcription factor genes in petunia during flower senescence and in response to hormone treatments. J. Exp. Bot. 2011, 62, 825–840. [Google Scholar] [CrossRef] [PubMed]
- Hartig, M.; Prokisch, H.; Meitinger, T.; Klopstock, T. Mitochondrial membrane protein-associated neurodegeneration (MPAN). Int. Rev. Neurobiol. 2013, 110, 73–84. [Google Scholar]
- Hartig, M.B.; Iuso, A.; Haack, T.; Kmiec, T.; Jurkiewicz, E.; Heim, K.; Roeber, S.; Tarabin, V.; Dusi, S.; Krajewska-Walasek, M.; et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2011, 89, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Gregory, A.; Lotia, M.; Jeong, S.Y.; Fox, R.; Zhen, D.; Sanford, L.; Hamada, J.; Jahic, A.; Beetz, C.; Freed, A.; et al. Autosomal dominant mitochondrial membrane protein-associated neurodegeneration (MPAN). Mol. Genet. Genom. Med. 2019, 7, e00736. [Google Scholar] [CrossRef] [Green Version]
- Meilleur, K.G.; Traore, M.; Sangare, M.; Britton, A.; Landoure, G.; Coulibaly, S.; Niare, B.; Mochel, F.; La Pean, A.; Rafferty, I.; et al. Hereditary spastic paraplegia and amyotrophy associated with a novel locus on chromosome 19. Neurogenetics 2010, 11, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landoure, G.; Zhu, P.P.; Lourenco, C.M.; Johnson, J.O.; Toro, C.; Bricceno, K.V.; Rinaldi, C.; Meilleur, K.G.; Sangare, M.; Diallo, O.; et al. Hereditary spastic paraplegia type 43 (SPG43) is caused by mutation in C19orf12. Hum. Mutat. 2013, 34, 1357–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschauer, M.; Gaul, C.; Behrmann, C.; Prokisch, H.; Zierz, S.; Haack, T.B. C19orf12 mutations in neurodegeneration with brain iron accumulation mimicking juvenile amyotrophic lateral sclerosis. J. Neurol. 2012, 259, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Salih, M.A.; Mooney, C.; Alzahrani, J.; Elmalik, S.A.; Kabiraj, M.M.; Khan, A.O.; Paudel, R.; Houlden, H.; Azzedine, H.; et al. C19orf12 mutation leads to a pallido-pyramidal syndrome. Gene 2014, 537, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venco, P.; Bonora, M.; Giorgi, C.; Papaleo, E.; Iuso, A.; Prokisch, H.; Pinton, P.; Tiranti, V. Mutations of C19orf12, coding for a transmembrane glycine zipper containing mitochondrial protein, cause mis-localization of the protein, inability to respond to oxidative stress and increased mitochondrial Ca(2)(+). Front. Genet. 2015, 6, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iuso, A.; Sibon, O.C.; Gorza, M.; Heim, K.; Organisti, C.; Meitinger, T.; Prokisch, H. Impairment of Drosophila orthologs of the human orphan protein C19orf12 induces bang sensitivity and neurodegeneration. PLoS ONE 2014, 9, e89439. [Google Scholar] [CrossRef] [Green Version]
- Olgiati, S.; Dogu, O.; Tufekcioglu, Z.; Diler, Y.; Saka, E.; Gultekin, M.; Kaleagasi, H.; Kuipers, D.; Graafland, J.; Breedveld, G.J.; et al. The p.Thr11Met mutation in c19orf12 is frequent among adult Turkish patients with MPAN. Parkinsonism Relat. Disord. 2017, 39, 64–70. [Google Scholar] [CrossRef]
- Gore, E.; Appleby, B.S.; Cohen, M.L.; DeBrosse, S.D.; Leverenz, J.B.; Miller, B.L.; Siedlak, S.L.; Zhu, X.; Lerner, A.J. Clinical and imaging characteristics of late onset mitochondrial membrane protein-associated neurodegeneration (MPAN). Neurocase 2016, 22, 476–483. [Google Scholar] [CrossRef] [Green Version]
- Kolisek, M.; Sponder, G.; Mastrototaro, L.; Smorodchenko, A.; Launay, P.; Vormann, J.; Schweigel-Rontgen, M. Substitution p.A350V in Na(+)/Mg(2)(+) exchanger SLC41A1, potentially associated with Parkinson’s disease, is a gain-of-function mutation. PLoS ONE 2013, 8, e71096. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.B.; Liu, J.Y.; Xu, X.J.; Mao, X.Y.; Zhang, W.; Zhou, H.H.; Liu, Z.Q. Neurodegeneration with brain iron accumulation: Insights into the mitochondria dysregulation. Biomed. Pharmacother. 2019, 118, 109068. [Google Scholar] [CrossRef]
- Kruer, M.C.; Paisan-Ruiz, C.; Boddaert, N.; Yoon, M.Y.; Hama, H.; Gregory, A.; Malandrini, A.; Woltjer, R.L.; Munnich, A.; Gobin, S.; et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann. Neurol. 2010, 68, 611–618. [Google Scholar] [CrossRef]
- Rattay, T.W.; Lindig, T.; Baets, J.; Smets, K.; Deconinck, T.; Sohn, A.S.; Hortnagel, K.; Eckstein, K.N.; Wiethoff, S.; Reichbauer, J.; et al. FAHN/SPG35: A narrow phenotypic spectrum across disease classifications. Brain 2019, 142, 1561–1572. [Google Scholar] [CrossRef]
- Eckhardt, M.; Yaghootfam, A.; Fewou, S.N.; Zoller, I.; Gieselmann, V. A mammalian fatty acid hydroxylase responsible for the formation of alpha-hydroxylated galactosylceramide in myelin. Biochem. J. 2005, 388, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Zoller, I.; Meixner, M.; Hartmann, D.; Bussow, H.; Meyer, R.; Gieselmann, V.; Eckhardt, M. Absence of 2-hydroxylated sphingolipids is compatible with normal neural development but causes late-onset axon and myelin sheath degeneration. J. Neurosci. 2008, 28, 9741–9754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, K.A.; Kern, M.J.; Fullbright, G.; Bielawski, J.; Scherer, S.S.; Yum, S.W.; Li, J.J.; Cheng, H.; Han, X.; Venkata, J.K.; et al. Central nervous system dysfunction in a mouse model of FA2H deficiency. Glia 2011, 59, 1009–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mari, F.; Berti, B.; Romano, A.; Baldacci, J.; Rizzi, R.; Grazia Alessandri, M.; Tessa, A.; Procopio, E.; Rubegni, A.; Lourenco, C.M.; et al. Clinical and neuroimaging features of autosomal recessive spastic paraplegia 35 (SPG35): Case reports, new mutations, and brief literature review. Neurogenetics 2018, 19, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Hogarth, P.; Kruer, M.C.; Gregory, A.; Wieland, T.; Schwarzmayr, T.; Graf, E.; Sanford, L.; Meyer, E.; Kara, E.; et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 2012, 91, 1144–1149. [Google Scholar] [CrossRef] [Green Version]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013, 45, 445–449. [Google Scholar] [CrossRef]
- Hayflick, S.J.; Kruer, M.C.; Gregory, A.; Haack, T.B.; Kurian, M.A.; Houlden, H.H.; Anderson, J.; Boddaert, N.; Sanford, L.; Harik, S.I.; et al. beta-Propeller protein-associated neurodegeneration: A new X-linked dominant disorder with brain iron accumulation. Brain 2013, 136, 1708–1717. [Google Scholar] [CrossRef]
- Maeda, S.; Otomo, C.; Otomo, T. The autophagic membrane tether ATG2A transfers lipids between membranes. Elife 2019, 8, e45777. [Google Scholar] [CrossRef]
- Lu, Q.; Yang, P.; Huang, X.; Hu, W.; Guo, B.; Wu, F.; Lin, L.; Kovacs, A.L.; Yu, L.; Zhang, H. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev. Cell. 2011, 21, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Bozic, M.; van den Bekerom, L.; Milne, B.A.; Goodman, N.; Roberston, L.; Prescott, A.R.; Macartney, T.J.; Dawe, N.; McEwan, D.G. A conserved ATG2-GABARAP family interaction is critical for phagophore formation. EMBO Rep. 2020, 21, e48412. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Sun, L.; Miao, G.; Ji, C.; Zhao, H.; Sun, H.; Miao, L.; Yoshii, S.R.; Mizushima, N.; Wang, X.; et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 2015, 11, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; Wang, Q.; Chen, X.; Zeng, Q.; Shao, Y.; Fang, H.; Liao, X.; Li, H.S.; Liu, M.G.; Xu, T.L.; et al. WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy 2020, 16, 531–547. [Google Scholar] [CrossRef]
- Ingrassia, R.; Memo, M.; Garavaglia, B. Ferrous Iron Up-regulation in Fibroblasts of Patients with Beta Propeller Protein-Associated Neurodegeneration (BPAN). Front. Genet. 2017, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibler, P.; Burbulla, L.F.; Dulovic, M.; Zittel, S.; Heine, J.; Schmidt, T.; Rudolph, F.; Westenberger, A.; Rakovic, A.; Munchau, A.; et al. Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. Brain 2018, 141, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- Najim al-Din, A.S.; Wriekat, A.; Mubaidin, A.; Dasouki, M.; Hiari, M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol. Scand. 1994, 89, 347–352. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Bras, J.; Verloes, A.; Schneider, S.A.; Mole, S.E.; Guerreiro, R.J. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum. Mol. Genet. 2012, 21, 2646–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada-Cuzcano, A.; Martin, S.; Chamova, T.; Synofzik, M.; Timmann, D.; Holemans, T.; Andreeva, A.; Reichbauer, J.; De Rycke, R.; Chang, D.I.; et al. Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 2017, 140, 287–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, M.I.; Bruggemann, N.; Chana, P.; Venegas, P.; Kagi, M.; Parrao, T.; Orellana, P.; Garrido, C.; Rojas, C.V.; Hauke, J.; et al. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov. Disord. 2010, 25, 1929–1937. [Google Scholar] [CrossRef]
- Schneider, S.A.; Paisan-Ruiz, C.; Quinn, N.P.; Lees, A.J.; Houlden, H.; Hardy, J.; Bhatia, K.P. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov. Disord. 2010, 25, 979–984. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Rane, A.; Chinta, S.J.; Andersen, J.K. Regulation of ATP13A2 via PHD2-HIF1alpha Signaling Is Critical for Cellular Iron Homeostasis: Implications for Parkinson’s Disease. J. Neurosci. 2016, 36, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- de Tezanos Pinto, F.; Adamo, H.P. The strategic function of the P5-ATPase ATP13A2 in toxic waste disposal. Neurochem. Int. 2018, 112, 108–113. [Google Scholar] [CrossRef]
- Park, J.S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef] [Green Version]
- Gusdon, A.M.; Zhu, J.; Van Houten, B.; Chu, C.T. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol. Dis. 2012, 45, 962–972. [Google Scholar] [CrossRef] [Green Version]
- Tsunemi, T.; Hamada, K.; Krainc, D. ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J. Neurosci. 2014, 34, 15281–15287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunemi, T.; Perez-Rosello, T.; Ishiguro, Y.; Yoroisaka, A.; Jeon, S.; Hamada, K.; Rammonhan, M.; Wong, Y.C.; Xie, Z.; Akamatsu, W.; et al. Increased Lysosomal Exocytosis Induced by Lysosomal Ca(2+) Channel Agonists Protects Human Dopaminergic Neurons from alpha-Synuclein Toxicity. J. Neurosci. 2019, 39, 5760–5772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemann, N.; Jankovic, J. Juvenile parkinsonism: Differential diagnosis, genetics, and treatment. Parkinsonism Relat. Disord. 2019, 67, 74–89. [Google Scholar] [CrossRef]
- Levi, S.; Finazzi, D. Neurodegeneration with brain iron accumulation: Update on pathogenic mechanisms. Front. Pharmacol. 2014, 5, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, G.; Busti, F.; Lira Zidanes, A.; Castagna, A.; Girelli, D. Aceruloplasminemia: A Severe Neurodegenerative Disorder Deserving an Early Diagnosis. Front. Neurosci. 2019, 13, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeill, A.; Pandolfo, M.; Kuhn, J.; Shang, H.; Miyajima, H. The neurological presentation of ceruloplasmin gene mutations. Eur. Neurol. 2008, 60, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Kono, S.; Miyajima, H. Molecular and pathological basis of aceruloplasminemia. Biol. Res. 2006, 39, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Cuyar, L.F.; Perry, G.; Miyajima, H.; Atwood, C.S.; Riveros-Angel, M.; Lyons, P.F.; Siedlak, S.L.; Smith, M.A.; Castellani, R.J. Redox active iron accumulation in aceruloplasminemia. Neuropathology 2008, 28, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Kono, S. Aceruloplasminemia: An update. Int. Rev. Neurobiol. 2013, 110, 125–151. [Google Scholar] [PubMed]
- Oshiro, S.; Kawahara, M.; Kuroda, Y.; Zhang, C.; Cai, Y.; Kitajima, S.; Shirao, M. Glial cells contribute more to iron and aluminum accumulation but are more resistant to oxidative stress than neuronal cells. Biochim. Biophys. Acta 2000, 1502, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Yoshida, K.; Miyagoe, Y.; Ishikawa, A.; Hanaoka, K.; Nomoto, S.; Kaneko, K.; Ikeda, S.; Takeda, S. Quantitative evaluation of expression of iron-metabolism genes in ceruloplasmin-deficient mice. Biochim. Biophys. Acta 2002, 1588, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grenier, D.; Huot, M.P.; Mayrand, D. Iron-chelating activity of tetracyclines and its impact on the susceptibility of Actinobacillus actinomycetemcomitans to these antibiotics. Antimicrob. Agents Chemother. 2000, 44, 763–766. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, M.; Hashioka, S.; Miki, H.; Nagahama, M.; Wake, R.; Miyaoka, T.; Horiguchi, J. Aceruloplasminemia With Psychomotor Excitement and Neurological Sign Was Improved by Minocycline (Case Report). Medicine 2016, 95, e3594. [Google Scholar] [CrossRef]
- Kuhn, J.; Bewermeyer, H.; Miyajima, H.; Takahashi, Y.; Kuhn, K.F.; Hoogenraad, T.U. Treatment of symptomatic heterozygous aceruloplasminemia with oral zinc sulphate. Brain Dev. 2007, 29, 450–453. [Google Scholar] [CrossRef]
- Muhoberac, B.B.; Vidal, R. Iron, Ferritin, Hereditary Ferritinopathy, and Neurodegeneration. Front. Neurosci. 2019, 13, 1195. [Google Scholar] [CrossRef] [Green Version]
- Muhoberac, B.B.; Vidal, R. Abnormal iron homeostasis and neurodegeneration. Front. Aging Neurosci. 2013, 5, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbeito, A.G.; Levade, T.; Delisle, M.B.; Ghetti, B.; Vidal, R. Abnormal iron metabolism in fibroblasts from a patient with the neurodegenerative disease hereditary ferritinopathy. Mol. Neurodegener. 2010, 5, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbeito, A.G.; Garringer, H.J.; Baraibar, M.A.; Gao, X.; Arredondo, M.; Nunez, M.T.; Smith, M.A.; Ghetti, B.; Vidal, R. Abnormal iron metabolism and oxidative stress in mice expressing a mutant form of the ferritin light polypeptide gene. J. Neurochem. 2009, 109, 1067–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garringer, H.J.; Irimia, J.M.; Li, W.; Goodwin, C.B.; Richine, B.; Acton, A.; Chan, R.J.; Peacock, M.; Muhoberac, B.B.; Ghetti, B.; et al. Effect of Systemic Iron Overload and a Chelation Therapy in a Mouse Model of the Neurodegenerative Disease Hereditary Ferritinopathy. PLoS ONE 2016, 11, e0161341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alazami, A.M.; Al-Saif, A.; Al-Semari, A.; Bohlega, S.; Zlitni, S.; Alzahrani, F.; Bavi, P.; Kaya, N.; Colak, D.; Khalak, H.; et al. Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am. J. Hum. Genet. 2008, 83, 684–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodhouse, N.J.; Sakati, N.A. A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities. J. Med. Genet. 1983, 20, 216–219. [Google Scholar] [CrossRef] [Green Version]
- Koshy, G.; Danda, S.; Thomas, N.; Mathews, V.; Viswanathan, V. Three siblings with Woodhouse-Sakati syndrome in an Indian family. Clin. Dysmorphol. 2008, 17, 57–60. [Google Scholar] [CrossRef]
- Alazami, A.M.; Schneider, S.A.; Bonneau, D.; Pasquier, L.; Carecchio, M.; Kojovic, M.; Steindl, K.; de Kerdanet, M.; Nezarati, M.M.; Bhatia, K.P.; et al. C2orf37 mutational spectrum in Woodhouse-Sakati syndrome patients. Clin. Genet. 2010, 78, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Steindl, K.; Alazami, A.M.; Bhatia, K.P.; Wuerfel, J.T.; Petersen, D.; Cartolari, R.; Neri, G.; Klein, C.; Mongiardo, B.; Alkuraya, F.S.; et al. A novel C2orf37 mutation causes the first Italian cases of Woodhouse Sakati syndrome. Clin. Genet. 2010, 78, 594–597. [Google Scholar] [CrossRef]
- Ben-Omran, T.; Ali, R.; Almureikhi, M.; Alameer, S.; Al-Saffar, M.; Walsh, C.A.; Felie, J.M.; Teebi, A. Phenotypic heterogeneity in Woodhouse-Sakati syndrome: Two new families with a mutation in the C2orf37 gene. Am. J. Med. Genet. A 2011, 155, 2647–2653. [Google Scholar] [CrossRef]
- Agopiantz, M.; Corbonnois, P.; Sorlin, A.; Bonnet, C.; Klein, M.; Hubert, N.; Pascal-Vigneron, V.; Jonveaux, P.; Cuny, T.; Leheup, B.; et al. Endocrine disorders in Woodhouse-Sakati syndrome: A systematic review of the literature. J. Endocrinol. Invest. 2014, 37, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.H.; Shah, K.; Nasir, A.; Steyaert, W.; Coucke, P.J.; Ahmad, W. Exome sequencing revealed a novel biallelic deletion in the DCAF17 gene underlying Woodhouse Sakati syndrome. Clin. Genet. 2016, 90, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Gurbuz, F.; Desai, S.; Diao, F.; Turkkahraman, D.; Wranitz, F.; Wood-Trageser, M.; Shin, Y.H.; Kotan, L.D.; Jiang, H.; Witchel, S.; et al. Novel inactivating mutations of the DCAF17 gene in American and Turkish families cause male infertility and female subfertility in the mouse model. Clin. Genet. 2018, 93, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhou, P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell. 2007, 26, 775–780. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene (OMIM*) | NBIA form (OMIM#)—Inheritance Frequency | Clinical and MRI Findings | Protein | Function |
|---|---|---|---|---|
| PANK2 (606157) | Pantothenate kinase-associated neurodegeneration (PKAN) (234200)—AR 35–50% | Onset is in early childhood, although atypical forms may occur at adolescence or early adulthood. Dystonia (oromandibular dystonia is prominent), parkinsonism, spasticity, pigmentary retinal degeneration, acanthocytosis, cognitive decline, neuropsychiatric disturbances. Hypointensity with central hyperintensity of the GP (“eye of the tiger”). | Pantothenate kinase 2 | PANK2 takes part in the first step of the CoA synthesis: PANK2 catalyzes the ATP-dependent phosphorylation of pantothenate to 4-phosphopantethenate. |
| PLA2G6 (603604) | PLA2G6-associated neurodegeneration (PLAN) (610217)—AR ~20% | Classical forms begin at infancy with progressive motor and mental retardation, marked truncal hypotonia, optic atrophy, pyramidal signs, and seizures. Atypical forms have an onset at adolescence or early adulthood, and include dystonia-parkinsonism, cerebellar ataxia, eye movement abnormalities, cognitive decline, and psychiatric disturbances. Hypointensity of the GP and in SN (in <50% of patients). | Phospholipase A2, group VI | PLA2G6 hydrolyses glycerophospholipids at the sn-2 position of acyl chains to produce lysophospholipids and free fatty acids, which participates in essential functions such as membrane remodelling, fatty acid oxidation, cell signaling, apoptosis, etc. |
| C19ORF12 (614297) | Mitochondrial membrane protein-associated neurodegeneration (MPAN) (614298)—AR 6–10% | Childhood to early adulthood-onset with prominent neuropathy. Patients present with spastic para-or tetraparesis with muscle atrophy. Parkinsonism and dystonia are frequent. Optic atrophy, dementia, and psychiatric symptoms may happen. Hypointensity of the GP and SN plus hyperintensity in the GP. | Chromosome 19 open reading frame 12 | Unknown function. Mitochondrial protein linked to lipid homeostasis because its expression follows fatty acid metabolism. |
| WDR45 (300526) | β-propeller-associated neurodegeneration (BPAN) (300894)—XD 1–2% | Childhood type shows developmental delay and intellectual disability, mimicking an atypical Rett syndrome. In adulthood, patients present with dystonia-parkinsonism and dementia, together with seizures, and ataxia. Hypointensity of the GP and SN with central hyperintense line. | WD (tryptophan-aspartic acid) repeat domain 45 | Member of the family of the WD40 proteins that promote protein-protein interactions and plays a role in autophagy, cell cycle and transcriptional control, and transduction. WDR45 is critical for autophagosome formation. |
| FA2H (611026) | Fatty acid hydroxylase-associated neurodegeneration (FANH) (612319)—AR Rare | Childhood-onset gait impairment caused by spastic paraplegia, ataxia and dystonia. Seizures and optic atrophy may be present. Hypointensity of the GP in some patients. | Fatty acid 2 hydroxylase | Enzyme that catalyzes the hydroxylation of fatty acids at position 2 of the N-acyl chain; 2-hydroxy-fatty acids are a precursor for ceramide synthesis (component of myelin sheaths). |
| FTL (134790) | Neuroferritinopathy (NF) (606159)—AD Rare | Adult-onset chorea with cognitive defects and psychiatric features. Cerebellar ataxia, parkinsonism, blepharospam and orolingual-mandibular dyskinesia. Hypointensity in basal ganglia, mainly GP and SN. | Ferritin | Major storage protein for cellular iron (up to 4500 iron atoms) consisting of a heavy chain with ferroxidase activity and a light chain that aids mineralization within the ferritin structure. |
| CP (117700) | Aceruloplasminemia (604290)—AR Rare | Adult-onset form with cognitive decline, cerebellar ataxia, and craniofacial dyskinesia, besides of retinal degeneration, and even, diabetes caused by the accumulation of iron. Hypointense striatum, thalamus and dentate. | Ceruloplasmin | Ferroxidase that facilitates ferroportin-mediated cellular iron export |
| DCAF17 612515 | Woodhouse-Sakati syndrome (WSS) (241080)—AR Rare | Multisystem disorder that presents hypogonadism, hair thinning-to-alopecia, diabetes mellitus, deafness, intellectual disability, extrapyramidal features. Additional symptoms are seizures, polyneuropathy, thyroid dysfunction, keratoconus, and syndactyly. Hypointensity of the GP and SN. | Db1 and Cul4-associated factor 17 | Unclear function. DCAF17 seems to be associated with protein ubiquitination involved in DNA damage and cell cycle control. |
| ATP13A2 (610513) | Kufor-Rakeb syndrome (KRS) (606693)—AR Two probands | Onset is usually in adolescence. KRS is characterized by juvenile parkinsonism, dystonia, eye movement abnormalities, autonomic dysfunction, dementia, and psychiatric features, Hypointensity in the basal ganglia/caudo-putamen. | ATPase cation transporting 13A2 | P-type ATPase associated with membranes of lysosomes, which is a divalent cation transporter that uses the energy stored in ATP to transport ions across membranes. Mislocalization of ATP13A2 to the endoplasmic reticulum and Zn2+ dyshomeostasis are crucial processes. |
| COASY (609855) | COASY protein-associated neurodegeneration (CoPAN) (615643)—AR Five probands | Early onset gait impairment and learning difficulties, with further dystonia, and spasticity. Hypointensity with central hyperintensity of the GP. | CoA synthase | Bifunctional enzyme that catalyzes the final two steps of the CoA synthesis. |
| Gene (OMIM*) | Disease (OMIM#) Inheritance | Clinical and MRI Findings | Protein | Function | References |
|---|---|---|---|---|---|
| FBX07 | Parkinson disease 15 (260300) AR | A girl with a parkinsonian-pyramidal syndrome and brain iron accumulation. Light vermian atrophy progressing to cerebral and cerebellar atrophy, and a hot cross bun sign suggesting ponto-cerebellar tract degeneration. Excessive brain iron deposition in the GP and SN appeared as hypointense signal on iron-sensitive sequences | F-box only protein 7 | Ubiquitin protein ligase involved in ubiquitin proteasome activity and parkin-mediated mitophagy. | [2] |
| FUCA1 (612280) | Fucosidosis (230000) AR | A 14 year-old girl with progressive fixed dystonia and red spots on the skin. Axial T2-weighted images showed slightly hyperintense spot within a hypointense GP and mild hyperintensity of the subcortical white matter consistent with diffuse mild hypomyelination. Over the years, brain atrophy was noticed. | α-L-fucosidase | Lysosomal enzyme involved in the degradation of fucose-containing glycoproteins and glycolipids. | [3] |
| GLB1 (611458) | GM1-gangliosidosis type I (infantile form; 230500) type II (juvenile form; 230600) type III (adult form; 8230650) AR | Two brothers with the juvenile form of GM1 gangliosidosis. A T2-weighted image revealed significant cerebral atrophy with barely noticeable hypointense signals in GP in the proband (9 years old). The cerebral atrophy and paramagnetic signals in the basal ganglia became more pronounced over the years. His affected brother presented a similar clinical course. | β-galactosidase-1 | Lysosomal hydrolase that cleaves the terminal β-galactose from ganglioside substrates and other glycoconjugates. | [4] |
| A 12 year-old boy with GM1 gangliosidosis type II. MRI revealed a hypointense T2 signal in the SN and in the GP, with marked hypointensity in susceptibility-weighted imaging and slight T2 hyperintensity in the posterior part of the putamen. | [5] | ||||
| GTPBP2 (607434) | Jaberi-Elahi syndrome (617988) AR | Three affected siblings who presented a complex neurologic disorder accompanied with mental deficiency and ataxia and dystonia features. Brain MRI showed cerebellar vermian atrophy, and susceptibility weighted imaging revealed hypointensity signal in the GP and SN suggesting NBIA. | GTP-binding protein 2 | GTP-binding protein that belongs to the GTPase superfamily whose members play a role in a wide variety of biological process (cell proliferation and differentiation, intracellular transport, regulation of cytoskeleton, and protein synthesis). | [6] |
| SPC2 (184755) | Leukoencephalopathy with dystonia and motor neuropathy (613724) AR | Adult-onset disease with brain MRI characteristic of NBIA: T2-weighted imaging showed signal change in the subcortical white matter, thalamus, globus pallidus cerebral peduncles, and pons. Susceptibility-weighted imaging showed signal dropout in the GP, SN, red nuclei, and dentate nuclei | Sterol carrier protein-2 | Peroxisomal enzyme with thiolase activity that is required for the breakdown of branched-chain fatty acids. | [7] |
| TBCE (604934) | Encephalopathy with amyotrophy and optic atrophy (617207) AR | Patients suffering from an early-onset and progressive neurodegenerative encephalopathy with distal spinal muscular atrophy, and brain iron accumulation in the 2nd decade at the level of the GP and SN. | Tubulin folding cofactor E | Chaperone required for the proper tubulin folding and polymerization. | [8] |
| KMT2B (606834) | Dystonia 28, childhood-onset (617284) AD | Several patients suffering from an early-onset progressive dystonia, with subtle, low pallidal signal on T2*, diffusion and susceptibility weighted sequences, particularly affecting the lateral aspect of the GP externa. | Lysine-specific methyltransferase 2B | Histone lysine methyltransferase involved in methylation of histone H3 at lysine 4 (H3K4) | [9] |
| DDHD1 (614603) | Spastic paraplegia 28 (609340) AR | A 55 year-old man with a complex form of spastic paraplegia with retinal dystrophy. MRI revealed a thin corpus callosum, a billateral T2 hyposignal of the internal pallidi and a hypersignal of the supratentorial white matter. | DDHD domain-containing protein 1 | Phospholipase that hydrolyzes phosphatidic acid and attenuates cell activation | [10] |
| AP4M1 (602296) | Spastic paraplegia 50 (612936) AR | Three affected relatives who exhibited very early psychomotor retardation with spasticity and severe mental deficiency. MRI showed global cerebral atrophy, white matter loss asymmetric ventriculomegaly and thining of the splenium of the corpus callosum. T2 sequences revealed symmetric mild hypointensity of the GP. | Adaptor-related protein complex 4 subunit MU-1 | Subunit of the heterotetrameric adaptor protein (AP) complex, which plays a role in the recognition and sorting of cargo proteins with tyrosine-based motifs from the trans-golgi network to the endosomal-lysosomal system | [11] |
| AP4S1 (607243) | Spastic paraplegia 52 (614067) AR | AP4 deficiency with ataxia and optic atrophy in a girl who presented with bilateral hypointense signal alterations in the GP in T2-weighted, suggesting abnormal iron accumulation. | Adaptor-related protein complex 4 σ-subunit MU-1 | Subunit of the heterotetrameric adaptor protein (AP) complex, which plays a role in the recognition and sorting of cargo proteins with tyrosine-based motifs from the trans-golgi network to the endosomal-lysosomal system. | [12] |
| RNASEH2B (610326) | Aicardi-Goutieres syndrome 2 (610181) AR | Two affected siblings who manifested encephalopathy with spastic quadriplegia and anarthria but preserved intellect. MRI revealed hypointensity of GP on T2, consistent with iron deposition | Ribonuclease H2 subunit B | Subunit of the human ribonuclease H2 enzyme complex that cleaves ribonucleotides from RNA-DNA duplexes. | [13] |
| LAMAN (609458) | α-Mannosidosis types I and II (248500) AR | A 34 year-old man with juvenile onset of α-mannosidosis type II with cerebral and cerebellar atrophy and bilateral hypointensities in the thalamus, GP and putamen on T2-(turbo spin echo; TSE)-weighted images, suggestive of iron accumulation. The SN and red nucleus were also hypointense on T2-(TSE)-weighted images. The basal ganglia were isointense on T1-weighted images. | α-mannosidase | Lysosomal hydrolase that cleaves α-linked mannose residues from the nonreducting end of N-linked glycoproteins. | [14] |
| REPS1 (614825) | NBIA 7 (617916) AR | Two sisters affected by an early-onset condition (trunk hypotonia, progressive cerebellar ataxia, pyramidal syndrome). MRI of the proband at age of 15 showed progressive cerebellar and cerebral atrophy, and hypointensity of the SN and GP consistent with iron deposition. Her affected sister at age of 2 showed no alterations. | RALBP1-associated Eps domain-containing protein 1 | Protein involved in endocytosis and vesicular transport. REPS1 interacts with endocytic scaffold intersectin 1, an essential player in clathrin-mediated endocytosis. | [15] |
| CRAT (600184) | NBIA 8 (617917) AR | A girl suffering from an early-onset condition with slowly progressive spinocerebellar degeneration. MRI showed cerebellar atrophy and posterior leukodystrophy at 15.5 years old, besides of hypointensity of the SN and GP consistent with iron deposition. | Carnitine acetyltransferase | Enzyme involved in the control of the acyl-CoA/CoA ratio in mitochondria, peroxisomes, and endoplasmic reticulum. | [15] |
| MANEAL (NA) OSTM1 (607649) | NA Osteopetrosis (259720) AR | Boy, carrier of homozygous mutations in MANEAL and OSTM1, with an early-onset complex neurodegenerative disorder. MRI demonstrated massive brain atrophy and symmetrical iron accumulation in GP, corpora mamillaria and cerebral peduncles, which may due to defects on MANEAL. | Mannosidase endo-α like protein Ostepetrosis-associated transmembrane protein 1 | Unknown function/OSTM1 plays a role in subcellular endosome/lysosome dispersion and autophagy processes, which may be related to lysosomal dysfunction. | [16] |
| MTSS1L (616957) | NA AR | A 10 year-old girl with microcephaly, intellectual disability, developmental delay, and hypotonia. Brain MRI at 5 years of age was unremarkable, and when repeated at age of 10, showed iron accumulation in GP bilaterally. Her sister (7 years) is similarly affected; her MRI at age of 14 months did not show iron deposition. | Metastasis suppressor 1-like | Protein involved in plasma membrane dynamics that has a conserved N-terminal domain (IRSP53/MIM homology domain or an inverse BAR domain). | [17] |
| VAC14 (604632) | Striatonigral degeneration childhood-onset (617054) AR | Two siblings affected by retinitis pigmentosa and an early childhood severe progressive spastic paraparesis with learning disabilities. MRI revealed at age of 9, decreased signal of the GP and SN (axial T2*) and striatal T2 prolongation (axial T2). | VAC14 or ArPIKfyve (associated regulator of the PIKfyve complex) | Component of the PIKfyve complex that tightly regulates the level of PtdIns(3,5)P2 in endosomal membranes. | [18] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinarejos, I.; Machuca, C.; Sancho, P.; Espinós, C. Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA). Antioxidants 2020, 9, 1020. https://doi.org/10.3390/antiox9101020
Hinarejos I, Machuca C, Sancho P, Espinós C. Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA). Antioxidants. 2020; 9(10):1020. https://doi.org/10.3390/antiox9101020
Chicago/Turabian StyleHinarejos, Isabel, Candela Machuca, Paula Sancho, and Carmen Espinós. 2020. "Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA)" Antioxidants 9, no. 10: 1020. https://doi.org/10.3390/antiox9101020
APA StyleHinarejos, I., Machuca, C., Sancho, P., & Espinós, C. (2020). Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA). Antioxidants, 9(10), 1020. https://doi.org/10.3390/antiox9101020