Esculetin as a Bifunctional Antioxidant Prevents and Counteracts the Oxidative Stress and Neuronal Death Induced by Amyloid Protein in SH-SY5Y Cells





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture and Preparation of Coumarin Solutions
2.3. Determination of Neuronal Viability
2.4. Determination of Intrinsic Antioxidant Activity
2.5. Determination of Direct and Indirect Antioxidant Activity
2.6. Determination of Antioxidant Coumarins in Membrane and Cytosolic Fractions
2.7. Determination of GSH Levels
2.8. Nuclear Extraction and Determination of Nrf2 Nuclear Levels
2.9. Determination of Erk, Akt and GSK3β Protein Phosphorylation
2.10. Aβ1–42 oligomer Preparation
2.11. MTT Formazan Exocytosis Assay
2.12. Determination of ROS Formation Induced by Aβ1–42 Oligomers
2.13. Determination of Neuronal Death Induced by Aβ1–42 Oligomers
2.14. Statistical Analysis
3. Results and Discussion
3.1. Direct and Indirect Antioxidant Activity of Coumarins
3.2. Effects of ESC and DAPH on Neuronal Antioxidant Response
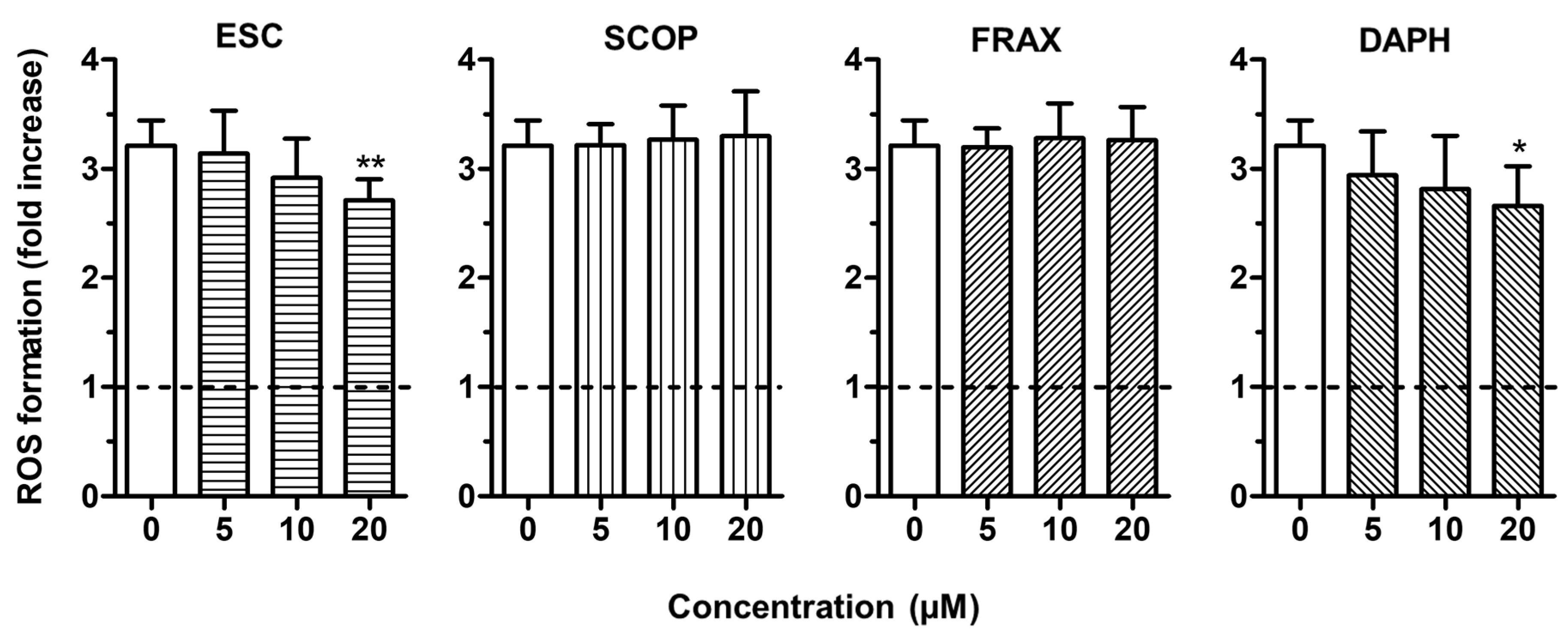
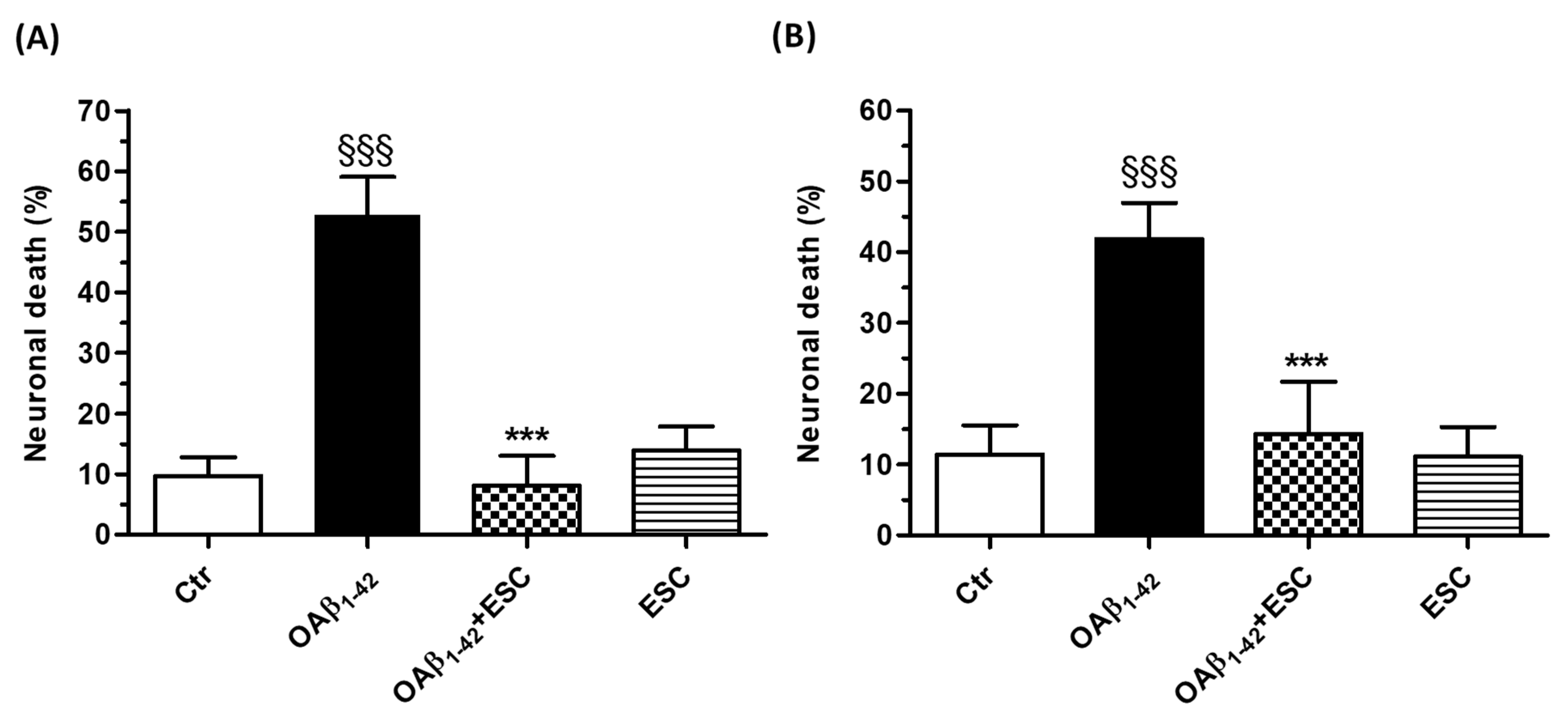
3.3. Neuroprotective Effects of ESC Against Aβ1–42 Oligomer-Induced Neuronal Death
3.4. Effects of ESC on Survival Kinase Pathways
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meraz-Ríos, M.A.; Franco-Bocanegra, D.; Rios, D.T.; Campos-Peña, V. Early onset Alzheimer’s disease and oxidative stress. Oxid. Med. Cell Longev. 2014, 2014, 375968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervellati, C.; Wood, P.L.; Romani, A.; Valacchi, G.; Squerzanti, M.; Sanz, J.M.; Ortolani, B.; Zuliani, G. Oxidative challenge in Alzheimer’s disease: State of knowledge and future needs. J. Investig. Med. 2016, 64, 21–32. [Google Scholar] [CrossRef]
- Sutherland, G.T.; Chami, B.; Youssef, P.; Witting, P.K. Oxidative stress in Alzheimer’s disease: Primary villain or physiological by-product? Redox Rep. 2013, 18, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Wojsiat, J.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. Oxidant/antioxidant imbalance in Alzheimer’s disease: Therapeutic and diagnostic prospects. Oxid. Med. Cell Longev. 2018, 2018, 6435861. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain glutathione levels—A novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef]
- Chiang, G.C.; Mao, X.; Kang, G.; Chang, E.; Pandya, S.; Vallabhajosula, S.; Isaacson, R.; Ravdin, L.D. Alzheimer’s Disease Neuroimaging Initiative; Shungu, D.C. Relationships among cortical glutathione levels, brain amyloidosis, and memory in healthy older adults investigated in vivo with (1)H-MRS and pittsburgh Compound-B PET. AJNR Am. J. Neuroradiol. 2017, 38, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Prasad, K.N. Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer’s disease. Mech. Ageing Dev. 2016, 153, 41–47. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, S.; Yan, J.; Li, Y.; Xin, Z.; Lin, Y.; Qu, Y. An overview of the molecular mechanisms and novel roles of Nrf2 in neurodegenerative disorders. Cytokine Growth Factor Rev. 2015, 26, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnside, S.W.; Hardingham, G.E. Transcriptional regulators of redox balance and other homeostatic processes with the potential to alter neurodegenerative disease trajectory. Biochem. Soc. Trans. 2017, 45, 1295–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Talalay, P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol. Nutr. Food Res. 2008, 52, S128–S138. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Rashmi, V.; Odhav, B. Review on natural coumarin lead compounds for their pharmacological activity. Biomed. Res. Int. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Thuong, P.T.; Hung, T.M.; Ngoc, T.M.; Ha, D.T.; Min, B.S.; Kwack, S.J.; Kang, T.S.; Choi, J.S.; Bae, K. Antioxidant activities of coumarins from Korean medicinal plants and their structure-activity relationships. Phyther. Res. 2010, 24, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kadakol, A.; Sharma, N.; Kulkarni, Y.A.; Gaikwad, A.B. Esculetin: A phytochemical endeavor fortifying effect against non-communicable diseases. Biomed. Pharmacother. 2016, 84, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Nang, C.; Luo, F.; Pan, H.; Zhang, K.; Liu, J.; Zhou, R.; Gao, J.; Chang, X.; He, H.; et al. Esculetin attenuates lipopolysaccharide (LPS)-induced neuroinflammatory processes and depressive-like behavior in mice. Physiol. Behav. 2016, 163, 184–192. [Google Scholar] [CrossRef]
- Sulakhiya, K.; Keshavlal, G.P.; Bezbaruah, B.B.; Dwivedi, S.; Gurjar, S.S.; Munde, N.; Jangra, A.; Lahkar, M.; Gogoi, R. Lipopolysaccharide induced anxiety- and depressive-like behaviour in mice are prevented by chronic pre-treatment of esculetin. Neurosci. Lett. 2016, 611, 106–111. [Google Scholar] [CrossRef]
- Martín-Aragón, S.; Villar, Á.; Benedí, J. Age-dependent effects of esculetin on mood-related behavior and cognition from stressed mice are associated with restoring brain antioxidant status. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 65, 1–16. [Google Scholar]
- Wang, C.; Pei, A.; Chen, J.; Yu, H.; Sun, M.L.; Liu, C.F.; Xu, X. A natural coumarin derivative esculetin offers neuroprotection on cerebral ischemia/reperfusion injury in mice. J. Neurochem. 2012, 121, 1007–1013. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Ellis, E.M. Neuroprotective effects of umbelliferone and esculetin in a mouse model of Parkinson’s disease. J. Neurosci. Res. 2013, 91, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Rampa, A.; Bartolini, M.; Pruccoli, L.; Naldi, M.; Iriepa, I.; Moraleda, I.; Belluti, F.; Gobbi, S.; Tarozzi, A.; Bisi, A. Exploiting the chalcone scaffold to develop multifunctional agents for Alzheimer’s disease. Molecules 2018, 23, 1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampa, A.; Tarozzi, A.; Mancini, F.; Pruccoli, L.; Di Martino, R.M.C.; Gobbi, S.; Bisi, A.; De Simone, A.; Palomba, F.; Zaccheroni, N.; et al. Naturally inspired molecules as multifunctional agents for Alzheimer’s disease treatment. Molecules 2016, 21, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampa, A.; Montanari, S.; Pruccoli, L.; Bartolini, M.; Falchi, F.; Feoli, A.; Cavalli, A.; Belluti, F.; Gobbi, S.; Tarozzi, A.; et al. Chalcone-based carbamates for Alzheimer’s disease treatment. Future Med. Chem. 2017, 9, 533–536. [Google Scholar] [CrossRef]
- Montanari, S.; Bartolini, M.; Neviani, P.; Belluti, F.; Gobbi, S.; Pruccoli, L.; Tarozzi, A.; Falchi, F.; Andrisano, V.; Miszta, P.; et al. Multitarget strategy to address Alzheimer’s disease: Design, synthesis, biological evaluation, and computational studies of coumarin-based derivatives. ChemMedChem 2016, 11, 1296–1308. [Google Scholar] [CrossRef]
- Tarozzi, A.; Morroni, F.; Hrelia, S.; Angeloni, C.; Marchesi, A.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effects of anthocyanins and their in vivo metabolites in SH-SY5Y cells. Neurosci. Lett. 2007, 424, 36–40. [Google Scholar] [CrossRef]
- Sestito, S.; Pruccoli, L.; Runfola, M.; Citi, V.; Martelli, A.; Saccomanni, G.; Calderone, V.; Tarozzi, A.; Rapposelli, S. Design and synthesis of H(2)S-donor hybrids: A new treatment for Alzheimer’s disease? Eur. J. Med. Chem. 2019, 184, 111745. [Google Scholar] [CrossRef]
- Morroni, F.; Sita, G.; Djemil, A.; D’Amico, M.; Pruccoli, L.; Cantelli-forti, G.; Hrelia, P.; Tarozzi, A. Comparison of adaptive neuroprotective mechanisms of sulforaphane and its interconversion product erucin in in vitro and in vivo models of Parkinson’s disease. J. Agric. Food Chem. 2018, 66, 856–865. [Google Scholar] [CrossRef]
- Tarozzi, A.; Bartolini, M.; Piazzi, L.; Valgimigli, L.; Amorati, R.; Bolondi, C.; Djemil, A.; Mancini, F.; Andrisano, V.; Rampa, A. From the dual function lead AP2238 to AP2469, a multi-target-directed ligand for the treatment of Alzheimer’s disease. Pharmacol. Res. Perspect. 2014, 2, e00023. [Google Scholar] [CrossRef]
- Kreutzmann, P.; Wolf, G.; Kupsch, K. Minocycline recovers MTT-formazan exocytosis impaired by amyloid beta peptide. Cell. Mol. Neurobiol. 2010, 30, 979–984. [Google Scholar] [CrossRef]
- Kaneko, T.; Baba, N.; Matsuo, M. Protection of coumarins against linoleic acid hydroperoxide induced cytotoxicity. Chem. Biol. Interact. 2003, 142, 239–254. [Google Scholar] [CrossRef]
- Wu, C.R.; Huang, M.Y.; Lin, Y.T.; Ju, H.Y.; Ching, H. Antioxidant properties of Cortex Fraxini and its simple coumarins. Food Chem. 2007, 104, 1464–1471. [Google Scholar] [CrossRef]
- Veselinović, J.B.; Veselinović, A.M.; Vitnik, Ž.J.; Vitnik, V.D.; Nikolić, G.M. Antioxidant properties of selected 4-phenyl hydroxycoumarins: Integrated in vitro and computational studies. Chem. Biol. Interact. 2014, 214, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Bano, S.; Parihar, M.S. Reduction of lipid peroxidation in different brain regions by a combination of alpha-tocopherol and ascorbic acid. J. Neural Transm. 1997, 104, 1277–1286. [Google Scholar] [CrossRef]
- Seong, S.H.; Ali, M.Y.; Jung, H.A.; Choi, J.S. Umbelliferone derivatives exert neuroprotective effects by inhibiting monoamine oxidase A, self-amyloidβ aggregation, and lipid peroxidation. Bioorg. Chem. 2019, 92, 103293. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Ellis, E.M. Esculetin-induced protection of human hepatoma HepG2 cells against hydrogen peroxide is associated with the Nrf2-dependent induction of the NAD(P)H: Quinone oxidoreductase 1 gene. Toxicol. Appl. Pharmacol. 2011, 250, 130–136. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, K.A.; Zhang, R.; Piao, M.J.; Ko, D.O.; Wang, Z.H.; Kang, S.S.; Lee, K.H.; Kang, H.K.; Kang, H.W.; et al. Protective effect of esculetin against oxidative stress-induced cell damage via scavenging reactive oxygen species. Acta Pharmacol. Sin. 2008, 29, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Zhu, L.; Chu, J.; Ma, Z.; Fu, Q.; Wei, W.; Deng, X.; Ma, S. Esculetin improves cognitive impairments induced by transient cerebral ischaemia and reperfusion in mice via regulation of mitochondrial fragmentation and mitophagy. Behav. Brain Res. 2019, 372, 112007. [Google Scholar] [CrossRef]
- Shinde, R.G.; Khan, A.A.; Kunwar, A.; Tripathi, V.S.; Barik, A. Fluorescence “off” and “on” signalling of esculetin in the presence of copper and thiol: A possible implication in cellular thiol sensing. Photochem. Photobiol. Sci. 2018, 17, 1197–1205. [Google Scholar] [CrossRef]
- Lee, C.R.; Shin, E.J.; Kim, H.C.; Choi, Y.S.; Shin, T.; Wie, M.B. Esculetin inhibits N-methyl-D-aspartate neurotoxicity via glutathione preservation in primary cortical cultures. Lab. Anim. Res. 2011, 27, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Martin-Aragón, S.; Benedi, J.M.; Villar, A.M. Effects of the antioxidant (6,7-dihydroxycoumarin) esculetin on the glutathione system and lipid peroxidation in mice. Gerontology 1998, 44, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Calabrese, V.; Giordano, J. The role of hormesis in the functional performance and protection of neural systems. Brain Circ. 2017, 3, 1–13. [Google Scholar] [PubMed]
- Han, M.H.; Park, C.; Lee, D.S.; Hong, S.H.; Choi, I.W.; Kim, G.Y.; Choi, S.H.; Shim, J.H.; Chae, J.I.; Yoo, Y.H.; et al. Cytoprotective effects of esculetin against oxidative stress are associated with the upregulation of Nrf2-mediated NQO1 expression via the activation of the ERK pathway. Int. J. Mol. Med. 2017, 39, 380–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarozzi, A.; Morroni, F.; Merlicco, A.; Bolondi, C.; Teti, G.; Falconi, M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effects of cyanidin 3-O-glucopyranoside on amyloid beta (25–35) oligomer-induced toxicity. Neurosci. Lett. 2010, 473, 72–76. [Google Scholar] [CrossRef]
- Liu, M.L.; Hong, S.T. Early phase of amyloid beta42-induced cytotoxicity in neuronal cells is associated with vacuole formation and enhancement of exocytosis. Exp. Mol. Med. 2005, 37, 559–566. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.S.; Maezawa, I.; Yao, N.; Xu, B.; Diaz-Avalos, R.; Rana, S.; Hua, D.H.; Cheng, R.H.; Lam, K.S.; Jin, L.W. Combining the rapid MTT formazan exocytosis assay and the MC65 protection assay led to the discovery of carbazole analogs as small molecule inhibitors of Aβ oligomer-induced cytotoxicity. Brain Res. 2007, 1130, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Nakano, M.; Imamura, H.; Sasaoka, N.; Yamamoto, M.; Uemura, N.; Shudo, T.; Fuchigami, T.; Takahashi, R.; Kakizuka, A. ATP maintenance via two types of ATP regulators mitigates pathological phenotypes in mouse models of Parkinson’s disease. EBioMedicine 2017, 22, 225–241. [Google Scholar] [CrossRef] [Green Version]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Kim, S.R.; An, H.J.; Kim, W.J.; Choe, M.; Han, J.A. Esculetin promotes type I procollagen expression in human dermal fibroblasts through MAPK and PI3K/Akt pathways. Mol. Cell. Biochem. 2012, 368, 61–67. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Dietary regulation of PI3K/AKT/GSK-3β pathway in Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Pitchumoni, S.S.; Doraiswamy, P.M. Current status of antioxidant therapy for Alzheimer’s Disease. J. Am. Geriatr. Soc. 1998, 46, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- LLoret, A.; Giraldo, E.; Viña, J. Is antioxidant therapy effective to treat alzheimer’s disease? Free Rad. Antiox. 2011, 1, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.Y.; Jannat, S.; Jung, H.A.; Choi, R.J.; Roy, A.; Choi, J.S. Anti-Alzheimer’s disease potential of coumarins from Angelica decursiva and Artemisia capillaris and structure-activity analysis. Asian Pac. J. Trop. Med. 2016, 9, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pruccoli, L.; Morroni, F.; Sita, G.; Hrelia, P.; Tarozzi, A. Esculetin as a Bifunctional Antioxidant Prevents and Counteracts the Oxidative Stress and Neuronal Death Induced by Amyloid Protein in SH-SY5Y Cells. Antioxidants 2020, 9, 551. https://doi.org/10.3390/antiox9060551
Pruccoli L, Morroni F, Sita G, Hrelia P, Tarozzi A. Esculetin as a Bifunctional Antioxidant Prevents and Counteracts the Oxidative Stress and Neuronal Death Induced by Amyloid Protein in SH-SY5Y Cells. Antioxidants. 2020; 9(6):551. https://doi.org/10.3390/antiox9060551
Chicago/Turabian StylePruccoli, Letizia, Fabiana Morroni, Giulia Sita, Patrizia Hrelia, and Andrea Tarozzi. 2020. "Esculetin as a Bifunctional Antioxidant Prevents and Counteracts the Oxidative Stress and Neuronal Death Induced by Amyloid Protein in SH-SY5Y Cells" Antioxidants 9, no. 6: 551. https://doi.org/10.3390/antiox9060551
APA StylePruccoli, L., Morroni, F., Sita, G., Hrelia, P., & Tarozzi, A. (2020). Esculetin as a Bifunctional Antioxidant Prevents and Counteracts the Oxidative Stress and Neuronal Death Induced by Amyloid Protein in SH-SY5Y Cells. Antioxidants, 9(6), 551. https://doi.org/10.3390/antiox9060551