Heme Oxygenase-1 in Gastrointestinal Tract Health and Disease


Abstract
:1. Introduction
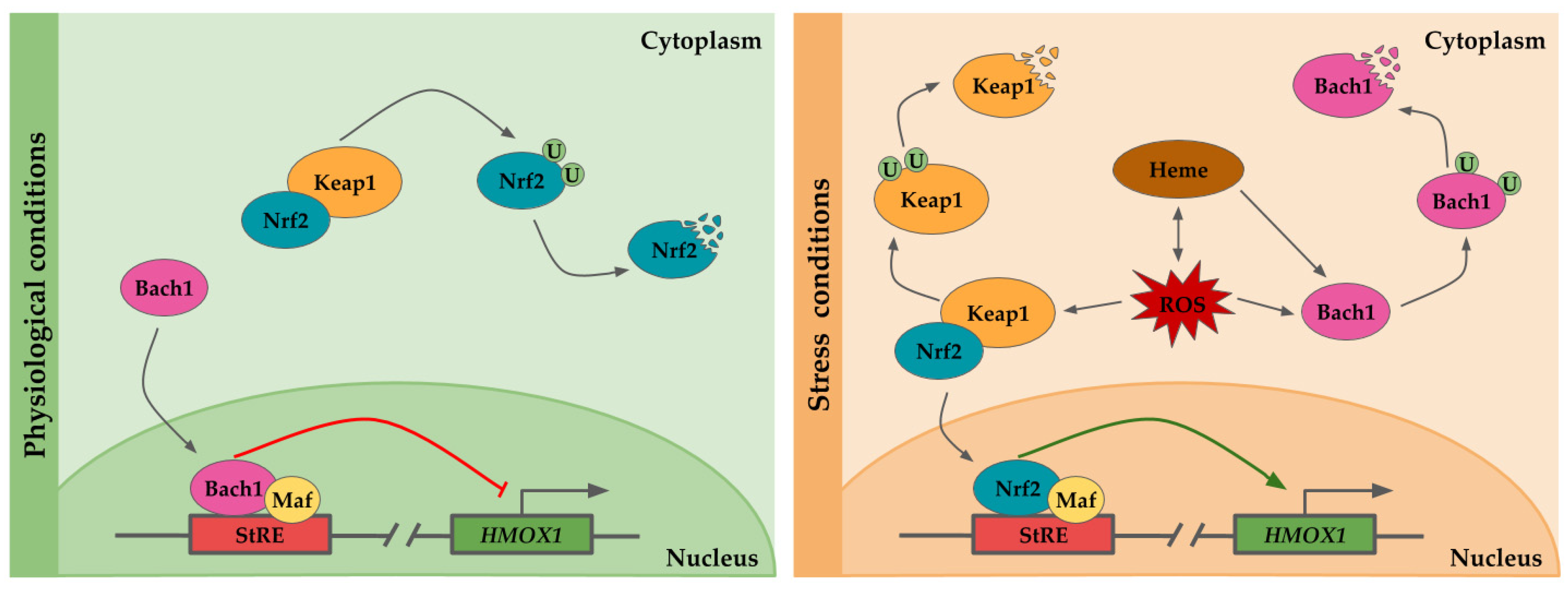
2. Molecular Regulation of HO-1 Expression
3. Cytoprotective and Deleterious Effects Mediated by HO-1
3.1. Biliverdin/Bilirubin
3.2. Carbon Monoxide
3.3. Iron and Ferroptosis
4. The Central Role of HO-1 in Gastrointestinal Tract Diseases
4.1. HO-1 and Esophagus
Esophagitis
4.2. HO-1 and Stomach
4.2.1. Peptic Ulcer Disease
4.2.2. Gastroparesis
4.3. HO-1 and Pancreas
4.3.1. Pancreatitis
4.3.2. Diabetes
4.4. HO-1 and Liver
4.4.1. Non-Alcoholic Fatty Liver Disease
4.4.2. Hepatic Ischemia–Reperfusion Injury
4.5. HO-1 and Small and Large Intestine
4.5.1. Inflammatory Bowel Disease
4.5.2. Post-Operative Ileus
5. HO-1 in Gastrointestinal Cancers
6. Clinical Trials Involving HO-1
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal heme oxygenase. Characterization of the enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar]
- Maines, M.D.; Trakshel, G.M. Purification and Characterization of Human Biliverdin Reductase. Arch. Biochem. Biophys. 1993, 300, 320–326. [Google Scholar] [CrossRef]
- Maines, M.D.; Trakshel, G.M.; Kutty, R.K. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 1986, 261, 411–419. [Google Scholar]
- Mccoubrey, W.K.; Huang, T.J.; Maines, M.D. Isolation and Characterization of a cDNA from the Rat Brain that Encodes Hemoprotein Heme Oxygenase-3. Eur. J. Biochem. 1997, 247, 725–732. [Google Scholar] [CrossRef]
- Maharshak, N.; Ryu, H.S.; Fan, T.-J.; Onyiah, J.C.; Schulz, S.; Otterbein, S.L.; Wong, R.; Hansen, J.J.; Otterbein, L.E.; Carroll, I.M.; et al. Escherichia coli heme oxygenase modulates host innate immune responses. Microbiol. Immunol. 2015, 59, 452–465. [Google Scholar] [CrossRef] [Green Version]
- Poss, K.D.; Tonegawa, S. Reduced Stress Defense in Heme Oxygenase 1-Deficient Cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef] [Green Version]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [Green Version]
- Bainbridge, S.A.; Smith, G.N. HO in pregnancy. Free Radic. Biol. Med. 2005, 38, 979–988. [Google Scholar] [CrossRef]
- Bishop, A.; Yet, S.F.; Lee, M.E.; Perrella, M.A.; Demple, B. A key role for heme oxygenase-1 in nitric oxide resistance in murine motor neurons and glia. Biochem. Biophys. Res. Commun. 2004, 325, 3–9. [Google Scholar] [CrossRef]
- Poss, K.D.; Thomas, M.J.; Ebralidze, A.K.; O’Dell, T.J.; Tonegawa, S. Hippocampal long-term potentiation is normal in heme oxygenase-2 mutant mice. Neuron 1995, 15, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Rogers, B.; Yakopson, V.; Teng, Z.P.; Guo, Y.; Regan, R.F. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Radic. Biol. Med. 2003, 35, 872–881. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar] [CrossRef]
- Braggins, P.E.; Trakshel, G.M.; Kutty, R.K.; Maines, M.D. Characterization of two heme oxygenase isoforms in rat spleen: Comparison with the hematin-induced and constitutive isoforms of the liver. Biochem. Biophys. Res. Commun. 1986, 141, 528–533. [Google Scholar] [CrossRef]
- Bauer, I.; Wanner, G.A.; Rensing, H.; Alte, C.; Miescher, E.A.; Wolf, B.; Pannen, B.H.J.; Clemens, M.G.; Bauer, M. Expression pattern of heme oxygenase isoenzymes 1 and 2 in normal and stress-exposed rat liver. Hepatology 1998, 27, 829–838. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Gottlieb, Y.; Truman, M.; Cohen, L.A.; Leichtmann-Bardoogo, Y.; Meyron-Holtz, E.G. Endoplasmic reticulum anchored heme-oxygenase 1 faces the cytosol. Haematologica 2012, 97, 1489–1493. [Google Scholar] [CrossRef] [Green Version]
- Bindu, S.; Pal, C.; Dey, S.; Goyal, M.; Alam, A.; Iqbal, M.S.; Dutta, S.; Sarkar, S.; Kumar, R.; Maity, P.; et al. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J. Biol. Chem. 2011, 286, 39387–39402. [Google Scholar] [CrossRef] [Green Version]
- Slebos, D.J.; Ryter, S.W.; Van Der Toorn, M.; Liu, F.; Guo, F.; Baty, C.J.; Karlsson, J.M.; Watkins, S.C.; Kim, H.P.; Wang, X.; et al. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am. J. Respir. Cell Mol. Biol. 2007, 36, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Nitti, M.; Piras, S.; Marinari, U.; Moretta, L.; Pronzato, M.; Furfaro, A. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A. Heme Oxygenase-1 as a Therapeutic Target in Neurodegenerative Diseases and Brain Infections. Curr. Pharm. Des. 2008, 14, 429–442. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Foresti, R.; Motterlini, R. Heme Oxygenase-1 and Carbon Monoxide in the Heart: The Balancing Act between Danger Signaling and Pro-Survival. Circ. Res. 2016, 118, 1940–1959. [Google Scholar] [CrossRef] [Green Version]
- Barton, S.G.R.G.; Rampton, D.S.; Winrow, V.R.; Domizio, P.; Feakins, R.M. Expression of heat shock protein 32 (hemoxygenase-1) in the normal and inflamed human stomach and colon: An immunohistochemical study. Cell Stress Chaperones 2003, 8, 329. [Google Scholar] [CrossRef] [Green Version]
- Alam, J.; Camhi, S.; Choi, A.M.K. Identification of a second region upstream of the mouse heme oxygenase-1 gene that functions as a basal level and inducer-dependent transcription enhancer. J. Biol. Chem. 1995, 270, 11977–11984. [Google Scholar] [CrossRef] [Green Version]
- Alam, J.; Igarashi, K.; Immenschuh, S.; Shibahara, S.; Tyrrell, R.M. Regulation of heme oxygenase-1 gene transcription: Recent advances and highlights from the international conference (Uppsala, 2003) on heme oxygenase. Antioxid. Redox Signal. 2004, 6, 924–933. [Google Scholar] [CrossRef]
- Lavrovsky, Y.; Schwartzman, M.L.; Levere, R.D.; Kappas, A.; Abraham, N.G. Identification of binding sites for transcription factors NF-κB and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc. Natl. Acad. Sci. USA 1994, 91, 5987–5991. [Google Scholar] [CrossRef] [Green Version]
- Inamdar, N.M.; Ahn, Y.I.; Alam, J. The heme-responsive element of the mouse heme oxygenase-1 gene is an extended AP-1 binding site that resembles the recognition sequences for MAF and NF-E2 transcription factors. Biochem. Biophys. Res. Commun. 1996, 221, 570–576. [Google Scholar] [CrossRef]
- Alam, J.; Cook, J.L. How Many Transcription Factors Does It Take to Turn On the Heme Oxygenase-1 Gene? Am. J. Respir. Cell Mol. Biol. 2007, 36, 166–174. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Brand, M.; Zenke, Y.; Tashiro, S.; Groudine, M.; Igarashi, K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc. Natl. Acad. Sci. USA 2004, 101, 1461–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007, 35, 7074–7086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme Induces Ubiquitination and Degradation of the Transcription Factor Bach1. Mol. Cell. Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, M.; Numazawa, S.; Yoshida, T. Redox regulation of the transcriptional repressor Bach1. Free Radic. Biol. Med. 2005, 38, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.C.; Sun, Z.; Habib, G.M.; Lieberman, M.W.; Hannink, M. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J. Biol. Chem. 2005, 280, 30091–30099. [Google Scholar] [CrossRef] [Green Version]
- Zuckerbraun, B.S.; Billiar, T.R.; Otterbein, S.L.; Kim, P.K.M.; Liu, F.; Choi, A.M.K.; Bach, F.H.; Otterbein, L.E. Carbon Monoxide Protects against Liver Failure through Nitric Oxide-induced Heme Oxygenase 1. J. Exp. Med. 2003, 198, 1707–1716. [Google Scholar] [CrossRef]
- Liu, X.; Peyton, K.J.; Ensenat, D.; Wang, H.; Hannink, M.; Alam, J.; Durante, W. Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc. Res. 2007, 75, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Iwata, M.; Inoue, T.; Asai, Y.; Hori, K.; Fujiwara, M.; Matsuo, S.; Tsuchida, W.; Suzuki, S. The protective role of localized nitric oxide production during inflammation may be mediated by the heme oxygenase-1/carbon monoxide pathway. Biochem. Biophys. Rep. 2020, 23, 100790. [Google Scholar] [CrossRef]
- Kuwano, Y.; Rabinovic, A.; Srikantan, S.; Gorospe, M.; Demple, B. Analysis of Nitric Oxide-Stabilized mRNAs in Human Fibroblasts Reveals HuR-Dependent Heme Oxygenase 1 Upregulation. Mol. Cell. Biol. 2009, 29, 2622–2635. [Google Scholar] [CrossRef] [Green Version]
- Exner, M.; Minar, E.; Wagner, O.; Schillinger, M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic. Biol. Med. 2004, 37, 1097–1104. [Google Scholar] [CrossRef]
- Dutra, F.F.; Bozza, M.T. Heme on innate immunity and inflammation. Front. Pharmacol. 2014, 5, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, R. Antioxidant activities of bile pigments. Antioxid. Redox Signal. 2004, 6, 841–849. [Google Scholar] [CrossRef]
- Figueiredo-Pereira, C.; Dias-Pedroso, D.; Soares, N.L.; Vieira, H.L.A. CO-mediated cytoprotection is dependent on cell metabolism modulation. Redox Biol. 2020, 32, 101470. [Google Scholar] [CrossRef]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 783–792. [Google Scholar] [CrossRef]
- Papanikolaou, G.; Pantopoulos, K. Iron metabolism and toxicity. Toxicol. Appl. Pharmacol. 2005, 202, 199–211. [Google Scholar] [CrossRef]
- Olusanya, B.O.; Kaplan, M.; Hansen, T.W.R. Neonatal hyperbilirubinaemia: A global perspective. Lancet Child Adolesc. Health 2018, 2, 610–620. [Google Scholar] [CrossRef] [Green Version]
- Barañano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef] [Green Version]
- Jansen, T.; Daiber, A. Direct Antioxidant Properties of Bilirubin and Biliverdin. Is there a Role for Biliverdin Reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Wu, T.W.; Fung, K.P.; Yang, C.C. Unconjugated bilirubin inhibits the oxidation of human low density lipoprotein better than trolox. Life Sci. 1994, 54. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Saleh, M.; Higginson, D.S.; Paul, B.D.; Juluri, K.R.; Snyder, S.H. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. USA 2009, 106, 5171–5176. [Google Scholar] [CrossRef] [Green Version]
- Lanone, S.; Bloc, S.; Foresti, R.; Almolki, A.; Taillé, C.; Callebert, J.; Conti, M.; Goven, D.; Aubier, M.; Dureuil, B.; et al. Bilirubin decreases NOS2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J. 2005, 19, 1890–1892. [Google Scholar] [CrossRef] [Green Version]
- Bokoch, G.M.; Knaus, U.G. NADPH oxidases: Not just for leukocytes anymore! Trends Biochem. Sci. 2003, 28, 502–508. [Google Scholar] [CrossRef]
- Keshavan, P.; Deem, T.L.; Schwemberger, S.J.; Babcock, G.F.; Cook-Mills, J.M.; Zucker, S.D. Unconjugated Bilirubin Inhibits VCAM-1-Mediated Transendothelial Leukocyte Migration. J. Immunol. 2005, 174, 3709–3718. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Lee, S.J.; Spiller, W.; Jung, K.J.; Lee, J.Y.; Kimm, H.; Back, J.H.; Lee, S.; Jee, S.H. Causal associations between serum bilirubin levels and decreased stroke risk a two-sample Mendelian randomization study. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Wu, X.; Bi, Y.; Yang, Y. Effect of bilirubin concentration on the risk of diabetic complications: A meta-analysis of epidemiologic studies. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Kunutsor, S.K.; Frysz, M.; Verweij, N.; Kieneker, L.M.; Bakker, S.J.L.; Dullaart, R.P.F. Circulating total bilirubin and risk of non-alcoholic fatty liver disease in the PREVEND study: Observational findings and a Mendelian randomization study. Eur. J. Epidemiol. 2020, 35, 123–137. [Google Scholar] [CrossRef] [Green Version]
- Boczkowski, J.; Poderoso, J.J.; Motterlini, R. CO-metal interaction: Vital signaling from a lethal gas. Trends Biochem. Sci. 2006, 31, 614–621. [Google Scholar] [CrossRef]
- Parfenova, H.; Leffler, C.W.; Basuroy, S.; Liu, J.; Fedinec, A.L. Antioxidant roles of heme oxygenase, carbon monoxide, and bilirubin in cerebral circulation during seizures. J. Cereb. Blood Flow Metab. 2012, 32, 1024–1034. [Google Scholar] [CrossRef] [Green Version]
- Basuroy, S.; Bhattacharya, S.; Leffler, C.W.; Parfenova, H. Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-α in cerebral vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2009, 296, C422. [Google Scholar] [CrossRef] [Green Version]
- Zuckerbraun, B.S.; Chin, B.Y.; Bilban, M.; d’Avila, J.d.C.; Rao, J.; Billiar, T.R.; Otterbein, L.E. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007, 21, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Silva, G.; Cunha, A.; Grégoire, I.P.; Seldon, M.P.; Soares, M.P. The Antiapoptotic Effect of Heme Oxygenase-1 in Endothelial Cells Involves the Degradation of p38α MAPK Isoform. J. Immunol. 2006, 177, 1894–1903. [Google Scholar] [CrossRef] [Green Version]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Lu, H.T.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M.K. Carbon monoxide has anti-inflammatory effects involving the mitogen- activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef]
- Song, R.; Mahidhara, R.S.; Liu, F.; Ning, W.; Otterbein, L.E.; Choi, A.M.K. Carbon monoxide inhibits human airway smooth muscle cell proliferation via mitogen-activated protein kinase pathway. Am. J. Respir. Cell Mol. Biol. 2002, 27, 603–610. [Google Scholar] [CrossRef]
- Soni, H.; Jain, M.; Mehta, A.A. Investigation into the mechanism(s) of antithrombotic effects of carbon monoxide releasing molecule-3 (CORM-3). Thromb. Res. 2011, 127, 551–559. [Google Scholar] [CrossRef]
- Morita, T.; Mitsialis, S.A.; Koike, H.; Liu, Y.; Kourembanas, S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J. Biol. Chem. 1997, 272, 32804–32809. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Kim, C.K.; Lee, H.; Jeoung, D.; Ha, K.S.; Kwon, Y.G.; Kim, K.W.; Kim, Y.M. Carbon monoxide promotes VEGF expression by increasing HIF-1α protein level via two distinct mechanisms, translational activation and stabilization of HIF-1α protein. J. Biol. Chem. 2010, 285, 32116–32125. [Google Scholar] [CrossRef] [Green Version]
- Hailemariam, K.; Iwasaki, K.; Huang, B.W.; Sakamoto, K.; Tsuji, Y. Transcriptional regulation of ferritin and antioxidant genes by HIPK2 under genotoxic stress. J. Cell Sci. 2010, 123, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Torti, F.M.; Torti, S.V. Regulation of ferritin genes and protein. Blood 2002, 99, 3505–3516. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Alkhateeb, A.A.; Connor, J.R. Nuclear ferritin: A new role for ferritin in cell biology. Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 793–797. [Google Scholar] [CrossRef]
- Kubilus, J.K.; Beazley, K.E.; Talbot, C.J.; Linsenmayer, T.F. Nuclear ferritin mediated regulation of JNK signaling in corneal epithelial cells. Exp. Eye Res. 2016, 145, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11–7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef] [Green Version]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayir, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Husseini, M.; Wang, G.-S.; Patrick, C.; Crookshank, J.A.; MacFarlane, A.J.; Noel, J.A.; Strom, A.; Scott, F.W. Heme Oxygenase-1 Induction Prevents Autoimmune Diabetes in Association With Pancreatic Recruitment of M2-Like Macrophages, Mesenchymal Cells, and Fibrocytes. Endocrinology 2015, 156, 3937–3949. [Google Scholar] [CrossRef] [Green Version]
- Takagi, T.; Naito, Y.; Mizushima, K.; Nukigi, Y.; Okada, H.; Suzuki, T.; Hirata, I.; Omatsu, T.; Okayama, T.; Handa, O.; et al. Increased intestinal expression of heme oxygenase-1 and its localization in patients with ulcerative colitis. J. Gastroenterol. Hepatol. 2008, 23, S229–S233. [Google Scholar] [CrossRef]
- Malaguarnera, L.; Madeddu, R.; Palio, E.; Arena, N.; Malaguarnera, M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J. Hepatol. 2005, 42, 585–591. [Google Scholar] [CrossRef]
- Rieder, F.; Biancani, P.; Harnett, K.; Yerian, L.; Falk, G.W. Inflammatory mediators in gastroesophageal reflux disease: Impact on esophageal motility, fibrosis, and carcinogenesis. Am. J. Physiol. Liver Physiol. 2010, 298, G571–G581. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Kim, E.-H.; Hahm, K.B. Oxidative stress in inflammation-based gastrointestinal tract diseases: Challenges and opportunities. J. Gastroenterol. Hepatol. 2012, 27, 1004–1010. [Google Scholar] [CrossRef]
- Kwon, O.J.; Choo, B.K.; Lee, J.Y.; Kim, M.Y.; Shin, S.H.; Seo, B.I.; Seo, Y.B.; Rhee, M.H.; Shin, M.R.; Kim, G.N.; et al. Protective effect of Rhei Rhizoma on reflux esophagitis in rats via Nrf2-mediated inhibition of NF-κB signaling pathway. BMC Complement. Altern. Med. 2016, 16, 7. [Google Scholar] [CrossRef] [Green Version]
- Magierowska, K.; Bakalarz, D.; Wójcik, D.; Korbut, E.; Danielak, A.; Głowacka, U.; Pajdo, R.; Buszewicz, G.; Ginter, G.; Surmiak, M.; et al. Evidence for Cytoprotective Effect of Carbon Monoxide Donor in the Development of Acute Esophagitis Leading to Acute Esophageal Epithelium Lesions. Cells 2020, 9, 1203. [Google Scholar] [CrossRef]
- Liu, G.; Jiang, C.; Li, D.; Yao, L.; Lin, Y.; Wang, B.; Qiu, J.; Wang, W.; Wang, W. Isorhamnetin alleviates esophageal mucosal injury in a chronic model of reflux esophagitis. Eur. J. Pharmacol. 2019, 864, 172720. [Google Scholar] [CrossRef]
- Kuna, L.; Jakab, J.; Smolic, R.; Raguz-Lucic, N.; Vcev, A.; Smolic, M. Peptic Ulcer Disease: A Brief Review of Conventional Therapy and Herbal Treatment Options. J. Clin. Med. 2019, 8, 179. [Google Scholar] [CrossRef] [Green Version]
- Uc, A.; Zhu, X.; Wagner, B.A.; Buettner, G.R.; Berg, D.J. Heme Oxygenase-1 Is Protective Against Nonsteroidal Anti-inflammatory Drug–induced Gastric Ulcers. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 471–476. [Google Scholar] [CrossRef]
- Becker, J.C.; Grosser, N.; Waltke, C.; Schulz, S.; Erdmann, K.; Domschke, W.; Schröder, H.; Pohle, T. Beyond gastric acid reduction: Proton pump inhibitors induce heme oxygenase-1 in gastric and endothelial cells. Biochem. Biophys. Res. Commun. 2006, 345, 1014–1021. [Google Scholar] [CrossRef]
- Han, Y.M.; Park, J.M.; Kang, J.X.; Cha, J.Y.; Lee, H.J.; Jeong, M.; Go, E.J.; Hahm, K.B. Mitigation of indomethacin-induced gastrointestinal damages in fat-1 transgenic mice via gate-keeper action of ω-3-polyunsaturated fatty acids. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Aburaya, M.; Tanaka, K.I.; Hoshino, T.; Tsutsumi, S.; Suzuki, K.; Makise, M.; Akagi, R.; Mizushima, T. Heme oxygenase-1 protects gastric mucosal cells against non-steroidal anti-inflammatory drugs. J. Biol. Chem. 2006, 281, 33422–33432. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-M.; Han, Y.-M.; Kangwan, N.; Lee, S.-Y.; Jung, M.-K.; Kim, E.-H.; Hahm, K.-B. S-allyl cysteine alleviates nonsteroidal anti-inflammatory drug-induced gastric mucosal damages by increasing cyclooxygenase-2 inhibition, heme oxygenase-1 induction, and histone deacetylation inhibition. J. Gastroenterol. Hepatol. 2014, 29, 80–92. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, A.; Qi, B.; Ma, Z.; Xiong, Y.; Dou, J.; Wang, J. Resveratrol Protects against Helicobacter pylori-Associated Gastritis by Combating Oxidative Stress. Int. J. Mol. Sci. 2015, 16, 27757–27769. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Paek, N.S.; Kwon, O.S.; Hahm, K.B. Anti-inflammatory actions of probiotics through activating suppressor of cytokine signaling (SOCS) expression and signaling in Helicobacter pylori infection: A novel mechanism. J. Gastroenterol. Hepatol. 2010, 25, 194–202. [Google Scholar] [CrossRef]
- Jeong, M.; Park, J.M.; Han, Y.M.; Park, K.Y.; Lee, D.H.; Yoo, J.H.; Cho, J.Y.; Hahm, K.B. Dietary prevention of Helicobacter pylori-associated gastric cancer with kimchi. Oncotarget 2015, 6, 29513–29526. [Google Scholar] [CrossRef] [Green Version]
- Camilleri, M.; Chedid, V.; Ford, A.C.; Haruma, K.; Horowitz, M.; Jones, K.L.; Low, P.A.; Park, S.Y.; Parkman, H.P.; Stanghellini, V. Gastroparesis. Nat. Rev. Dis. Prim. 2018, 4, 1–19. [Google Scholar] [CrossRef]
- Grover, M.; Farrugia, G.; Lurken, M.S.; Bernard, C.E.; Faussonepellegrini, M.S.; Smyrk, T.C.; Parkman, H.P.; Abell, T.L.; Snape, W.J.; Hasler, W.L.; et al. Cellular changes in diabetic and idiopathic gastroparesis. Gastroenterology 2011, 140, 1575–1585.e8. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.M.; Gibbons, S.J.; Nguyen, T.V.; Stoltz, G.J.; Lurken, M.S.; Ordog, T.; Szurszewski, J.H.; Farrugia, G. Heme Oxygenase-1 Protects Interstitial Cells of Cajal From Oxidative Stress and Reverses Diabetic Gastroparesis. Gastroenterology 2008, 135, 2055–2064.e2. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.M.; Kashyap, P.C.; Dutta, N.; Stoltz, G.J.; Ordog, T.; Shea Donohue, T.; Bauer, A.J.; Linden, D.R.; Szurszewski, J.H.; Gibbons, S.J.; et al. CD206-Positive M2 Macrophages That Express Heme Oxygenase-1 Protect Against Diabetic Gastroparesis in Mice. Gastroenterology 2010, 138, 2399–2409.e1. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, P.C.; Choi, K.M.; Dutta, N.; Linden, D.R.; Szurszewski, J.H.; Gibbons, S.J.; Farrugia, G. Carbon monoxide reverses diabetic gastroparesis in NOD mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G1013. [Google Scholar] [CrossRef]
- Bharucha, A.E.; Daley, S.L.; Low, P.A.; Gibbons, S.J.; Choi, K.M.; Camilleri, M.; Saw, J.J.; Farrugia, G.; Zinsmeister, A.R. Effects of hemin on heme oxygenase-1, gastric emptying, and symptoms in diabetic gastroparesis. Neurogastroenterol. Motil. 2016, 28, 1731–1740. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H. Oxidative Stress in Pancreatic Beta Cell Regeneration. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.P.; Mourad, M.M.; Bramhall, S.R. Acute pancreatitis: Current perspectives on diagnosis and management. J. Inflamm. Res. 2018, 11, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Kleeff, J.; Whitcomb, D.C.; Shimosegawa, T.; Esposito, I.; Lerch, M.M.; Gress, T.; Mayerle, J.; Drewes, A.M.; Rebours, V.; Akisik, F.; et al. Chronic pancreatitis. Nat. Rev. Dis. Prim. 2017, 3, 1–18. [Google Scholar] [CrossRef]
- Fu, K.; Sarras, M.P.; De Lisle, R.C.; Andrews, G.K. Expression of oxidative stress-responsive genes and cytokine genes during caerulein-induced acute pancreatitis. Am. J. Physiol. 1997, 273. [Google Scholar] [CrossRef]
- Habtezion, A.; Kwan, R.; Yang, A.L.; Morgan, M.E.; Akhtar, E.; Wanaski, S.P.; Collins, S.D.; Butcher, E.C.; Kamal, A.; Omary, M.B. Heme oxygenase-1 is induced in peripheral blood mononuclear cells of patients with acute pancreatitis: A potential therapeutic target. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G12–G20. [Google Scholar] [CrossRef] [Green Version]
- Gulla, A.; Evans, B.J.; Navenot, J.M.; Pundzius, J.; Barauskas, G.; Gulbinas, A.; Dambrauskas, Z.; Arafat, H.; Wang, Z.-X. Heme Oxygenase-1 Gene Promoter Polymorphism Is Associated With the Development of Necrotizing Acute Pancreatitis. Pancreas 2014, 43, 1271–1276. [Google Scholar] [CrossRef]
- Kylänpää, L.; Rakonczay, Z.; O’Reilly, D.A. The Clinical Course of Acute Pancreatitis and the Inflammatory Mediators That Drive It. Int. J. Inflam. 2012, 2012, 360685. [Google Scholar] [CrossRef] [Green Version]
- Nakamichi, I.; Habtezion, A.; Zhong, B.; Contag, C.H.; Butcher, E.C.; Omary, M.B. Hemin-activated macrophages home to the pancreas and protect from acute pancreatitis via heme oxygenase-1 induction. J. Clin. Investig. 2005, 115, 3007–3014. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.H.; Sun, Y.H.; Fan, K.L.; Dong, X.B.; Han, N.; Zhao, H.; Kong, L. Protective effects of heme oxygenase-1 against severe acute pancreatitis via inhibition of tumor necrosis factor-α and augmentation of interleukin-10. BMC Gastroenterol. 2017, 17. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Jiang, X.; Zhai, Y.Y.; Luo, L.Z.; Xu, H.L.; Xiao, J.; Kou, L.; Zhao, Y.Z. Protective effects and mechanisms of bilirubin nanomedicine against acute pancreatitis. J. Control. Release 2020, 322, 312–325. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, R.; Hu, G.; Zhu, H.H.; Gao, W.-Q.; Xue, J. Carbon Monoxide Impairs CD11b + Ly-6C hi Monocyte Migration from the Blood to Inflamed Pancreas via Inhibition of the CCL2/CCR2 Axis. J. Immunol. 2018, 200, 2104–2114. [Google Scholar] [CrossRef] [Green Version]
- Kharroubi, A.T.; Darwish, H.M. Diabetes mellitus: The epidemic of the century. World J. Diabetes 2015, 6, 850. [Google Scholar] [CrossRef]
- Tan, S.Y.; Mei Wong, J.L.; Sim, Y.J.; Wong, S.S.; Mohamed Elhassan, S.A.; Tan, S.H.; Ling Lim, G.P.; Rong Tay, N.W.; Annan, N.C.; Bhattamisra, S.K.; et al. Type 1 and 2 diabetes mellitus: A review on current treatment approach and gene therapy as potential intervention. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 364–372. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Prim. 2015, 1, 1–22. [Google Scholar] [CrossRef]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Böni-Schnetzler, M.; Donath, M.Y. The Role of Inflammation in β-cell Dedifferentiation. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Bao, W.; Song, F.; Li, X.; Rong, S.; Yang, W.; Wang, D.; Xu, J.; Fu, J.; Zhao, Y.; Liu, L. Association Between Heme Oxygenase-1 Gene Promoter Polymorphisms and Type 2 Diabetes Mellitus: A HuGE Review and Meta-Analysis. Am. J. Epidemiol. 2010, 172, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Cheriyath, P.; Gorrepati, V.S.; Peters, I.; Nookala, V.; Murphy, M.E.; Srouji, N.; Fischman, D. High Total Bilirubin as a Protective Factor for Diabetes Mellitus: An Analysis of NHANES Data From 1999–2006. J. Clin. Med. Res. 2010, 2, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Ndisang, J.F.; Jadhav, A. Up-regulating the hemeoxygenase system enhances insulin sensitivity and improves glucose metabolism in insulin-resistant diabetes in Goto-Kakizaki rats. Endocrinology 2009, 150, 2627–2636. [Google Scholar] [CrossRef]
- Pogu, J.; Tzima, S.; Kollias, G.; Anegon, I.; Blancou, P.; Simon, T. Genetic Restoration of Heme Oxygenase-1 Expression Protects from Type 1 Diabetes in NOD Mice. Int. J. Mol. Sci. 2019, 20, 1676. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Peng, S.Y.; Hong, S.; Chen, Q.W.; Zeng, X.; Rong, L.; Zhong, Z.L.; Zhang, X.Z. Biomimetic carbon monoxide nanogenerator ameliorates streptozotocin induced type 1 diabetes in mice. Biomaterials 2020, 245, 119986. [Google Scholar] [CrossRef]
- Stefan, N.; Häring, H.U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Brunt, E.M.; Wong, V.W.S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Prim. 2015, 1, 1–22. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Yu, J.; Chu, E.S.H.; Wang, R.; Wang, S.; Wu, C.W.; Wong, V.W.S.; Chan, H.L.Y.; Farrell, G.C.; Sung, J.J.Y. Heme Oxygenase-1 Protects Against Steatohepatitis in Both Cultured Hepatocytes and Mice. Gastroenterology 2010, 138, 694–704.e1. [Google Scholar] [CrossRef]
- Li, D.; Zhao, D.; Du, J.; Dong, S.; Aldhamin, Z.; Yuan, X.; Li, W.; Du, H.; Zhao, W.; Cui, L.; et al. Heme oxygenase-1 alleviated non-alcoholic fatty liver disease via suppressing ROS-dependent endoplasmic reticulum stress. Life Sci. 2020, 253, 117678. [Google Scholar] [CrossRef]
- Yang, H.; Zhao, L.F.; Zhao, Z.F.; Wang, Y.; Zhao, J.J.; Zhang, L. Heme oxygenase-1 prevents liver fibrosis in rats by regulating the expression of PPARγ and NF-κB. World J. Gastroenterol. 2012, 18, 1680–1688. [Google Scholar] [CrossRef]
- Yang, H.; Chen, B.; Zhao, Z.; Zhang, L.; Zhang, Y.; Chen, J.; Zhang, X.; Zhang, X.; Zhao, L. Heme oxygenase-1 exerts pro-apoptotic effects on hepatic stellate cells in vitro through regulation of nuclear factor-κB. Exp. Ther. Med. 2018, 16, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Petrowsky, H.; Hong, J.C.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Ischaemia-reperfusion injury in liver transplantation-from bench to bedside. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 79–89. [Google Scholar] [CrossRef]
- Fang, J.; Qin, H.; Seki, T.; Nakamura, H.; Tsukigawa, K.; Shin, T.; Maeda, H. Therapeutic potential of pegylated hemin for reactive oxygen species-related diseases via induction of heme oxygenase-1: Results from a rat hepatic ischemia/reperfusion injury model. J. Pharmacol. Exp. Ther. 2011, 339, 779–789. [Google Scholar] [CrossRef]
- Geuken, E.; Buis, C.I.; Visser, D.S.; Blokzijl, H.; Moshage, H.; Nemes, B.; Leuvenink, H.G.D.; de Jong, K.P.; Peeters, P.M.J.G.; Slooff, M.J.H.; et al. Expression of Heme Oxygenase-1 in Human Livers Before Transplantation Correlates with Graft Injury and Function After Transplantation. Am. J. Transplant. 2005, 5, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Zhang, M.; Kageyama, S.; Ke, B.; Fujii, T.; Sosa, R.A.; Reed, E.F.; Datta, N.; Zarrinpar, A.; Busuttil, R.W.; et al. Macrophage heme oxygenase-1-SIRT1-p53 axis regulates sterile inflammation in liver ischemia-reperfusion injury. J. Hepatol. 2017, 67, 1232–1242. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Nakamura, K.; Kageyama, S.; Lawal, A.O.; Gong, K.W.; Bhetraratana, M.; Fujii, T.; Sulaiman, D.; Hirao, H.; Bolisetty, S.; et al. Myeloid HO-1 modulates macrophage polarization and protects against ischemia-reperfusion injury. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Tsuchihashi, S.; Zhai, Y.; Bo, Q.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Heme Oxygenase-1 Mediated Cytoprotection Against Liver Ischemia and Reperfusion Injury: Inhibition of Type-1 Interferon Signaling. Transplantation 2007, 83, 1628–1634. [Google Scholar] [CrossRef]
- Rao, J.; Qian, X.; Li, G.; Pan, X.; Zhang, C.; Zhang, F.; Zhai, Y.; Wang, X.; Lu, L. ATF3-Mediated NRF2/HO-1 Signaling Regulates TLR4 Innate Immune Responses in Mouse Liver Ischemia/Reperfusion Injury. Am. J. Transplant. 2015, 15, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Shi, T.; Qiao, H.; Jiang, X.; Jiang, H.; Krissansen, G.W.; Sun, X. Hepatic overexpression of heme oxygenase-1 improves liver allograft survival by expanding t regulatory cells. J. Surg. Res. 2011, 166, e187–e194. [Google Scholar] [CrossRef]
- Shen, X.-D.; Ke, B.; Uchida, Y.; Ji, H.; Gao, F.; Zhai, Y.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Native macrophages genetically modified to express heme oxygenase 1 protect rat liver transplants from ischemia/reperfusion injury. Liver Transplant. 2011, 17, 201–210. [Google Scholar] [CrossRef]
- Nakamura, K.; Kageyama, S.; Yue, S.; Huang, J.; Fujii, T.; Ke, B.; Sosa, R.A.; Reed, E.F.; Datta, N.; Zarrinpar, A.; et al. Heme oxygenase-1 regulates sirtuin-1-autophagy pathway in liver transplantation: From mouse to human. Am. J. Transplant. 2018, 18, 1110–1121. [Google Scholar] [CrossRef] [Green Version]
- Lazar, V.; Ditu, L.M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of gut microbiota and immune system interactions in infectious diseases, immunopathology, and cancer. Front. Immunol. 2018, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 7247238. [Google Scholar] [CrossRef] [Green Version]
- Paul, G.; Bataille, F.; Obermeier, F.; Bock, J.; Klebl, F.; Strauch, U.; Lochbaum, D.; Rümmele, P.; Farkas, S.; Schölmerich, J.; et al. Analysis of intestinal haem-oxygenase-1 (HO-1) in clinical and experimental colitis. Clin. Exp. Immunol. 2005, 140, 547–555. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, M.T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.N. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef] [Green Version]
- Harusato, A.; Naito, Y.; Takagi, T.; Uchiyama, K.; Mizushima, K.; Hirai, Y.; Higashimura, Y.; Katada, K.; Handa, O.; Ishikawa, T.; et al. BTB and CNC Homolog 1 (Bach1) Deficiency Ameliorates TNBS Colitis in Mice. Inflamm. Bowel Dis. 2013, 19, 740–753. [Google Scholar] [CrossRef]
- Zheng, J.-D.; He, Y.; Yu, H.-Y.; Liu, Y.-L.; Ge, Y.-X.; Li, X.-T.; Li, X.; Wang, Y.; Guo, M.-R.; Qu, Y.-L.; et al. Unconjugated bilirubin alleviates experimental ulcerative colitis by regulating intestinal barrier function and immune inflammation. World J. Gastroenterol. 2019, 25, 1865–1878. [Google Scholar] [CrossRef]
- Horváth, K.; Varga, C.; Berkó, A.; Pósa, A.; László, F.; Whittle, B.J.R. The involvement of heme oxygenase-1 activity in the therapeutic actions of 5-aminosalicylic acid in rat colitis. Eur. J. Pharmacol. 2008, 581, 315–323. [Google Scholar] [CrossRef]
- Yan, Y.X.; Shao, M.J.; Qi, Q.; Xu, Y.S.; Yang, X.Q.; Zhu, F.H.; He, S.J.; He, P.L.; Feng, C.L.; Wu, Y.W.; et al. Artemisinin analogue SM934 ameliorates DSS-induced mouse ulcerative colitis via suppressing neutrophils and macrophages. Acta Pharmacol. Sin. 2018, 39, 1633–1644. [Google Scholar] [CrossRef]
- Yan, S.C.; Wang, Y.J.; Li, Y.J.; Cai, W.Y.; Weng, X.G.; Li, Q.; Chen, Y.; Yang, Q.; Zhu, X.X. Dihydroartemisinin Regulates the Th/Treg Balance by Inducing Activated CD4+ T cell Apoptosis via Heme Oxygenase-1 Induction in Mouse Models of Inflammatory Bowel Disease. Molecules 2019, 24, 2475. [Google Scholar] [CrossRef] [Green Version]
- Han, K.H.; Park, J.M.; Jeong, M.; Han, Y.M.; Go, E.J.; Park, J.; Kim, H.; Han, J.G.; Kwon, O.; Hahm, K.B. Heme oxygenase-1 induction and anti-inflammatory actions of atractylodes macrocephala and taraxacum herba extracts prevented colitis and was more effective than sulfasalazine in preventing relapse. Gut Liver 2017, 11, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.; Kwon, J.; Cho, Y.; Kim, I.; Kang, S. Anti-Inflammatory Effect of Angelica gigas via Heme Oxygenase (HO)-1 Expression. Nutrients 2015, 7, 4862–4874. [Google Scholar] [CrossRef] [Green Version]
- Thapa, B.; Pak, S.; Kwon, H.J.; Lee, K. Decursinol angelate ameliorates dextran sodium sulfate-induced colitis by modulating type 17 helper T cell responses. Biomol. Ther. 2019, 27, 466–473. [Google Scholar] [CrossRef]
- Kim, K.J.; Park, J.M.; Lee, J.S.; Kim, Y.S.; Kangwan, N.; Han, Y.M.; Kang, E.A.; An, J.M.; Park, Y.K.; Hahm, K.B. Oligonol prevented the relapse of dextran sulfate sodium-ulcerative colitis through enhancing NRF2-mediated antioxidative defense mechanism. J. Physiol. Pharmacol. 2018, 69. [Google Scholar] [CrossRef]
- Li, Z.; Ma, T.; Zhang, W.; Shang, Y.; Zhang, Y.; Ma, Y. Genipin attenuates dextran sulfate sodium-induced colitis via suppressing inflammatory and oxidative responses. Inflammopharmacology 2020, 28, 333–339. [Google Scholar] [CrossRef]
- Fan, H.; Chen, W.; Zhu, J.; Zhang, J.; Peng, S. Toosendanin alleviates dextran sulfate sodium-induced colitis by inhibiting M1 macrophage polarization and regulating NLRP3 inflammasome and Nrf2/HO-1 signaling. Int. Immunopharmacol. 2019, 76, 105909. [Google Scholar] [CrossRef]
- Chi, J.H.; Kim, Y.H.; Sohn, D.H.; Seo, G.S.; Lee, S.H. Ameliorative effect of Alnus japonica ethanol extract on colitis through the inhibition of inflammatory responses and attenuation of intestinal barrier disruption in vivo and in vitro. Biomed. Pharmacother. 2018, 108, 1767–1774. [Google Scholar] [CrossRef]
- Mei, Y.; Wang, Z.; Zhang, Y.; Wan, T.; Xue, J.; He, W.; Luo, Y.; Xu, Y.; Bai, X.; Wang, Q.; et al. FA-97, a New Synthetic Caffeic Acid Phenethyl Ester Derivative, Ameliorates DSS-Induced Colitis Against Oxidative Stress by Activating Nrf2/HO-1 Pathway. Front. Immunol. 2020, 10, 2969. [Google Scholar] [CrossRef] [Green Version]
- Sangaraju, R.; Nalban, N.; Alavala, S.; Rajendran, V.; Jerald, M.K.; Sistla, R. Protective effect of galangin against dextran sulfate sodium (DSS)-induced ulcerative colitis in Balb/c mice. Inflamm. Res. 2019, 68, 691–704. [Google Scholar] [CrossRef]
- Bragg, D.; El-Sharkawy, A.M.; Psaltis, E.; Maxwell-Armstrong, C.A.; Lobo, D.N. Postoperative ileus: Recent developments in pathophysiology and management. Clin. Nutr. 2015, 34, 367–376. [Google Scholar] [CrossRef]
- Moore, B.A.; Otterbein, L.E.; Türler, A.; Choi, A.M.K.; Bauer, A.J. Inhaled carbon monoxide suppresses the development of postoperative ileus in the murine small intestine. Gastroenterology 2003, 124, 377–391. [Google Scholar] [CrossRef]
- De Backer, O.; Elinck, E.; Blanckaert, B.; Leybaert, L.; Motterlini, R.; Lefebvre, R.A. Water-soluble CO-releasing molecules reduce the development of postoperative ileus via modulation of MAPK/HO-1 signalling and reduction of oxidative stress. Gut 2009, 58, 347–356. [Google Scholar] [CrossRef]
- Van Dingenen, J.; Pieters, L.; Van Nuffel, E.; Lefebvre, R.A. Hemin reduces postoperative ileus in a heme oxygenase 1-dependent manner while dimethyl fumarate does without heme oxygenase 1-induction. Neurogastroenterol. Motil. 2020, 32. [Google Scholar] [CrossRef]
- Yin, H.; Fang, J.; Liao, L.; Maeda, H.; Su, Q. Upregulation of heme oxygenase-1 in colorectal cancer patients with increased circulation carbon monoxide levels, potentially affects chemotherapeutic sensitivity. BMC Cancer 2014, 14, 436. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Liu, Q.; Wang, B.; Chen, G.; Xu, L.; Zhou, H. Expression and function of heme oxygenase-1 in human gastric cancer. Exp. Biol. Med. 2012, 237, 362–371. [Google Scholar] [CrossRef]
- Berberat, P.O.; Dambrauskas, Z.; Gulbinas, A.; Giese, T.; Giese, N.; Künzli, B.; Autschbach, F.; Meuer, S.; Büchler, M.W.; Friess, H. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin. Cancer Res. 2005, 11, 3790–3798. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.C.; Fukui, H.; Imai, Y.; Sekikawa, A.; Kimura, T.; Yamagishi, H.; Yoshitake, N.; Pohle, T.; Domschke, W.; Fujimori, T. Colonic expression of heme oxygenase-1 is associated with a better long-term survival in patients with colorectal cancer. Scand. J. Gastroenterol. 2007, 42, 852–858. [Google Scholar] [CrossRef]
- Seo, G.S.; Jiang, W.Y.; Chi, J.H.; Jin, H.; Park, W.C.; Sohn, D.H.; Park, P.H.; Lee, S.H. Heme oxygenase-1 promotes tumor progression and metastasis of colorectal carcinoma cells by inhibiting antitumor immunity. Oncotarget 2015, 6, 19792–19806. [Google Scholar] [CrossRef] [Green Version]
- Andrés, N.C.; Fermento, M.E.; Gandini, N.A.; Romero, A.L.; Ferro, A.; Donna, L.G.; Curino, A.C.; Facchinetti, M.M. Heme oxygenase-1 has antitumoral effects in colorectal cancer: Involvement of p53. Exp. Mol. Pathol. 2014, 97, 321–331. [Google Scholar] [CrossRef]
- Hsu, F.F.; Yeh, C.T.; Sun, Y.J.; Chiang, M.T.; Lan, W.M.; Li, F.A.; Lee, W.H.; Chau, L.Y. Signal peptide peptidase-mediated nuclear localization of heme oxygenase-1 promotes cancer cell proliferation and invasion independent of its enzymatic activity. Oncogene 2015, 34, 2360–2370. [Google Scholar] [CrossRef]
- Birrane, G.; Li, H.; Yang, S.; Tachado, S.D.; Seng, S. Cigarette smoke induces nuclear translocation of heme oxygenase 1 (HO-1) in prostate cancer cells: Nuclear HO-1 promotes vascular endothelial growth factor secretion. Int. J. Oncol. 2013, 42, 1919–1928. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO system in tumor growth, angiogenesis and metabolism—Targeting HO-1 as an anti-tumor therapy. Vascul. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef]
- Vítek, L.; Gbelcová, H.; Muchová, L.; Váňová, K.; Zelenka, J.; Koníčková, R.; Šuk, J.; Zadinova, M.; Knejzlík, Z.; Ahmad, S.; et al. Antiproliferative effects of carbon monoxide on pancreatic cancer. Dig. Liver Dis. 2014, 46, 369–375. [Google Scholar] [CrossRef]
- Lv, C.; Su, Q.; Fang, J.; Yin, H. Styrene-maleic acid copolymer-encapsulated carbon monoxide releasing molecule-2 (SMA/CORM-2) suppresses proliferation, migration and invasion of colorectal cancer cells in vitro and in vivo. Biochem. Biophys. Res. Commun. 2019, 520, 320–326. [Google Scholar] [CrossRef]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [Green Version]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Hanada, N.; Takahata, T.; Zhou, Q.; Ye, X.; Sun, R.; Itoh, J.; Ishiguro, A.; Kijima, H.; Mimura, J.; Itoh, K.; et al. Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 2012, 12, 66. [Google Scholar] [CrossRef] [Green Version]
- Siveen, K.S.; Kuttan, G. Role of macrophages in tumour progression. Immunol. Lett. 2009, 123, 97–102. [Google Scholar] [CrossRef]
- Weis, N.; Weigert, A.; Von Knethen, A.; Brüne, B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol. Biol. Cell 2009, 20, 1280–1288. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Aung, N.Y.; Ohe, R.; Yano, M.; Hashimoto, T.; Fujishima, T.; Kimura, W.; Yamakawa, M. Increasing Heme Oxygenase-1-Expressing Macrophages Indicates a Tendency of Poor Prognosis in Advanced Colorectal Cancer. Digestion 2019, 1–10. [Google Scholar] [CrossRef]
- Blancou, P.; Anegon, I. Editorial: Heme oxygenase-1 and dendritic cells: What else? J. Leukoc. Biol. 2010, 87, 185–187. [Google Scholar] [CrossRef]
- Han, L.; Jiang, J.; Ma, Q.; Wu, Z.; Wang, Z. The inhibition of heme oxygenase-1 enhances the chemosensitivity and suppresses the proliferation of pancreatic cancer cells through the SHH signaling pathway. Int. J. Oncol. 2018, 52, 2101–2109. [Google Scholar] [CrossRef]
- Fang, J.; Sawa, T.; Akaike, T.; Greish, K.; Maeda, H. Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. Int. J. Cancer 2004, 109, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.S.; Li, H.S.; Qi, D.F.; Zhang, J.; Jiang, X.C.; Shi, K.; Zhang, X.J.; Zhang, X.H. Zinc protoporphyrin IX enhances chemotherapeutic response of hepatoma cells to cisplatin. World J. Gastroenterol. 2014, 20, 8572–8582. [Google Scholar] [CrossRef]
- Shang, F.T.; Hui, L.L.; An, X.S.; Zhang, X.C.; Guo, S.G.; Kui, Z. ZnPPIX inhibits peritoneal metastasis of gastric cancer via its antiangiogenic activity. Biomed. Pharmacother. 2015, 71, 240–246. [Google Scholar] [CrossRef]
- Wu, M.; Chien, C.; Chang, J.; Chen, Y. Pro-apoptotic effect of haem oxygenase-1 in human colorectal carcinoma cells via endoplasmic reticular stress. J. Cell. Mol. Med. 2019, 23, 5692–5704. [Google Scholar] [CrossRef]
- DiMarco-Crook, C.; Rakariyatham, K.; Li, Z.; Du, Z.; Zheng, J.; Wu, X.; Xiao, H. Synergistic anticancer effects of curcumin and 3′,4′-didemethylnobiletin in combination on colon cancer cells. J. Food Sci. 2020, 85, 1292–1301. [Google Scholar] [CrossRef]
- Han, Y.; Huang, M.; Li, L.; Cai, X.; Gao, Z.; Li, F.; Rakariyatham, K.; Song, M.; Fernández Tomé, S.; Xiao, H. Non-extractable polyphenols from cranberries: Potential anti-inflammation and anti-colon-cancer agents. Food Funct. 2019, 10, 7714–7723. [Google Scholar] [CrossRef]
- Bi, W.; He, C.N.; Li, X.X.; Zhou, L.Y.; Liu, R.J.; Zhang, S.; Li, G.Q.; Chen, Z.C.; Zhang, P.F. Ginnalin A from Kujin tea (: Acer tataricum subsp. ginnala) exhibits a colorectal cancer chemoprevention effect via activation of the Nrf2/HO-1 signaling pathway. Food Funct. 2018, 9, 2809–2819. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, B.; Feng, X.; Qu, F.; Wang, S.; Wu, L.; Wang, X.; Liu, Q.; Wang, P.; Zhang, K. Apoptosis and autophagy induced by DVDMs-PDT on human esophageal cancer Eca-109 cells. Photodiagn. Photodyn. Ther. 2018, 24, 198–205. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Klickovic, U.; Doberer, D.; Gouya, G.; Aschauer, S.; Weisshaar, S.; Storka, A.; Bilban, M.; Wolzt, M. Human pharmacokinetics of high dose oral curcumin and its effect on heme oxygenase-1 expression in healthy male subjects. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Kimpara, T.; Takeda, A.; Watanabe, K.; Itoyama, Y.; Ikawa, S.; Watanabe, M.; Arai, H.; Sasaki, H.; Higuchi, S.; Okita, N.; et al. Microsatellite polymorphism in the human heme oxygenase-1 gene promoter and its application in association studies with Alzheimer and Parkinson disease. Hum. Genet. 1997, 100, 145–147. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Choi, A.M.K. Heme oxygenase: Colors of defense against cellular stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, M.; Akagi, R.; Morita, K.; Takeuchi, M.; Morimatsu, H.; Toda, Y.; Shimizu, H.; Maeshima, K.; Sasaki, T.; Takahashi, T. Heme Arginate Pretreatment Attenuates Pulmonary NF-κB and AP-1 Activation Induced by Hemorrhagic Shock via Heme Oxygenase-1 Induction. Med. Chem. 2006, 2, 272–274. [Google Scholar] [CrossRef]
- Mustajoki, P.; Nordmann, Y. Early Administration of Heme Arginate for Acute Porphyric Attacks. Arch. Intern. Med. 1993, 153, 2004–2008. [Google Scholar] [CrossRef]
- Anderson, K.E.; Bloomer, J.R.; Bonkovsky, H.L.; Kushner, J.P.; Pierach, C.A.; Pimstone, N.R.; Desnick, R.J. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann. Intern. Med. 2005, 142, 439–450. [Google Scholar] [CrossRef]
- Bharucha, A.E.; Kulkarni, A.; Choi, K.M.; Camilleri, M.; Lempke, M.; Brunn, G.J.; Gibbons, S.J.; Zinsmeister, A.R.; Farrugia, G. First-in-human study demonstrating pharmacological activation of heme oxygenase-1 in humans. Clin. Pharmacol. Ther. 2010, 87, 187–190. [Google Scholar] [CrossRef] [Green Version]
- Doberer, D.; Haschemi, A.; Andreas, M.; Zapf, T.C.; Clive, B.; Jeitler, M.; Heinzl, H.; Wagner, O.; Wolzt, M.; Bilban, M. Haem arginate infusion stimulates haem oxygenase-1 expression in healthy subjects. Br. J. Pharmacol. 2010, 161, 1751–1762. [Google Scholar] [CrossRef] [Green Version]
- Bharucha, A.E.; Choi, K.M.; Saw, J.J.; Gibbons, S.J.; Farrugia, G.F.; Carlson, D.A.; Zinsmeister, A.R. Effects of aspirin & simvastatin and aspirin, simvastatin, & lipoic acid on heme oxygenase-1 in healthy human subjects. Neurogastroenterol. Motil. 2014, 26, 1437–1442. [Google Scholar] [CrossRef]
- Urbánek, P.; Leníček, M.; Muchová, L.; Subhanová, I.; Dušek, L.; Kaspříková, N.; Hrabal, P.; Brůha, R.; Vítek, L. No association of promoter variations of HMOX1 and UGT1A1 genes with liver injury in chronic hepatitis C. Ann. Hepatol. 2011, 10, 445–451. [Google Scholar] [CrossRef]
- Subhanova, I.; Muchova, L.; Lenicek, M.; Vreman, H.J.; Luksan, O.; Kubickova, K.; Kreidlova, M.; Zima, T.; Vitek, L.; Urbanek, P. Expression of Biliverdin Reductase A in Peripheral Blood Leukocytes Is Associated with Treatment Response in HCV-Infected Patients. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northgraves, M.J.; Arunachalam, L.; Madden, L.A.; Marshall, P.; Hartley, J.E.; MacFie, J.; Vince, R.V. Feasibility of a novel exercise prehabilitation programme in patients scheduled for elective colorectal surgery: A feasibility randomised controlled trial. Support. Care Cancer 2020, 28, 3197–3206. [Google Scholar] [CrossRef] [Green Version]
- Habtezion, A.; Kwan, R.; Akhtar, E.; Wanaski, S.P.; Collins, S.D.; Wong, R.J.; Stevenson, D.K.; Butcher, E.C.; Omary, M.B. Panhematin provides a therapeutic benefit in experimental pancreatitis. Gut 2011, 60, 671–679. [Google Scholar] [CrossRef]
- Wang, H.; Gou, W.; Strange, C.; Wang, J.; Nietert, P.J.; Cloud, C.; Owzarski, S.; Shuford, B.; Duke, T.; Luttrell, L.; et al. Islet Harvest in Carbon Monoxide-Saturated Medium for Chronic Pancreatitis Patients Undergoing Islet Autotransplantation. Cell Transplant. 2019, 28, 25S–36S. [Google Scholar] [CrossRef] [Green Version]
- Korbut, E.; Brzozowski, T.; Magierowski, M. Carbon Monoxide Being Hydrogen Sulfide and Nitric Oxide Molecular Sibling, as Endogenous and Exogenous Modulator of Oxidative Stress and Antioxidative Mechanisms in the Digestive System. Oxid. Med. Cell. Longev. 2020, 2, 1–14. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| ID 1 | Phase | Intervention Model | Status | Interventions | Ref. |
|---|---|---|---|---|---|
| NCT00895167 | 1 | Single Group Assignment | Completed | Dietary Supplement: Curcumin | [192] |
| NCT01768507 | N/A 2 | Parallel Assignment | Completed | Dietary Supplement: Resveratrol | - |
| NCT02213523 | N/A 2 | Parallel Assignment | Completed |
| - |
| NCT00882804 | 1 | Parallel Assignment | Completed |
| [193] |
| NCT00682370 | 1 | Crossover Assignment | Completed |
| [194] |
| Non-registered | 1 | Parallel Assignment | Completed |
| [195] |
| NCT02232308 | 1 | Parallel Assignment | Completed |
| - |
| Organ | ID 1 | Disease | Phase | Intervention Model | Status | Interventions | Ref. |
|---|---|---|---|---|---|---|---|
| Liver | NCT04195438 | Liver cancer | N/A 2 | Single Group Assignment | Recruiting |
| - |
| NCT00842205 |
| data | Parallel Assignment | Unknown |
| [201] | |
| NCT01112033 3 | HCV | N/A 2 | N/A 2 | Completed | None | [202] | |
| Intestine/ colon | NCT03494764 | Colitis, ulcerative | 2 | Crossover Assignment | Active, not recruiting | Hyperbaric Oxygen Therapy | - |
| NCT02264496 | Colorectal cancer | N/A 2 | Parallel Assignment | Completed | Exercise | [203] |
| Treatment-Based | Organ | ID 1 | Disease | Phase | Intervention Model | Status | Interventions | Ref. |
|---|---|---|---|---|---|---|---|---|
| CO | Intestine/ colon | NCT01050712 2 | Post-operative ileus | 2 | Parallel Assignment | Terminated |
| -- |
| Pancreas | NCT02567240 | Chronic pancreatitis | 2 | Parallel Assignment | Completed | Carbon monoxide-bubbled mediums | [204] | |
| Hemin | Stomach | NCT01206582 | Gastroparesis | 2 | Parallel Assignment | Completed |
| [102] |
| Pancreas | NCT01855841 | Acute pancreatitis | 2 | Parallel Assignment | Recruiting |
| - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puentes-Pardo, J.D.; Moreno-SanJuan, S.; Carazo, Á.; León, J. Heme Oxygenase-1 in Gastrointestinal Tract Health and Disease. Antioxidants 2020, 9, 1214. https://doi.org/10.3390/antiox9121214
Puentes-Pardo JD, Moreno-SanJuan S, Carazo Á, León J. Heme Oxygenase-1 in Gastrointestinal Tract Health and Disease. Antioxidants. 2020; 9(12):1214. https://doi.org/10.3390/antiox9121214
Chicago/Turabian StylePuentes-Pardo, Jose D., Sara Moreno-SanJuan, Ángel Carazo, and Josefa León. 2020. "Heme Oxygenase-1 in Gastrointestinal Tract Health and Disease" Antioxidants 9, no. 12: 1214. https://doi.org/10.3390/antiox9121214
APA StylePuentes-Pardo, J. D., Moreno-SanJuan, S., Carazo, Á., & León, J. (2020). Heme Oxygenase-1 in Gastrointestinal Tract Health and Disease. Antioxidants, 9(12), 1214. https://doi.org/10.3390/antiox9121214