Thioredoxin and Glutaredoxin Systems as Potential Targets for the Development of New Treatments in Friedreich’s Ataxia

 ,
, 
 , and
, and
Abstract
:
1. Introduction
Thioredoxin and Glutaredoxin Systems in Redox Biology
2. Thioredoxins and Glutaredoxins in Friedreich’s Ataxia
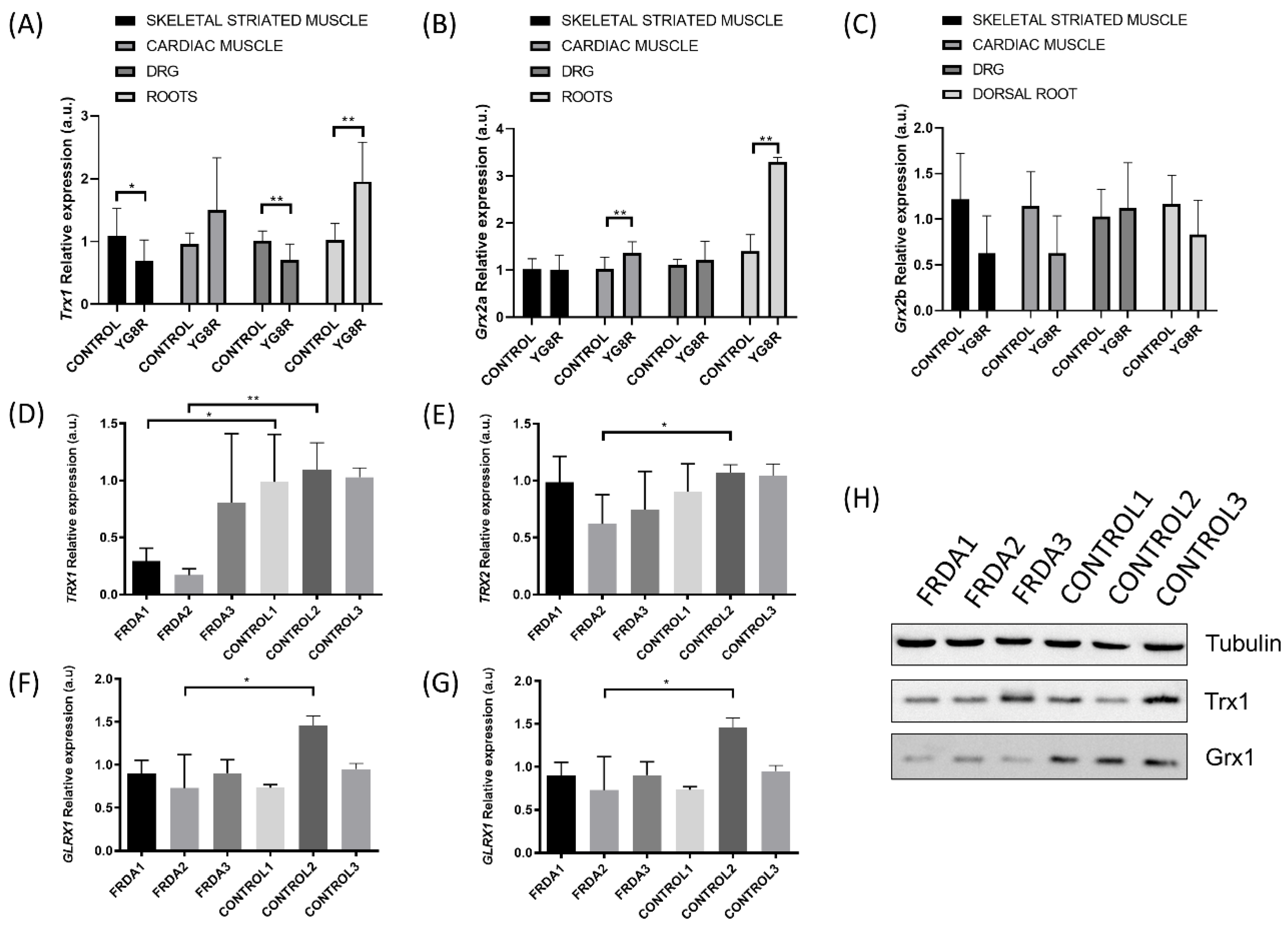
2.1. TRXs and GLRXs Are Downregulated in FRDA Models
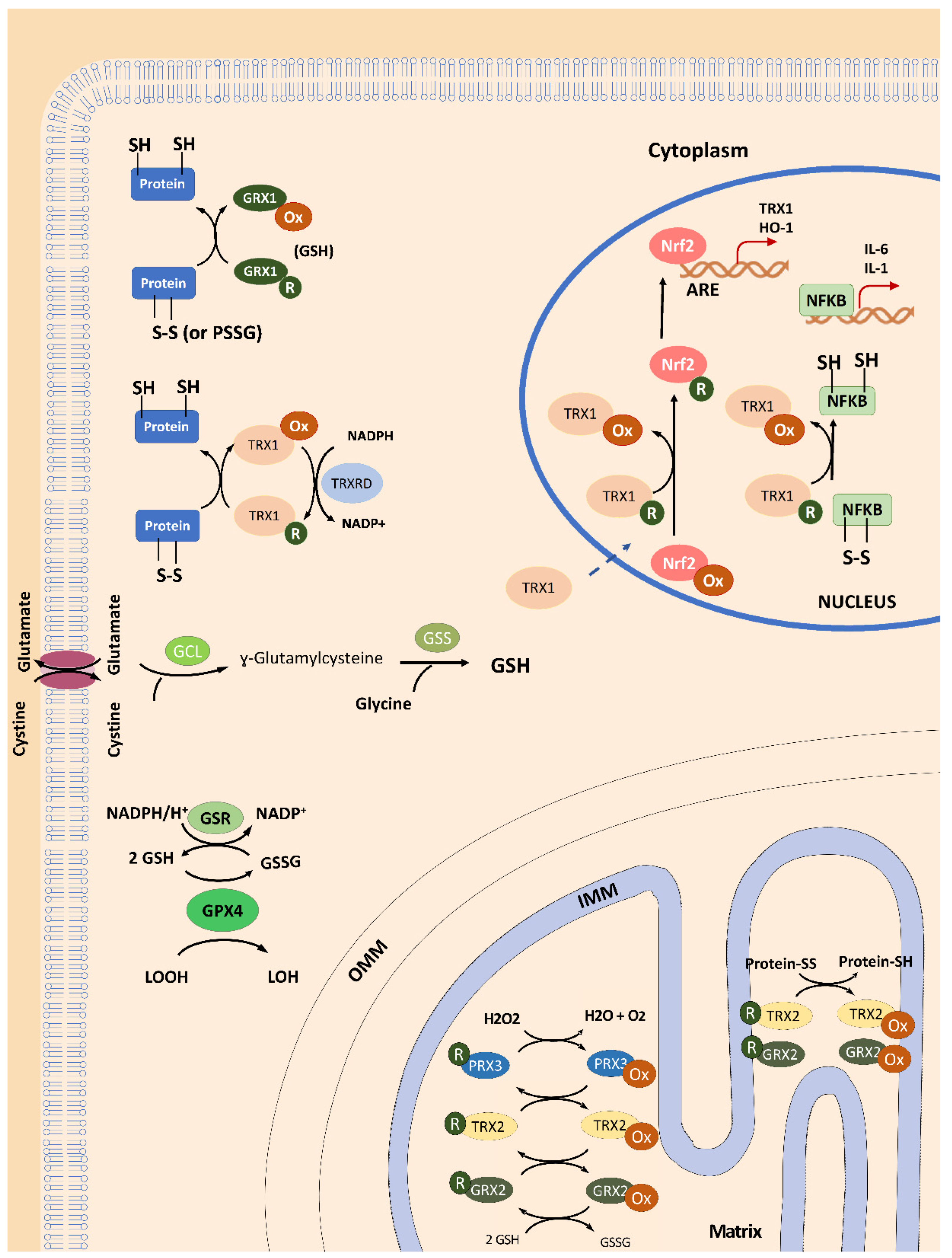
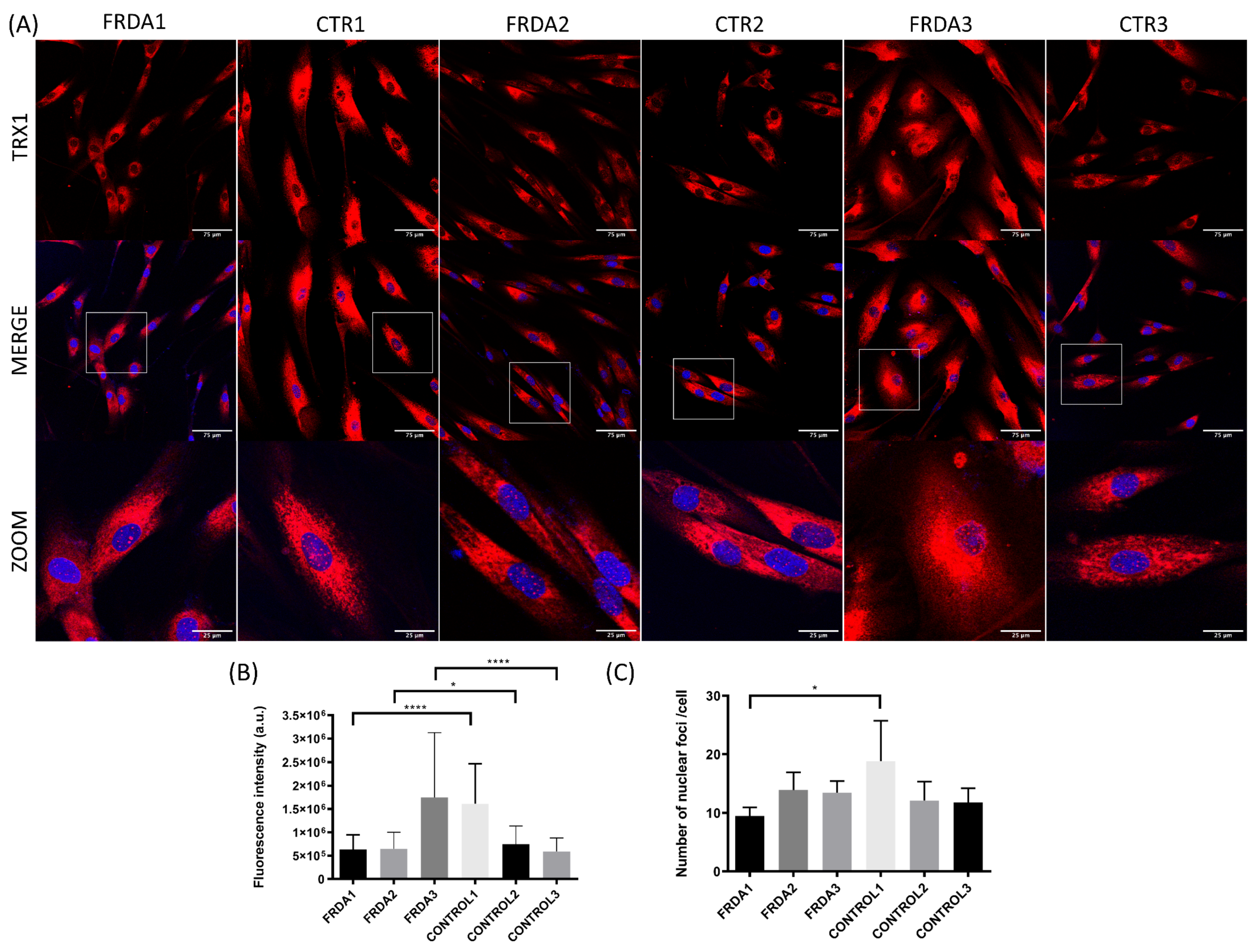
2.2. TRX1 Nuclear Translocation Is Altered in FRDA Fibroblasts
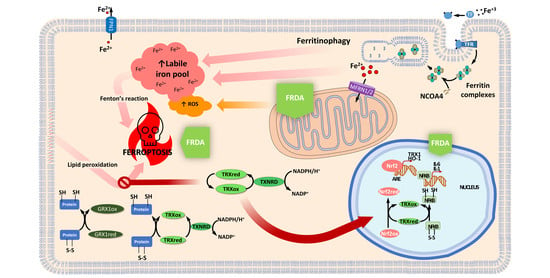
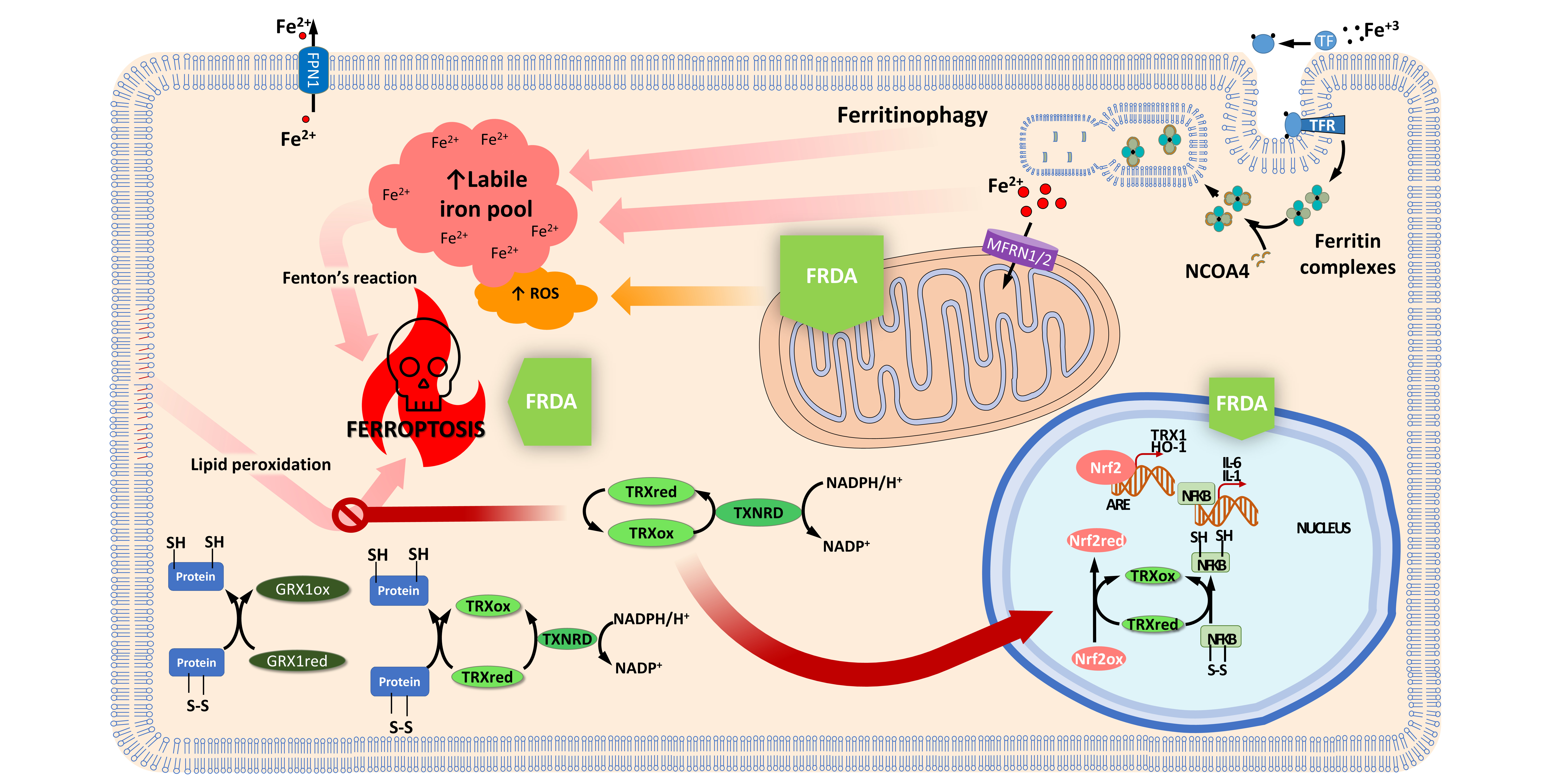
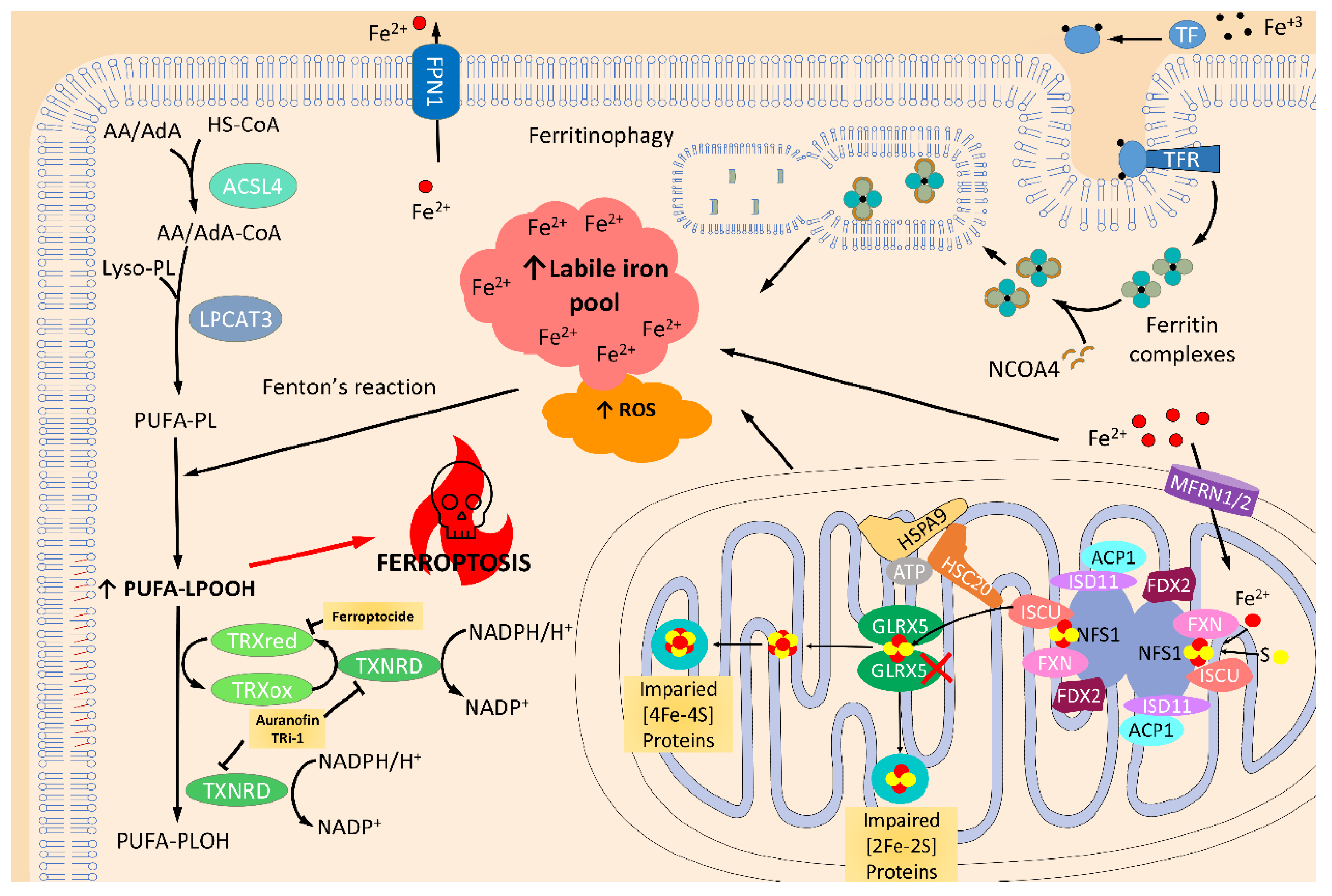
2.3. Thioredoxin and Glutaredoxin Systems in Iron and Iron-Sulfur Cluster Metabolism
2.4. Thioredoxin and Glutaredoxin Systems Regulating Ferroptosis
3. Activation of the Thioredoxin Family by NRF2 Activators as Therapeutic Options in FRDA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lu, J.; Holmgren, A. Thioredoxin system in cell death progression. Antioxid. Redox Signal. 2012, 17, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, D.F.; Abderrazak, A.; El Hadri, K.; Simmet, T.; Rouis, M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid. Redox Signal. 2013, 19, 1266–1303. [Google Scholar] [CrossRef] [PubMed]
- Vlamis-Gardikas, A.; Holmgren, A. Thioredoxin and Glutaredoxin Isoforms. In Methods in Enzymology; Sies, H., Packer, L., Eds.; Academic Press: Cambridge, MA, USA, 2002; Volume 347, pp. 286–296. [Google Scholar]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef]
- Holmgren, A. Thioredoxin. Annu. Rev. Biochem. 1985, 54, 237–271. [Google Scholar] [CrossRef]
- Arai, R.J.; Masutani, H.; Yodoi, J.; Debbas, V.; Laurindo, F.R.; Stern, A.; Monteiro, H.P. Nitric oxide induces thioredoxin-1 nuclear translocation: Possible association with the p21Ras survival pathway. Biochem. Biophys. Res. Commun. 2006, 348, 1254–1260. [Google Scholar] [CrossRef]
- Akamatsu, Y.; Ohno, T.; Hirota, K.; Kagoshima, H.; Yodoi, J.; Shigesada, K. Redox regulation of the DNA binding activity in transcription factor PEBP2. The roles of two conserved cysteine residues. J. Biol. Chem. 1997, 272, 14497–14500. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K.; Matsui, M.; Iwata, S.; Nishiyama, A.; Mori, K.; Yodoi, J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc. Natl. Acad. Sci. USA 1997, 94, 3633–3638. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gimenez, J.L.; Seco-Cervera, M.; Aguado, C.; Roma-Mateo, C.; Dasi, F.; Priego, S.; Markovic, J.; Knecht, E.; Sanz, P.; Pallardo, F.V. Lafora disease fibroblasts exemplify the molecular interdependence between thioredoxin 1 and the proteasome in mammalian cells. Free Radic. Biol. Med. 2013, 65, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Léveillard, T.; Aït-Ali, N. Cell Signaling with Extracellular Thioredoxin and Thioredoxin-Like Proteins: Insight into Their Mechanisms of Action. Oxidative Med. Cell. Longev. 2017, 2017, 8475125. [Google Scholar] [CrossRef] [Green Version]
- Bertini, R.; Howard, O.M.; Dong, H.F.; Oppenheim, J.J.; Bizzarri, C.; Sergi, R.; Caselli, G.; Pagliei, S.; Romines, B.; Wilshire, J.A.; et al. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J. Exp. Med. 1999, 189, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Yodoi, J.; Matsuo, Y.; Tian, H.; Masutani, H.; Inamoto, T. Anti-Inflammatory Thioredoxin Family Proteins for Medicare, Healthcare and Aging Care. Nutrients 2017, 9, 1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, K.; Murata, M.; Sachi, Y.; Nakamura, H.; Takeuchi, J.; Mori, K.; Yodoi, J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 1999, 274, 27891–27897. [Google Scholar] [CrossRef] [Green Version]
- Makino, Y.; Yoshikawa, N.; Okamoto, K.; Hirota, K.; Yodoi, J.; Makino, I.; Tanaka, H. Direct association with thioredoxin allows redox regulation of glucocorticoid receptor function. J. Biol. Chem. 1999, 274, 3182–3188. [Google Scholar] [CrossRef] [Green Version]
- Das, K.C.; Lewis-Molock, Y.; White, C.W. Elevation of manganese superoxide dismutase gene expression by thioredoxin. Am. J. Respir. Cell Mol. Biol. 1997, 17, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Perez, V.I.; Lew, C.M.; Cortez, L.A.; Webb, C.R.; Rodriguez, M.; Liu, Y.; Qi, W.; Li, Y.; Chaudhuri, A.; Van Remmen, H.; et al. Thioredoxin 2 haploinsufficiency in mice results in impaired mitochondrial function and increased oxidative stress. Free Radic. Biol. Med. 2008, 44, 882–892. [Google Scholar] [CrossRef]
- Tanaka, T.; Hosoi, F.; Yamaguchi-Iwai, Y.; Nakamura, H.; Masutani, H.; Ueda, S.; Nishiyama, A.; Takeda, S.; Wada, H.; Spyrou, G.; et al. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002, 21, 1695–1703. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, A.; Aslund, F. Glutaredoxin. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1995; Volume 252, pp. 283–292. [Google Scholar]
- Trnka, D.; Engelke, A.D.; Gellert, M.; Moseler, A.; Hossain, M.F.; Lindenberg, T.T.; Pedroletti, L.; Odermatt, B.; de Souza, J.V.; Bronowska, A.K.; et al. Molecular basis for the distinct functions of redox-active and FeS-transfering glutaredoxins. Nat. Commun. 2020, 11, 3445. [Google Scholar] [CrossRef]
- Bushweller, J.H.; Aslund, F.; Wuthrich, K.; Holmgren, A. Structural and functional characterization of the mutant Escherichia coli glutaredoxin (C14→S) and its mixed disulfide with glutathione. Biochemistry 1992, 31, 9288–9293. [Google Scholar] [CrossRef] [PubMed]
- Gravina, S.A.; Mieyal, J.J. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry 1993, 32, 3368–3376. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom-Ljung, J.; Holmgren, A. Glutaredoxin accelerates glutathione-dependent folding of reduced ribonuclease A together with protein disulfide-isomerase. J. Biol. Chem. 1995, 270, 7822–7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, U.; Mieyal, P.A.; Mieyal, J.J. pH profiles indicative of rate-limiting nucleophilic displacement in thioltransferase catalysis. Biochemistry 1997, 36, 3199–3206. [Google Scholar] [CrossRef]
- Prieto-Alamo, M.J.; Jurado, J.; Gallardo-Madueno, R.; Monje-Casas, F.; Holmgren, A.; Pueyo, C. Transcriptional regulation of glutaredoxin and thioredoxin pathways and related enzymes in response to oxidative stress. J. Biol. Chem. 2000, 275, 13398–13405. [Google Scholar] [CrossRef] [Green Version]
- Starke, D.W.; Chock, P.B.; Mieyal, J.J. Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J. Biol. Chem. 2003, 278, 14607–14613. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, A.; Johansson, C.; Berndt, C.; Lonn, M.E.; Hudemann, C.; Lillig, C.H. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem. Soc. Trans. 2005, 33, 1375–1377. [Google Scholar] [CrossRef] [Green Version]
- Wingert, R.A.; Galloway, J.L.; Barut, B.; Foott, H.; Fraenkel, P.; Axe, J.L.; Weber, G.J.; Dooley, K.; Davidson, A.J.; Schmid, B.; et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature 2005, 436, 1035–1039. [Google Scholar] [CrossRef]
- Holmgren, A. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 1989, 264, 13963–13966. [Google Scholar]
- Wells, W.W.; Xu, D.P.; Yang, Y.F.; Rocque, P.A. Mammalian thioltransferase (glutaredoxin) and protein disulfide isomerase have dehydroascorbate reductase activity. J. Biol. Chem. 1990, 265, 15361–15364. [Google Scholar] [PubMed]
- Lillig, C.H.; Berndt, C.; Vergnolle, O.; Lonn, M.E.; Hudemann, C.; Bill, E.; Holmgren, A. Characterization of human glutaredoxin 2 as iron-sulfur protein: A possible role as redox sensor. Proc. Natl. Acad. Sci. USA 2005, 102, 8168–8173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, M.; Johansson, C.; Chandra, J.; Enoksson, M.; Jacobsson, G.; Ljung, J.; Johansson, M.; Holmgren, A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J. Biol. Chem. 2001, 276, 26269–26275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladyshev, V.N.; Liu, A.; Novoselov, S.V.; Krysan, K.; Sun, Q.A.; Kryukov, V.M.; Kryukov, G.V.; Lou, M.F. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J. Biol. Chem. 2001, 276, 30374–30380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, C.; Hudemann, C.; Hanschmann, E.M.; Axelsson, R.; Holmgren, A.; Lillig, C.H. How does iron-sulfur cluster coordination regulate the activity of human glutaredoxin 2? Antioxid. Redox Signal. 2007, 9, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Kavanagh, K.L.; Gileadi, O.; Oppermann, U. Reversible sequestration of active site cysteines in a 2Fe-2S-bridged dimer provides a mechanism for glutaredoxin 2 regulation in human mitochondria. J. Biol. Chem. 2007, 282, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ouyang, B.; Li, Y.; Feng, Y.; Jacquot, J.P.; Rouhier, N.; Xia, B. Glutathione regulates the transfer of iron-sulfur cluster from monothiol and dithiol glutaredoxins to apo ferredoxin. Protein Cell 2012, 3, 714–721. [Google Scholar] [CrossRef] [Green Version]
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: Implications for mitochondrial redox regulation and antioxidant DEFENSE. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef] [Green Version]
- Johansson, C.; Lillig, C.H.; Holmgren, A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J. Biol. Chem. 2004, 279, 7537–7543. [Google Scholar] [CrossRef] [Green Version]
- Hanschmann, E.M.; Lonn, M.E.; Schutte, L.D.; Funke, M.; Godoy, J.R.; Eitner, S.; Hudemann, C.; Lillig, C.H. Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J. Biol. Chem. 2010, 285, 40699–40705. [Google Scholar] [CrossRef] [Green Version]
- Enoksson, M.; Fernandes, A.P.; Prast, S.; Lillig, C.H.; Holmgren, A.; Orrenius, S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem. Biophys. Res. Commun. 2005, 327, 774–779. [Google Scholar] [CrossRef]
- Takashima, Y.; Hirota, K.; Nakamura, H.; Nakamura, T.; Akiyama, K.; Cheng, F.S.; Maeda, M.; Yodoi, J. Differential expression of glutaredoxin and thioredoxin during monocytic differentiation. Immunol. Lett. 1999, 68, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Ohno, T.; Hirota, K.; Nishiyama, A.; Nakamura, H.; Wada, H.; Yodoi, J. Mouse glutaredoxin—cDNA cloning, high level expression in E. coli and its possible implication in redox regulation of the DNA binding activity in transcription factor PEBP2. Free Radic. Res. 1999, 31, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Starke, D.W.; Mieyal, J.J.; Gronostajski, R.M. Thioltransferase (glutaredoxin) reactivates the DNA-binding activity of oxidation-inactivated nuclear factor I. J. Biol. Chem. 1998, 273, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, K.; Matsui, M.; Murata, M.; Takashima, Y.; Cheng, F.S.; Itoh, T.; Fukuda, K.; Yodoi, J. Nucleoredoxin, glutaredoxin, and thioredoxin differentially regulate NF-kappaB, AP-1, and CREB activation in HEK293 cells. Biochem. Biophys. Res. Commun. 2000, 274, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Manzaneque, M.T.; Tamarit, J.; Belli, G.; Ros, J.; Herrero, E. Grx5 is a mitochondrial glutaredoxin required for the activity of iron/sulfur enzymes. Mol. Biol. Cell 2002, 13, 1109–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Zhang, H.; Lu, J.; Holmgren, A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 2012, 287, 38210–38219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Du, Y.; Zhang, X.; Lu, J.; Holmgren, A. Glutaredoxin 2 reduces both thioredoxin 2 and thioredoxin 1 and protects cells from apoptosis induced by auranofin and 4-hydroxynonenal. Antioxid. Redox Signal. 2014, 21, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Cebula, M.; Schmidt, E.E.; Arner, E.S. TrxR1 as a potent regulator of the Nrf2-Keap1 response system. Antioxid. Redox Signal. 2015, 23, 823–853. [Google Scholar] [CrossRef]
- Bidichandani, S.I.; Delatycki, M.B. Friedreich Ataxia. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Delatycki, M.B.; Corben, L.A. Clinical features of Friedreich ataxia. J. Child Neurol. 2012, 27, 1133–1137. [Google Scholar] [CrossRef]
- Cady, R.B.; Bobechko, W.P. Incidence, Natural History, and Treatment of Scoliosis in Friedreich’s Ataxia. J. Pediatric Orthop. 1984, 4, 673–676. [Google Scholar] [CrossRef]
- Cnop, M.; Mulder, H.; Igoillo-Esteve, M. Diabetes in Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 94–102. [Google Scholar] [CrossRef]
- Harding, A.E.; Hewer, R.L. The heart disease of Friedreich’s ataxia: A clinical and electrocardiographic study of 115 patients, with an analysis of serial electrocardiographic changes in 30 cases. Q. J. Med. 1983, 52, 489–502. [Google Scholar] [PubMed]
- Pandolfo, M. Friedreich ataxia: The clinical picture. J. Neurol. 2009, 256 (Suppl. 1), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; De Biase, I.; Gomez, M.; Delatycki, M.B.; Ashizawa, T.; Bidichandani, S.I. Friedreich ataxia in carriers of unstable borderline GAA triplet-repeat alleles. Ann. Neurol. 2004, 56, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Rotig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Puccio, H. Dysregulation of cellular iron metabolism in Friedreich ataxia: From primary iron-sulfur cluster deficit to mitochondrial iron accumulation. Front. Pharmacol. 2014, 5, 130. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gimenez, J.L.; Gimeno, A.; Gonzalez-Cabo, P.; Dasi, F.; Bolinches-Amoros, A.; Molla, B.; Palau, F.; Pallardo, F.V. Differential expression of PGC-1alpha and metabolic sensors suggest age-dependent induction of mitochondrial biogenesis in Friedreich ataxia fibroblasts. PLoS ONE 2011, 6, e20666. [Google Scholar] [CrossRef] [Green Version]
- Bolinches-Amoros, A.; Molla, B.; Pla-Martin, D.; Palau, F.; Gonzalez-Cabo, P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell. Neurosci. 2014, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 51. [Google Scholar] [CrossRef] [Green Version]
- Navarro, J.A.; Ohmann, E.; Sanchez, D.; Botella, J.A.; Liebisch, G.; Molto, M.D.; Ganfornina, M.D.; Schmitz, G.; Schneuwly, S. Altered lipid metabolism in a Drosophila model of Friedreich’s ataxia. Hum. Mol. Genet. 2010, 19, 2828–2840. [Google Scholar] [CrossRef] [Green Version]
- Chantrel-Groussard, K.; Geromel, V.; Puccio, H.; Koenig, M.; Munnich, A.; Rotig, A.; Rustin, P. Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum. Mol. Genet. 2001, 10, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Jiralerspong, S.; Ge, B.; Hudson, T.J.; Pandolfo, M. Manganese superoxide dismutase induction by iron is impaired in Friedreich ataxia cells. FEBS Lett. 2001, 509, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Vaubel, R.A.; Isaya, G. Iron–sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Mol. Cell. Neurosci. 2013, 55, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Jasoliya, M.J.; McMackin, M.Z.; Henderson, C.K.; Perlman, S.L.; Cortopassi, G.A. Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans. Hum. Mol. Genet. 2017, 26, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Bertino, F.; Tolosano, E. Hereditary Ataxia: A Focus on Heme Metabolism and Fe-S Cluster Biogenesis. Int. J. Mol. Sci. 2020, 21, 3760. [Google Scholar] [CrossRef]
- Yoon, T.; Cowan, J.A. Iron-sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef]
- Layer, G.; Ollagnier-de Choudens, S.; Sanakis, Y.; Fontecave, M. Iron-sulfur cluster biosynthesis: Characterization of Escherichia coli CYaY as an iron donor for the assembly of [2Fe-2S] clusters in the scaffold IscU. J. Biol. Chem. 2006, 281, 16256–16263. [Google Scholar] [CrossRef] [Green Version]
- Parent, A.; Elduque, X.; Cornu, D.; Belot, L.; Le Caer, J.P.; Grandas, A.; Toledano, M.B.; D’Autreaux, B. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 2015, 6, 5686. [Google Scholar] [CrossRef] [Green Version]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Muller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019, 10, 3566. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 2210. [Google Scholar] [CrossRef] [Green Version]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef] [PubMed]
- Llorens, J.V.; Navarro, J.A.; Martinez-Sebastian, M.J.; Baylies, M.K.; Schneuwly, S.; Botella, J.A.; Molto, M.D. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J. 2007, 21, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Magrane, J.; Rattelle, A.; Stepanova, A.; Galkin, A.; Clark, E.M.; Dong, Y.N.; Halawani, S.M.; Lynch, D.R. Early cerebellar deficits in mitochondrial biogenesis and respiratory chain complexes in the KIKO mouse model of Friedreich ataxia. Dis. Models Mech. 2017, 10, 1343–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef] [Green Version]
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the Mitochondrial Heme Metabolism Complex. PLoS ONE 2015, 10, e0135896. [Google Scholar] [CrossRef] [Green Version]
- Steinkellner, H.; Singh, H.N.; Muckenthaler, M.U.; Goldenberg, H.; Moganty, R.R.; Scheiber-Mojdehkar, B.; Sturm, B. No changes in heme synthesis in human Friedreich s ataxia erythroid progenitor cells. Gene 2017, 621, 5–11. [Google Scholar] [CrossRef]
- Martelli, A.; Schmucker, S.; Reutenauer, L.; Mathieu, J.R.R.; Peyssonnaux, C.; Karim, Z.; Puy, H.; Galy, B.; Hentze, M.W.; Puccio, H. Iron regulatory protein 1 sustains mitochondrial iron loading and function in frataxin deficiency. Cell Metab. 2015, 21, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J.L.; Blake, J.C.; Chamberlain, S.; Thomas, P.K.; Cooper, J.M.; Schapira, A.H. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet. 2000, 9, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Puccio, H.; Simon, D.; Cossee, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Huang, M.L.; Becker, E.M.; Whitnall, M.; Suryo Rahmanto, Y.; Ponka, P.; Richardson, D.R. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. USA 2009, 106, 16381–16386. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, A.H.; Michael, S.C.; Knutson, M.D.; Haile, D.J.; Qian, J.; Levi, S.; Santambrogio, P.; Garrick, M.D.; Lamarche, J.B. The dentate nucleus in Friedreich’s ataxia: The role of iron-responsive proteins. Acta Neuropathol. 2007, 114, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, I.H.; Raniga, P.; Delatycki, M.B.; Stagnitti, M.R.; Corben, L.A.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Tissue atrophy and elevated iron concentration in the extrapyramidal motor system in Friedreich ataxia: The IMAGE-FRDA study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1261–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeppen, A.H.; Morral, J.A.; Davis, A.N.; Qian, J.; Petrocine, S.V.; Knutson, M.D.; Gibson, W.M.; Cusack, M.J.; Li, D. The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol. 2009, 118, 763–776. [Google Scholar] [CrossRef]
- Llorens, J.V.; Soriano, S.; Calap-Quintana, P.; Gonzalez-Cabo, P.; Molto, M.D. The Role of Iron in Friedreich’s Ataxia: Insights From Studies in Human Tissues and Cellular and Animal Models. Front. Neurosci. 2019, 13, 75. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.Y.; Poddar, A.; Magtanong, L.; Lumb, J.H.; Mileur, T.R.; Reid, M.A.; Dovey, C.M.; Wang, J.; Locasale, J.W.; Stone, E.; et al. A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep. 2019, 26, 1544–1556.e8. [Google Scholar] [CrossRef] [Green Version]
- Llabani, E.; Hicklin, R.W.; Lee, H.Y.; Motika, S.E.; Crawford, L.A.; Weerapana, E.; Hergenrother, P.J. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat. Chem. 2019, 11, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Behnisch-Cornwell, S.; Bandaru, S.S.M.; Napierkowski, M.; Wolff, L.; Zubair, M.; Urbainsky, C.; Lillig, C.; Schulzke, C.; Bednarski, P.J. Pentathiepins: A Novel Class of Glutathione Peroxidase 1 Inhibitors that Induce Oxidative Stress, Loss of Mitochondrial Membrane Potential and Apoptosis in Human Cancer Cells. ChemMedChem 2020. [Google Scholar] [CrossRef] [PubMed]
- Codazzi, F.; Hu, A.; Rai, M.; Donatello, S.; Salerno Scarzella, F.; Mangiameli, E.; Pelizzoni, I.; Grohovaz, F.; Pandolfo, M. Friedreich ataxia-induced pluripotent stem cell-derived neurons show a cellular phenotype that is corrected by a benzamide HDAC inhibitor. Hum. Mol. Genet. 2016, 25, 4847–4855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.D.; Grivennikova, V.G. Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 863–871. [Google Scholar] [CrossRef]
- Balsa, E.; Perry, E.A.; Bennett, C.F.; Jedrychowski, M.; Gygi, S.P.; Doench, J.G.; Puigserver, P. Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nat. Commun. 2020, 11, 2714. [Google Scholar] [CrossRef]
- Heidari, M.M.; Houshmand, M.; Hosseinkhani, S.; Nafissi, S.; Khatami, M. Complex I and ATP content deficiency in lymphocytes from Friedreich’s ataxia. Can. J. Neurol. Sci. 2009, 36, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Salehi, M.H.; Kamalidehghan, B.; Houshmand, M.; Yong Meng, G.; Sadeghizadeh, M.; Aryani, O.; Nafissi, S. Gene expression profiling of mitochondrial oxidative phosphorylation (OXPHOS) complex I in Friedreich ataxia (FRDA) patients. PLoS ONE 2014, 9, e94069. [Google Scholar] [CrossRef] [Green Version]
- Turanov, A.A.; Kehr, S.; Marino, S.M.; Yoo, M.H.; Carlson, B.A.; Hatfield, D.L.; Gladyshev, V.N. Mammalian thioredoxin reductase 1: Roles in redox homoeostasis and characterization of cellular targets. Biochem. J. 2010, 430, 285–293. [Google Scholar] [CrossRef]
- Cozza, G.; Rossetto, M.; Bosello-Travain, V.; Maiorino, M.; Roveri, A.; Toppo, S.; Zaccarin, M.; Zennaro, L.; Ursini, F. Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: The polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic. Biol. Med. 2017, 112, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeti, R.; Parkinson, M.H.; Hargreaves, I.P.; Angelova, P.R.; Sandi, C.; Pook, M.A.; Giunti, P.; Abramov, A.Y. Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia. Cell Death Dis. 2016, 7, e2237. [Google Scholar] [CrossRef] [PubMed]
- Calap-Quintana, P.; Soriano, S.; Llorens, J.V.; Al-Ramahi, I.; Botas, J.; Molto, M.D.; Martinez-Sebastian, M.J. TORC1 Inhibition by Rapamycin Promotes Antioxidant Defences in a Drosophila Model of Friedreich’s Ataxia. PLoS ONE 2015, 10, e0132376. [Google Scholar] [CrossRef]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 2013, 19, 1481–1493. [Google Scholar] [CrossRef] [Green Version]
- Auchere, F.; Santos, R.; Planamente, S.; Lesuisse, E.; Camadro, J.M. Glutathione-dependent redox status of frataxin-deficient cells in a yeast model of Friedreich’s ataxia. Hum. Mol. Genet. 2008, 17, 2790–2802. [Google Scholar] [CrossRef] [Green Version]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Investig. 2001, 31, 1007–1011. [Google Scholar] [CrossRef]
- Bulteau, A.L.; Planamente, S.; Jornea, L.; Dur, A.; Lesuisse, E.; Camadro, J.M.; Auchere, F. Changes in mitochondrial glutathione levels and protein thiol oxidation in yfh1 yeast cells and the lymphoblasts of patients with Friedreich’s ataxia. Biochim. Biophys. Acta 2012, 1822, 212–225. [Google Scholar] [CrossRef]
- Lefevre, S.; Sliwa, D.; Auchere, F.; Brossas, C.; Ruckenstuhl, C.; Boggetto, N.; Lesuisse, E.; Madeo, F.; Camadro, J.M.; Santos, R. The yeast metacaspase is implicated in oxidative stress response in frataxin-deficient cells. FEBS Lett. 2012, 586, 143–148. [Google Scholar] [CrossRef]
- Bulteau, A.L.; Dancis, A.; Gareil, M.; Montagne, J.J.; Camadro, J.M.; Lesuisse, E. Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic. Biol. Med. 2007, 42, 1561–1570. [Google Scholar] [CrossRef]
- Tanito, M.; Agbaga, M.P.; Anderson, R.E. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic. Biol. Med. 2007, 42, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Telorack, M.; Meyer, M.; Ingold, I.; Conrad, M.; Bloch, W.; Werner, S. A Glutathione-Nrf2-Thioredoxin Cross-Talk Ensures Keratinocyte Survival and Efficient Wound Repair. PLoS Genet. 2016, 12, e1005800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, C.; Roos, A.K.; Montano, S.J.; Sengupta, R.; Filippakopoulos, P.; Guo, K.; von Delft, F.; Holmgren, A.; Oppermann, U.; Kavanagh, K.L. The crystal structure of human GLRX5: Iron-sulfur cluster co-ordination, tetrameric assembly and monomer activity. Biochem. J. 2011, 433, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMackin, M.Z.; Durbin-Johnson, B.; Napierala, M.; Napierala, J.S.; Ruiz, L.; Napoli, E.; Perlman, S.; Giulivi, C.; Cortopassi, G.A. Potential biomarker identification for Friedreich’s ataxia using overlapping gene expression patterns in patient cells and mouse dorsal root ganglion. PLoS ONE 2019, 14, e0223209. [Google Scholar] [CrossRef]
- Diwakar, L.; Kenchappa, R.S.; Annepu, J.; Ravindranath, V. Downregulation of glutaredoxin but not glutathione loss leads to mitochondrial dysfunction in female mice CNS: Implications in excitotoxicity. Neurochem. Int. 2007, 51, 37–46. [Google Scholar] [CrossRef]
- Kenchappa, R.S.; Ravindranath, V. Glutaredoxin is essential for maintenance of brain mitochondrial complex I: Studies with MPTP. FASEB J. 2003, 17, 717–719. [Google Scholar] [CrossRef] [Green Version]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; Di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem. 2003, 278, 42588–42595. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Gonzalez-Billault, C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: Implications for neuronal development and trafficking. Front. Cell. Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef] [Green Version]
- Haugen, A.C.; Di Prospero, N.A.; Parker, J.S.; Fannin, R.D.; Chou, J.; Meyer, J.N.; Halweg, C.; Collins, J.B.; Durr, A.; Fischbeck, K.; et al. Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: Cellular model of pathology. PLoS Genet. 2010, 6, e1000812. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate Regulation of Glutathione Biosynthesis and Release by Nrf2-Expressing Glia Potently Protects Neurons from Oxidative Stress. J. Neurosci. 2003, 23, 3394. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Talalay, P.; Dinkova-Kostova, A.T.; Holtzclaw, W.D. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv. Enzym. Regul. 2003, 43, 121–134. [Google Scholar] [CrossRef]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Masutani, H.; Yamaguchi, Y.; Itoh, K.; Yamamoto, M.; Yodoi, J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. J. Biol. Chem. 2001, 276, 18399–18406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, Y.; Taniguchi-Ueda, Y.; Mori, K.; Yodoi, J. A novel promoter sequence is involved in the oxidative stress-induced expression of the adult T-cell leukemia-derived factor (ADF)/human thioredoxin (Trx) gene. Nucleic Acids Res. 1996, 24, 2746–2752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzovino, A.; Chiang, S.; Brown, B.E.; Hawkins, C.L.; Richardson, D.R.; Huang, M.L. Molecular Alterations in a Mouse Cardiac Model of Friedreich Ataxia: An Impaired Nrf2 Response Mediated via Upregulation of Keap1 and Activation of the Gsk3beta Axis. Am. J. Pathol. 2017, 187, 2858–2875. [Google Scholar] [CrossRef] [Green Version]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchere, F.; Lonn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, e4253. [Google Scholar] [CrossRef] [Green Version]
- D′Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, P.; Petrillo, S.; Bertini, E.S.; Piemonte, F. Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement. Biomolecules 2020, 10, 702. [Google Scholar] [CrossRef]
- Hansen, J.M.; Watson, W.H.; Jones, D.P. Compartmentation of Nrf-2 redox control: Regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol. Sci. 2004, 82, 308–317. [Google Scholar] [CrossRef]
- Garcia-Gimenez, J.L.; Markovic, J.; Dasi, F.; Queval, G.; Schnaubelt, D.; Foyer, C.H.; Pallardo, F.V. Nuclear glutathione. Biochim. Biophys. Acta 2013, 1830, 3304–3316. [Google Scholar] [CrossRef] [PubMed]
- Markovic, J.; Borras, C.; Ortega, A.; Sastre, J.; Vina, J.; Pallardo, F.V. Glutathione is recruited into the nucleus in early phases of cell proliferation. J. Biol. Chem. 2007, 282, 20416–20424. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.D.; Tan, E.K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallander, M.L.; Leibold, E.A.; Eisenstein, R.S. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim. Biophys. Acta 2006, 1763, 668–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, S.; Petrocine, S.V.; Qian, J.; Lamarche, J.B.; Knutson, M.D.; Garrick, M.D.; Koeppen, A.H. Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum 2006, 5, 257–267. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich ataxia: Neuropathology revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Berndt, C.; Hanschmann, E.-M.; Urbainsky, C.; Jordt, L.M.; Müller, C.S.; Bodnar, Y.; Schipper, S.; Handorf, O.; Nowack, R.; Moulis, J.-M.; et al. FeS-cluster coordination of vertebrate thioredoxins regulates suppression of hypoxia-induced factor 2α through iron regulatory protein 1. bioRxiv 2020. [Google Scholar] [CrossRef]
- Haunhorst, P.; Berndt, C.; Eitner, S.; Godoy, J.R.; Lillig, C.H. Characterization of the human monothiol glutaredoxin 3 (PICOT) as iron-sulfur protein. Biochem. Biophys. Res. Commun. 2010, 394, 372–376. [Google Scholar] [CrossRef]
- Martelli, A.; Napierala, M.; Puccio, H. Understanding the genetic and molecular pathogenesis of Friedreich’s ataxia through animal and cellular models. Dis. Models Mech. 2012, 5, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Rouault, T.A. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 2010, 49, 4945–4956. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.X.; Sun, X.; Yan, X.L.; Guo, Z.N.; Yang, Y. Ferroptosis in Neurological Diseases. Front. Cell. Neurosci. 2020, 14, 218. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Y.; Wu, J.; Yang, J.; Li, M.; Chen, Q. The Potential Value of Targeting Ferroptosis in Early Brain Injury After Acute CNS Disease. Front. Mol. Neurosci. 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Zille, M.; Kumar, A.; Kundu, N.; Bourassa, M.W.; Wong, V.S.C.; Willis, D.; Karuppagounder, S.S.; Ratan, R.R. Ferroptosis in Neurons and Cancer Cells Is Similar But Differentially Regulated by Histone Deacetylase Inhibitors. eNeuro 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, H.; Yang, X.; Wu, Q.; An, P.; Jin, X.; Liu, W.; Huang, X.; Li, Y.; Yan, S.; et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct. Target Ther. 2020, 5, 138. [Google Scholar] [CrossRef]
- Chepikova, O.E.; Malin, D.; Strekalova, E.; Lukasheva, E.V.; Zamyatnin, A.A., Jr.; Cryns, V.L. Lysine oxidase exposes a dependency on the thioredoxin antioxidant pathway in triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020, 183, 549–564. [Google Scholar] [CrossRef]
- Cai, L.L.; Ruberto, R.A.; Ryan, M.J.; Eaton, J.K.; Schreiber, S.L.; Viswanathan, V.S. Modulation of ferroptosis sensitivity by TXNRD1 in pancreatic cancer cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.L.; Nydes, M.; Shanley, K.L.; Morales Pantoja, I.E.; Howard, T.A.; Bizzozero, O.A. Reduced expression of the ferroptosis inhibitor glutathione peroxidase-4 in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurochem. 2019, 148, 426–439. [Google Scholar] [CrossRef]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef] [Green Version]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Proneth, B. Selenium: Tracing Another Essential Element of Ferroptotic Cell Death. Cell Chem. Biol. 2020, 27, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; You, J.H.; Shin, D.; Roh, J.L. Inhibition of Glutaredoxin 5 predisposes Cisplatin-resistant Head and Neck Cancer Cells to Ferroptosis. Theranostics 2020, 10, 7775–7786. [Google Scholar] [CrossRef] [PubMed]
- Bjornstedt, M.; Hamberg, M.; Kumar, S.; Xue, J.; Holmgren, A. Human thioredoxin reductase directly reduces lipid hydroperoxides by NADPH and selenocystine strongly stimulates the reaction via catalytically generated selenols. J. Biol. Chem. 1995, 270, 11761–11764. [Google Scholar] [CrossRef] [Green Version]
- May, J.M.; Morrow, J.D.; Burk, R.F. Thioredoxin reductase reduces lipid hydroperoxides and spares alpha-tocopherol. Biochem. Biophys. Res. Commun. 2002, 292, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Lu, C.; Schoenfeld, R.; Shan, Y.; Tsai, H.J.; Hammock, B.; Cortopassi, G. Frataxin deficiency induces Schwann cell inflammation and death. Biochim. Biophys. Acta 2009, 1792, 1052–1061. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.M.; Santos, R. Neurodegeneration in Friedreich’s ataxia: From defective frataxin to oxidative stress. Oxidative Med. Cell. Longev. 2013, 2013, 487534. [Google Scholar] [CrossRef]
- Rodríguez, L.R.; Lapena, T.; Calap-Quintana, P.; Molto, M.D.; Gonzalez-Cabo, P.; Navarro Langa, J.A. Antioxidant Therapies and Oxidative Stress in Friedreich s Ataxia: The Right Path or Just a Diversion? Antioxidants 2020, 9, 664. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Bertini, E.S.; Piemonte, F. The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia. Int. J. Mol. Sci. 2020, 21, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L478–L488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaga, S.; Zhan, L.; Matsumoto, M.; Maulik, N. Resveratrol enhances neovascularization in the infarcted rat myocardium through the induction of thioredoxin-1, heme oxygenase-1 and vascular endothelial growth factor. J. Mol. Cell. Cardiol. 2005, 39, 813–822. [Google Scholar] [CrossRef]
- Thirunavukkarasu, M.; Penumathsa, S.V.; Koneru, S.; Juhasz, B.; Zhan, L.; Otani, H.; Bagchi, D.; Das, D.K.; Maulik, N. Resveratrol alleviates cardiac dysfunction in streptozotocin-induced diabetes: Role of nitric oxide, thioredoxin, and heme oxygenase. Free Radic. Biol. Med. 2007, 43, 720–729. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Voullaire, L.; Sandi, C.; Pook, M.A.; Ioannou, P.A.; Delatycki, M.B.; Sarsero, J.P. Pharmacological screening using an FXN-EGFP cellular genomic reporter assay for the therapy of Friedreich ataxia. PLoS ONE 2013, 8, e55940. [Google Scholar] [CrossRef] [Green Version]
- Yiu, E.M.; Tai, G.; Peverill, R.E.; Lee, K.J.; Croft, K.D.; Mori, T.A.; Scheiber-Mojdehkar, B.; Sturm, B.; Praschberger, M.; Vogel, A.P.; et al. An open-label trial in Friedreich ataxia suggests clinical benefit with high-dose resveratrol, without effect on frataxin levels. J. Neurol. 2015, 262, 1344–1353. [Google Scholar] [CrossRef]
- Georges, P.; Boza-Moran, M.G.; Gide, J.; Peche, G.A.; Foret, B.; Bayot, A.; Rustin, P.; Peschanski, M.; Martinat, C.; Aubry, L. Induced pluripotent stem cells-derived neurons from patients with Friedreich ataxia exhibit differential sensitivity to resveratrol and nicotinamide. Sci. Rep. 2019, 9, 14568. [Google Scholar] [CrossRef] [Green Version]
- Nivet-Antoine, V.; Cottart, C.H.; Lemarechal, H.; Vamy, M.; Margaill, I.; Beaudeux, J.L.; Bonnefont-Rousselot, D.; Borderie, D. trans-Resveratrol downregulates Txnip overexpression occurring during liver ischemia-reperfusion. Biochimie 2010, 92, 1766–1771. [Google Scholar] [CrossRef]
- Bedarida, T.; Baron, S.; Vibert, F.; Ayer, A.; Henrion, D.; Thioulouse, E.; Marchiol, C.; Beaudeux, J.L.; Cottart, C.H.; Nivet-Antoine, V. Resveratrol Decreases TXNIP mRNA and Protein Nuclear Expressions With an Arterial Function Improvement in Old Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 720–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and Other Nutrigenomic Nrf2 Activators: Can the Clinician’s Expectation Be Matched by the Reality? Oxidarive Med. Cell. Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef] [Green Version]
- Tanito, M.; Masutani, H.; Kim, Y.C.; Nishikawa, M.; Ohira, A.; Yodoi, J. Sulforaphane induces thioredoxin through the antioxidant-responsive element and attenuates retinal light damage in mice. Investig. Ophthalmol. Vis. Sci. 2005, 46, 979–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernandez-Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hintze, K.J.; Wald, K.A.; Zeng, H.; Jeffery, E.H.; Finley, J.W. Thioredoxin reductase in human hepatoma cells is transcriptionally regulated by sulforaphane and other electrophiles via an antioxidant response element. J. Nutr. 2003, 133, 2721–2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, S.; Huang, M.L.H.; Richardson, D.R. Treatment of dilated cardiomyopathy in a mouse model of Friedreich’s ataxia using N-acetylcysteine and identification of alterations in microRNA expression that could be involved in its pathogenesis. Pharmacol. Res. 2020, 159, 104994. [Google Scholar] [CrossRef]
- Petrillo, S.; Piermarini, E.; Pastore, A.; Vasco, G.; Schirinzi, T.; Carrozzo, R.; Bertini, E.; Piemonte, F. Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. Int. J. Mol. Sci. 2017, 18, 2173. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich’s Ataxia Neural Stem Cells. Front. Cell. Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; D’Amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the Treatment of Friedreich’s Ataxia: A Comparison among Drugs. Int. J. Mol. Sci. 2019, 20, 5211. [Google Scholar] [CrossRef] [Green Version]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef]
- Lynch, D.R.; Chin, M.P.; Delatycki, M.B.; Subramony, S.H.; Corti, M.; Hoyle, J.C.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Mathews, K.D.; et al. Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, Z.; Ashrafizadeh, M. Melatonin as a potential modulator of Nrf2. Fundam. Clin. Pharmacol. 2020, 34, 11–19. [Google Scholar] [CrossRef]
- Mahalanobish, S.; Dutta, S.; Saha, S.; Sil, P.C. Melatonin induced suppression of ER stress and mitochondrial dysfunction inhibited NLRP3 inflammasome activation in COPD mice. Food Chem. Toxicol. 2020, 144, 111588. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, T.; Lee, T.H. Cellular Mechanisms of Melatonin: Insight from Neurodegenerative Diseases. Biomolecules 2020, 10, 1158. [Google Scholar] [CrossRef] [PubMed]
- Nune, S.; Donald, R.; Shakkottai, V.; Hassan, F. 1242 A Case of Friedreich’s Ataxia and REM Sleep Behavior Disorder in a Teenager causing Suboptimal Non-Invasive Positive Pressure (NIPPV) Compliance. Sleep 2017, 40, A462. [Google Scholar] [CrossRef] [Green Version]
- Tai, G.; Corben, L.A.; Yiu, E.M.; Milne, S.C.; Delatycki, M.B. Progress in the treatment of Friedreich ataxia. Neurol. Neurochir. Pol. 2018, 52, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Strawser, C.; Schadt, K.; Hauser, L.; McCormick, A.; Wells, M.; Larkindale, J.; Lin, H.; Lynch, D.R. Pharmacological therapeutics in Friedreich ataxia: The present state. Expert Rev. Neurother. 2017, 17, 895–907. [Google Scholar] [CrossRef]
- Myers, L.; Farmer, J.M.; Wilson, R.B.; Friedman, L.; Tsou, A.; Perlman, S.L.; Subramony, S.H.; Gomez, C.M.; Ashizawa, T.; Wilmot, G.R.; et al. Antioxidant use in Friedreich ataxia. J. Neurol. Sci. 2008, 267, 174–176. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Pre-Clinical Studies in FRDA | Clinical Trials in FRDA | ||||
|---|---|---|---|---|---|---|
| Model | Doses/Treatment | Ref. | Nº Subjects | Doses/Treatment | Ref. | |
| Resveratrol | YG8R mouse | 200 mg/kg daily for 3 days. Subcutaneous injection. | [170] | 27 FRDA patients: 13 low dosis and 14 high dosis | 0.5 g or 2.5 g twice daily for 12 weeks. Capsules. | [171] |
| Human fibroblast MSCiPSC-derived neurons | 25 µM to 125 µM once 25 µM to 125 µM once 10 µM to 50 µM once | [172] | 40 patients (estimated) | 2 g daily for 24 weeks. Capsules. | ClinicalTrials.gov Id: NCT03933163 | |
| Sulforaphane (SFN) | Mouse NSC34 motor neurons Human fibroblasts | 5 µM for 24 h 10 µM for 24 h | [180] | |||
| Neural stem cells KIKO mouse | 5 µM for 2, 6, and 24 h | [181] | ||||
| Human fibroblast | 10 µM for 2, 6, and 24 h | [182] | ||||
| Omaveloxolone | Cerebellar Granule Neurons KIKO and YG8R mice Human Fibroblast | 50 nM for 24 h 50 nM for 24 h | [183] | 103 patients | 150 mg daily for 48 weeks. Capsules. | [184] |
| Melatonin | Case report: 1 FRDA patient | 5 mg and 10 mg | [188] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seco-Cervera, M.; González-Cabo, P.; Pallardó, F.V.; Romá-Mateo, C.; García-Giménez, J.L. Thioredoxin and Glutaredoxin Systems as Potential Targets for the Development of New Treatments in Friedreich’s Ataxia. Antioxidants 2020, 9, 1257. https://doi.org/10.3390/antiox9121257
Seco-Cervera M, González-Cabo P, Pallardó FV, Romá-Mateo C, García-Giménez JL. Thioredoxin and Glutaredoxin Systems as Potential Targets for the Development of New Treatments in Friedreich’s Ataxia. Antioxidants. 2020; 9(12):1257. https://doi.org/10.3390/antiox9121257
Chicago/Turabian StyleSeco-Cervera, Marta, Pilar González-Cabo, Federico V. Pallardó, Carlos Romá-Mateo, and José Luis García-Giménez. 2020. "Thioredoxin and Glutaredoxin Systems as Potential Targets for the Development of New Treatments in Friedreich’s Ataxia" Antioxidants 9, no. 12: 1257. https://doi.org/10.3390/antiox9121257
APA StyleSeco-Cervera, M., González-Cabo, P., Pallardó, F. V., Romá-Mateo, C., & García-Giménez, J. L. (2020). Thioredoxin and Glutaredoxin Systems as Potential Targets for the Development of New Treatments in Friedreich’s Ataxia. Antioxidants, 9(12), 1257. https://doi.org/10.3390/antiox9121257