The Impact of Sugar-Sweetened Beverage Consumption on the Liver: A Proteomics-Based Analysis


 and
and
Abstract
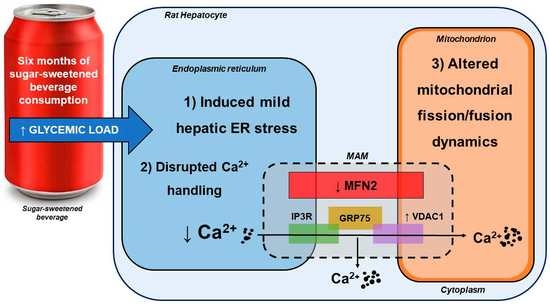
:
1. Introduction
2. Methods and Materials
2.1. Animals and Experimental Protocol
2.2. Tissue Collection
2.3. Proteomics
2.4. Isolation of the Endoplasmic Reticulum
2.5. Calcium Colorimetric Assay
2.6. Western Blotting
2.7. Oxidative Stress Analyses
2.7.1. Thiobarbituric Acid Reactive Substances (TBARS)
2.7.2. Glutathione Redox Status
2.7.3. Conjugated Dienes (CDs)
2.7.4. Oxygen Radical Absorbance Capacity (ORAC)
2.8. Statistical Analysis
3. Results
3.1. Proteomics Analysis of the Liver
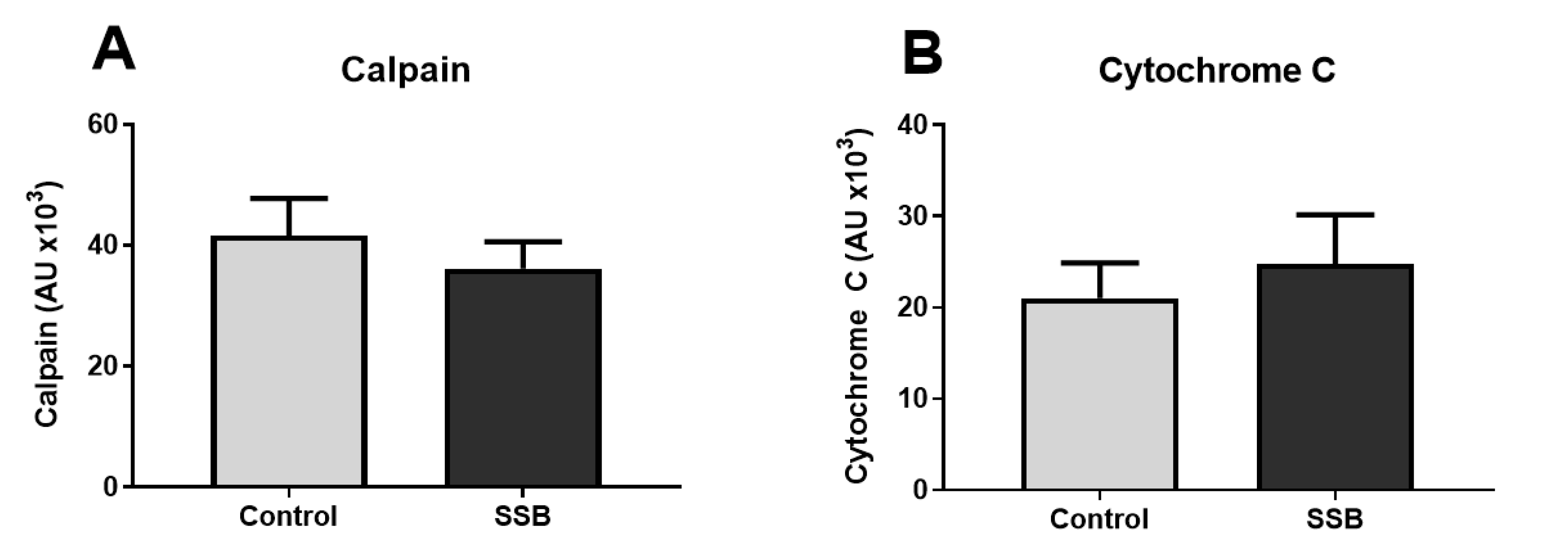
3.2. Markers of ER Stress
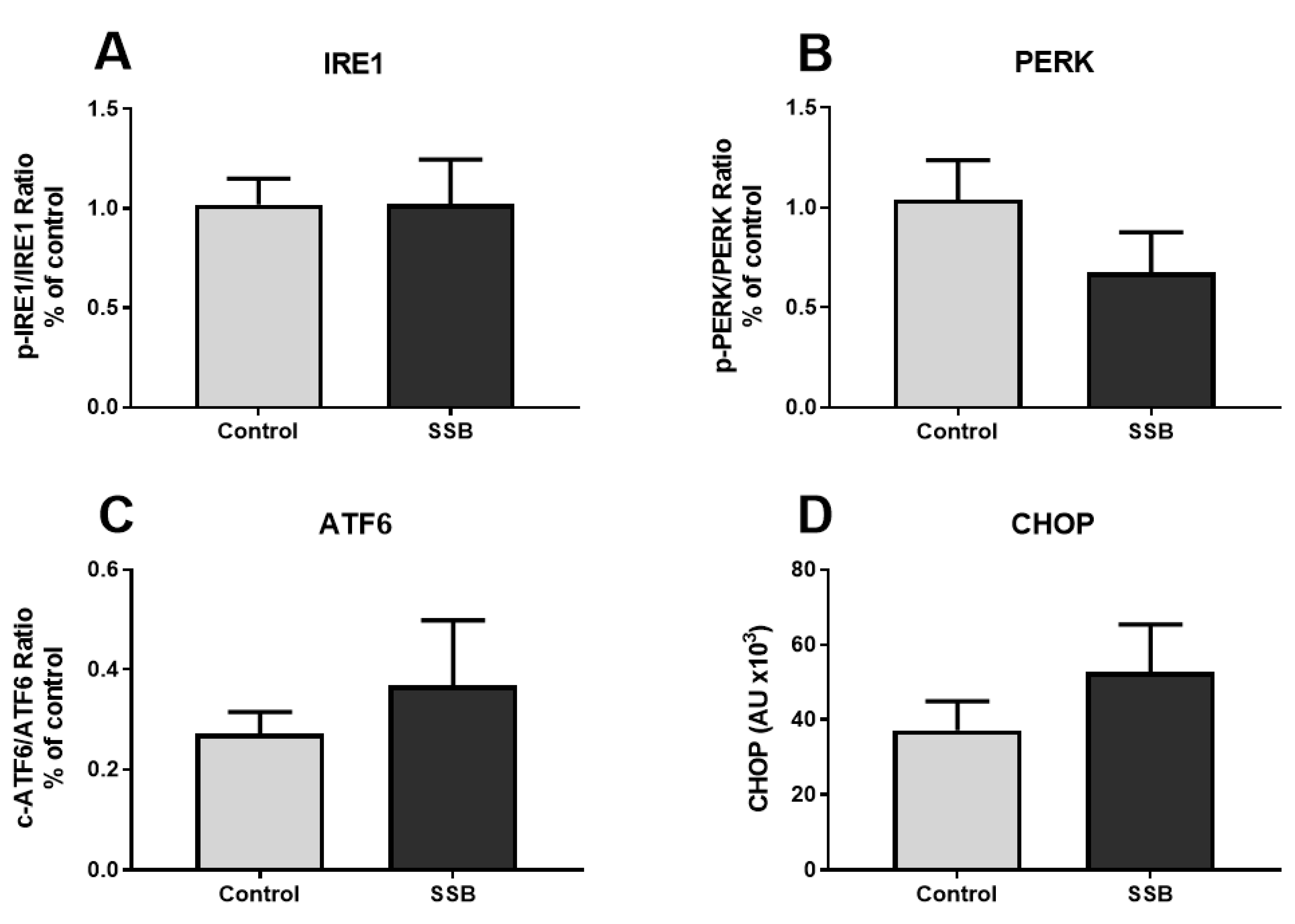
3.3. Markers of Mitochondrial Function and MAM Composition
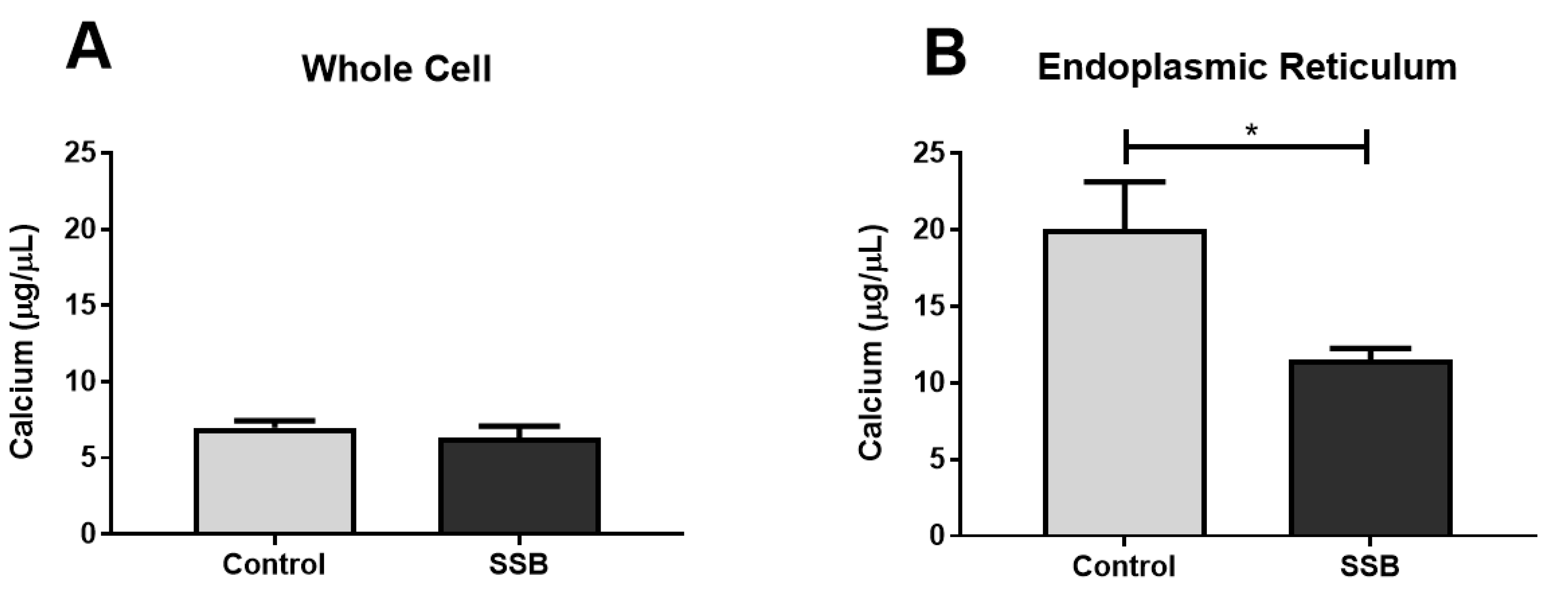
3.4. Calcium Assessments
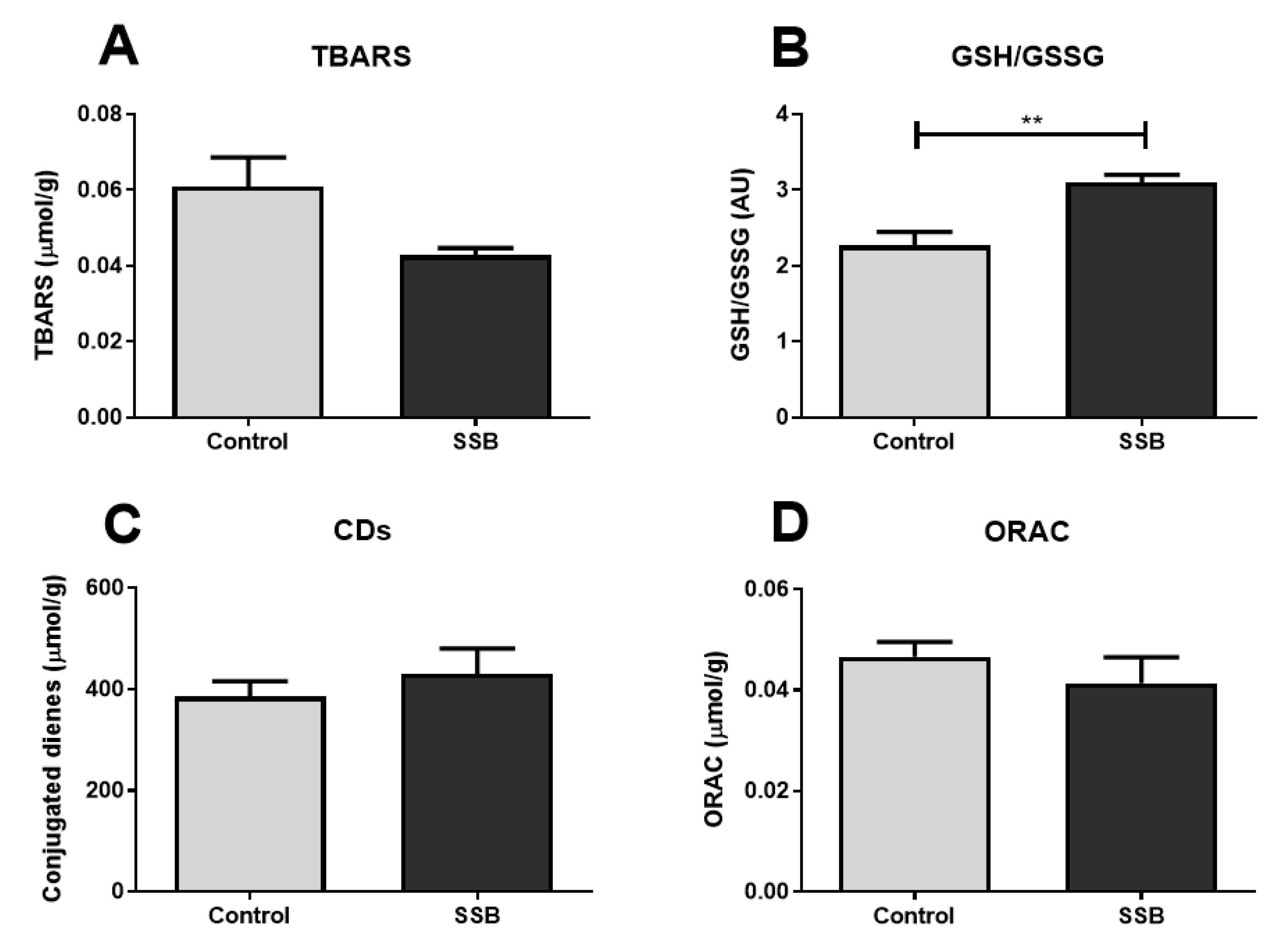
3.5. Oxidative Stress Analyses
4. Discussion
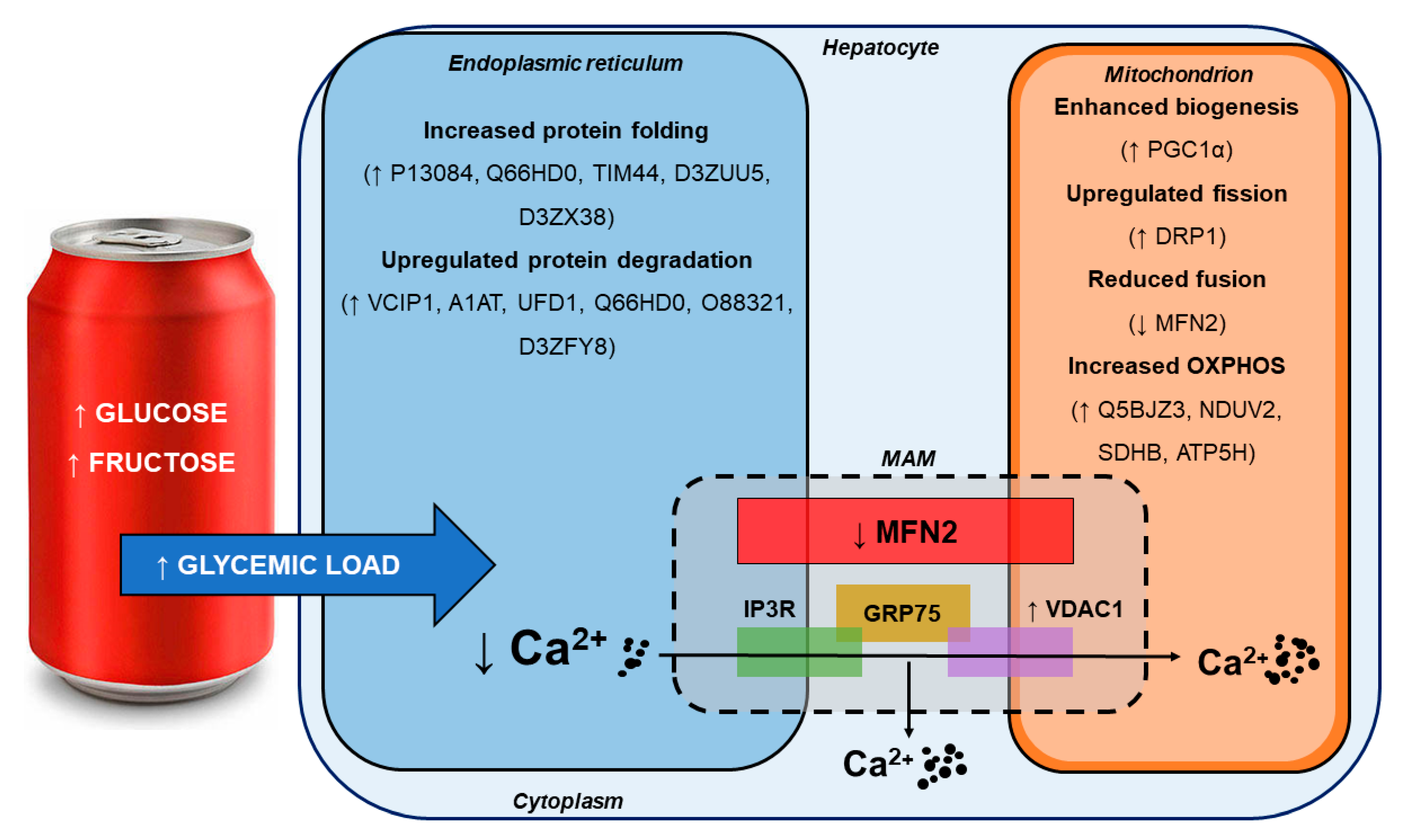
4.1. SSB Consumption Induced a Proteomic Phenotype Indicative of Mild Hepatic ER Stress
4.2. SSB Consumption Elicited Changes in Mitochondrial Dynamics
4.3. Moderate SSB Consumption Perturbed Calcium Handling Across MAMs
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Global Report on Diabetes; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Deshpande, G.; Mapanga, R.F.; Essop, M.F. Frequent sugar-sweetened beverage consumption and the onset of cardiometabolic diseases: Cause for concern? J. Endocr. Soc. 2017, 1, 1372–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekonnen, T.A.; Odden, M.C.; Coxson, P.G.; Guzman, D.; Lightwood, J.; Wang, Y.C.; Bibbins-Domingo, K. Health benefits of reducing sugar-sweetened beverage intake in high risk populations of California: Results from the cardiovascular disease (CVD) policy model. PLoS ONE 2013, 8, e81723. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Chu, A.Y.; Kang, J.H.; Jensen, M.K.; Curhan, G.C.; Pasquale, L.R.; Ridker, P.M.; Hunter, D.J.; Willett, W.C.; Rimm, E.B.; et al. Sugar-sweetened beverages and genetic risk of obesity. N. Engl. J. Med. 2012, 367, 1387–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornwell, T.B.; McAlister, A.R. Contingent choice. Exploring the relationship between sweetened beverages and vegetable consumption. Appetite 2013, 62, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, D.C.; Threapleton, D.E.; Evans, C.E.L.; Cleghorn, C.L.; Nykjaer, C.; Woodhead, C.; Burley, V.J. Association between sugar-sweetened and artificially sweetened soft drinks and type 2 diabetes: SSystematic review and dose-response meta-analysis of prospective studies. Br. J. Nutr. 2014, 112, 725–734. [Google Scholar] [CrossRef]
- Imamura, F.; O’Connor, L.; Ye, Z.; Mursu, J.; Hayashino, Y.; Bhupathiraju, S.N.; Forouhi, N.G. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: Systematic review, meta-analysis, and estimation of population attributable fraction. BMJ 2015, 351, h3576. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, B.; Lamarche, B.; Després, J.-P. Targeting Overconsumption of sugar-sweetened beverages vs. overall poor diet quality for cardiometabolic diseases risk prevention: Place your bets! Nutrients 2017, 9, 600. [Google Scholar] [CrossRef]
- Kim, O.-K.; Jun, W.; Lee, J. Mechanism of ER stress and inflammation for hepatic insulin resistance in obesity. Ann. Nutr. Metab. 2015, 67, 218–227. [Google Scholar] [CrossRef]
- Ye, R.; Ni, M.; Wang, M.; Luo, S.; Zhu, G.; Chow, R.H.; Lee, A.S. Inositol 1,4,5-trisphosphate receptor 1 mutation perturbs glucose homeostasis and enhances susceptibility to diet-induced diabetes. J. Endocrinol. 2011, 210, 209–217. [Google Scholar] [CrossRef]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Zeeshan, H.; Lee, G.H.; Kim, H.-R.; Chae, H.; Maher, H.; Zeeshan, A.; Lee, G.H.; Kim, H.-R.; Chae, H. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signaling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Samali, A.; FitzGerald, U.; Deegan, S.; Gupta, S. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int. J. Cell Biol. 2010, 2010, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The Role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Urano, F. Measuring ER Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 490, ISBN 9780123851147. [Google Scholar]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta—Mol. Cell Res. 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta—Bioenerg. 2014, 1837, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around mitochondria associated membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Medici, V.; Bremer, A.A.; Lee, V.; Lam, H.D.; Nunez, M.V.; Chen, G.X.; Keim, N.L.; Havel, P.J. A dose-response study of consuming high-fructose corn syrup–sweetened beverages on lipid/lipoprotein risk factors for cardiovascular disease in young adults. Am. J. Clin. Nutr. 2015, 101, 1144–1154. [Google Scholar] [CrossRef] [Green Version]
- Driescher, N.; Joseph, D.E.; Human, V.R.; Ojuka, E.; Cour, M.; Hadebe, N.; Bester, D.; Marnewick, J.L.; Lecour, S.; Lochner, A.; et al. The impact of sugar-sweetened beverage intake on rat cardiac function. Heliyon 2019, 5, e01357. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asensi, M.; Sastre, J.; Pallardo, F.V.; Lloret, A.; Lehner, M.; Garcia-de-la Asuncion, J.; Viña, J. Ratio of reduced to oxidized glutathione as indicator of oxidative stress status and DNA damage. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1999; Volume 299, pp. 267–276. [Google Scholar]
- Sebastián, D.; Hernández-Alvarez, M.I.; Segalés, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Lee, H.J.; Ho Jung, M.; Song, J. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: A molecular mechanism leading to hepatic insulin resistance. Cell. Signal. 2009, 21, 169–177. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef]
- Prasad, M.; Walker, A.N.; Kaur, J.; Thomas, J.L.; Powell, S.A.; Pandey, A.V.; Whittal, R.M.; Burak, W.E.; Petruzzelli, G.; Bose, H.S. Endoplasmic Reticulum Stress Enhances Mitochondrial Metabolic Activity in Mammalian Adrenals and Gonads. Mol. Cell. Biol. 2016, 36, 3058–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Xiang, W.; Terry, L.; Kretzschmar, H.A.; Windl, O. Transcriptional analysis implicates endoplasmic reticulum stress in bovine spongiform encephalopathy. PLoS ONE 2010, 5, e14207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Perdomo, G.; Kim, D.H.; Qu, S.; Ringquist, S.; Trucco, M.; Dong, H.H. Proteomic analysis of fructose-induced fatty liver in hamsters. Metabolism 2008, 57, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Jegatheesan, P.; Bandt, J. De Fructose and NAFLD: The multifaceted aspects of fructose metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanhope, K.L. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit. Rev. Clin. Lab. Sci. 2016, 53, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Morand, J.P.F.; Macri, J.; Adeli, K. Proteomic profiling of hepatic endoplasmic reticulum-associated proteins in an animal model of insulin resistance and metabolic dyslipidemia. J. Biol. Chem. 2005, 280, 17626–17633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.J.; Nie, S.; Dai, J.; Wu, J.R.; Zeng, R. Proteome, phosphoproteome, and hydroxyproteome of liver mitochondria in diabetic rats at early pathogenic stages. Mol. Cell. Proteom. 2010, 9, 100–116. [Google Scholar] [CrossRef] [Green Version]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Balakumar, M.; Raji, L.; Prabhu, D.; Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. High-fructose diet is as detrimental as high-fat diet in the induction of insulin resistance and diabetes mediated by hepatic/pancreatic endoplasmic reticulum (ER) stress. Mol. Cell. Biochem. 2016, 423, 93–104. [Google Scholar] [CrossRef]
- del Campo, A.; Parra, V.; Vásquez-Trincado, C.; Gutiérrez, T.; Morales, P.E.; López-Crisosto, C.; Bravo-Sagua, R.; Navarro-Marquez, M.F.; Verdejo, H.E.; Contreras-Ferrat, A.; et al. Mitochondrial fragmentation impairs insulin-dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. Am. J. Physiol.—Endocrinol. Metab. 2014, 306, E1–E13. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wei, Y.; Sowers, J.R. Role of Mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Salazar Hernández, M.A.; Auen, T.; Mucka, P.; Lee, J.; Ozcan, U. PGC-1α functions as a co-suppressor of XBP1s to regulate glucose metabolism. Mol. Metab. 2018, 7, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, K.; Lin, H.; Tavares, C.D.J.; Dominy, J.E.; Camporez, J.P.; Perry, R.J.; Schilling, R.; Rines, A.K.; Lee, J.; Hickey, M.; et al. Selective chemical inhibition of PGC-1α gluconeogenic activity ameliorates Type 2 diabetes. Cell 2017, 169, 148–160.e15. [Google Scholar] [CrossRef] [Green Version]
- Sebastián, D.; Zorzano, A. Mitochondrial dynamics and metabolic homeostasis. Curr. Opin. Physiol. 2018, 3, 34–40. [Google Scholar] [CrossRef]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial dynamics in diabetes. Antioxid. Redox Signal. 2011, 14, 439–457. [Google Scholar] [CrossRef]
- Schneeberger, M.; Dietrich, M.O.; Sebastián, D.; Imbernón, M.; Castaño, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodríguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 2013, 155, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [Green Version]
- Gan, K.X.; Wang, C.; Chen, J.H.; Zhu, C.J.; Song, G.Y. Mitofusin-2 ameliorates high-fat diet-induced insulin resistance in liver of rats. World J. Gastroenterol. 2013, 19, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, P.; Sieck, G.C. Endoplasmic reticulum stress and mitochondrial function in airway smooth muscle. Front. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, J.; Chen, X.; Delmotte, P.; Sieck, G.C. TNFα selectively activates the IRE1α/XBP1 endoplasmic reticulum stress pathway in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L483–L493. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Santella, L.; Branca, D.; Brini, M. Generation, control, and processing of cellular calcium signals. Crit. Rev. Biochem. Mol. Biol. 2001, 36, 107–260. [Google Scholar] [CrossRef] [PubMed]
- Gincel, D.; Zaid, H.; Shoshan-Barmatz, V. Calcium binding and translocation by the voltage-dependent anion channel: A possible regulatory mechanism in mitochondrial function. Biochem. J. 2001, 358, 147–155. [Google Scholar] [CrossRef]
- Giorgi, C.; De Stefani, D.; Bononi, A.; Rizzuto, R.; Pinton, P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 2009, 41, 1817–1827. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Golan, M. Mitochondrial VDAC1: Function in cell life and death and a target for cancer therapy. Curr. Med. Chem. 2012, 19, 714–735. [Google Scholar] [CrossRef]
- Boehning, D.; Patterson, R.L.; Sedaghat, L.; Glebova, N.O.; Kurosaki, T.; Snyder, S.H. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 2003, 5, 1051–1061. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (Accession Number) | Function; Location | SSB vs. Control |
|---|---|---|
| (A) Signs of ER stress (elevated protein folding, protein degradation and oxidative phosphorylation) | ||
| Cluster of DnaJ (Hsp40) homolog subfamily B member 1 (predicted) isoform CRA_a (D3ZUU5) | Chaperone cofactor-dependent protein folding; cytosol, extracellular vesicular exosome and nucleus (ScaffoldQ+). | Inf (absent in Control) |
| Prefoldin 1 (D3ZX38) | Binds specifically to cytosolic chaperonin to promote protein folding; ER (Prefoldin complex) (ScaffoldQ+). | 280% ↑ |
| Cluster of mitochondrial import inner membrane translocase subunit TIM44 (TIM44) | Involved in importing proteins from the mitochondrial inner membrane to the mitochondrial matrix (ATP-dependent), chaperone binding; mitochondrial inner membrane and matrix (ScaffoldQ+; UniProtKB). | 160% ↑ |
| Cluster of nucleophosmin (sp|P13084|NPM) | Involved in various cellular processes including protein chaperoning; cytoplasm and nucleus (UniProtKB). | 80% ↑ |
| Endoplasmin (sp|Q66HD0|ENPL) | Involved in protein chaperoning and ER-associated degradation; ER lumen (UniProtKB). | 40% ↑ |
| Ubiquitin fusion degradation protein 1 homolog (UFD1) | Partakes in the degradation ER-associated degradation and ubiquitin fusion degradation of misfolded proteins and the activation of certain transcription factors; cytosol and nucleus (UniProtKB). | 140% ↑ |
| Cluster of Protein LOC100912618 (tr|D3ZFY8|D3ZFY8) | Catalyzes the attachment of ubiquitin protein to proteins. Also involved in various steps of DNA replication; cytoplasm and nucleus (ScaffoldQ+). | 50% ↑ |
| Deubiquitinating protein VCIP135 (VCIP1) | Deubiquitination of proteins to prevent protein degradation; ER and golgi stacks (UniProtKB). | 95% ↓ |
| Alpha-1-antiproteinase (A1AT) | Associated with the acute-phase response and inhibits proteases activity; secreted into intracellular space (ScaffoldQ+; UniProtKB). | 30% ↓ |
| Nicotinamide nucleotide transhydrogenase (Q5BJZ3) | NADPH regeneration and proton transport; mitochondrial inner membrane (ScaffoldQ+). | 390% ↑ |
| Cluster of NADH dehydrogenase [ubiquinone] flavoprotein 2 mitochondrial (sp|P19234|NDUV2) | Mitochondrial electron transport, NADH to ubiquinone; mitochondrial ETC complex I (ScaffoldQ+). | 30% ↑ |
| Succinate dehydrogenase [ubiquinone] iron-sulfur subunit (SDHB) | Subunit of Complex II → transfer electrons from succinate to ubiquinone; mitochondrial inner membrane (UniProtKB). | 20% ↑ |
| ATP synthase subunit d (sp|P31399|ATP5H) | Maintenance of ATP synthase structure during ATP synthesis; mitochondrial inner membrane (UniProtKB). | 20% ↑ |
| (B) Possible mechanisms of ER stress (altered calcium handling, redox balance and mitochondrial function) | ||
| Cluster of translationally-controlled tumor protein (TCTP) | Involved in cellular calcium homeostasis and inhibits apoptotic process signaling; cytoplasm and extracellular exosome (UniProtKB). | 40% ↑ |
| Translocon-associated protein subunit β (B5DEQ0) | Involved in ER calcium homeostasis (STRING); ER membrane (ScaffoldQ+). | 100% ↓ |
| Phosphohistidine phosphatase 1 isoform CRA_a (D3ZP47) | Dephosphorylates proteins and inhibits calcium channels; cytosol and extracellular exosome (ScaffoldQ+). | 80% ↓ |
| Regucalcin (RGN) | Cellular calcium ion homeostasis, cytoplasm and nucleoplasm (ScaffoldQ+). | 20% ↓ |
| Cluster of thioredoxin domain-containing protein 12 (sp|Q498E0|TXD12) | Involved in redox homeostasis and inhibits ER stress-induced apoptosis; lumen of ER (UniProtKB). | 460% ↑ |
| Thioredoxin. mitochondrial (THIOM) | Involved in cellular redox homeostasis and mitochondrial membrane potential. Mitochondrial thioredoxin is also involved in inhibiting apoptosis; mitochondria (UniProtKB). | 50% ↑ |
| Cluster of protein RGD1565183 (D3ZJD3) | Translation - structural component of large ribosomal subunit (ScaffoldQ+); nucleolus (UniProtKB). | 80% ↓ |
| Cluster of glutathione S-transferase-α1 (GSTA1) | Protects against oxidative stress and products of lipid peroxidation through glutathione peroxidase activity, cytosol and extracellular exome (NCBI; UniProtKB). | 50% ↓ |
| Cluster of peroxiredoxin-5 [mitochondrial] (D3ZEN5) | Antioxidant properties; mitochondria (STRING). | 40% ↓ |
| Nicotinate-nucleotide pyrophosphorylase [carboxylating] (NADC) | Involved in quinolinate catabolism and nicotinamide adenine dinucleotide (NAD+) synthesis. NAD+ plays a role in cellular metabolism and mitochondrial health (ScaffoldQ+). | 60% ↓ |
| 3-mercaptopyruvate sulfurtransferase (THTM) | Involved in the production of the antioxidant hydrogen sulfide (H2S). Changes in enzyme activity and H2S is indicative of the development of T2DM and hyperglycemia-induced epithelial cell damage; cytoplasm and mitochondria (UniProtKB). | 40% ↓ |
| (C) Possible downstream consequences of ER stress - changes in pyruvate glucose and lipid metabolism | ||
| Cluster of Pdhx protein (Q5BJX2) | Important structural protein of pyruvate dehydrogenase complex; mitochondria (ScaffoldQ+). | 70% ↓ |
| Cluster of dimethylglycine dehydrogenase (tr|Q5RKL4) | Catabolizes dimethylglycine to glycine; mitochondrial matrix Dimethylglycine is involved in the regulation of glucose metabolism (UniProtKB) | 70% ↓ |
| Acyl-protein thioesterase 1 (LYPA1) | Involved in fatty acid metabolism and protein depalmitoylation; cytoplasm (ScaffoldQ+, UniProtKB). | 80% ↓ |
| Cluster of isoamyl acetate-hydrolyzing esterase 1 homolog (sp|Q711G3|IAH1) | Lipid catabolism (lipase) and hydrolase activity; extracellular exosome (UniProtKB). | 40% ↓ |
| Cluster of EH domain-containing protein 1 (sp|Q641Z6|EHD1) | Regulates cholesterol homeostasis and lipid droplet storage. Also involved in endocytosis; cytoplasm and endocytic vesicles (ScaffoldQ+). | 1000% ↑ |
| Rat apolipoprotein E protein (Q65ZS7) | Facilitates the binding and uptake of lipoprotein particles (LDLs in particular) to clear it out of the plasma (UniProtKB). | 100% ↑ |
| Hydroxymethylglutaryl-CoA synthase. cytoplasmic (HMCS1) | Involved in the initial phase of cholesterol biosynthesis; cytoplasm, nucleoplasm and plasma membrane (UniProtKB). | 40% ↑ |
| Cluster of non-specific lipid-transfer protein (sp|P11915|NLTP) | Regulates intracellular cholesterol transport; peroxisome (ScaffoldQ+). | 10% ↑ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benade, J.; Sher, L.; De Klerk, S.; Deshpande, G.; Bester, D.; Marnewick, J.L.; Sieck, G.; Laher, I.; Essop, M.F. The Impact of Sugar-Sweetened Beverage Consumption on the Liver: A Proteomics-Based Analysis. Antioxidants 2020, 9, 569. https://doi.org/10.3390/antiox9070569
Benade J, Sher L, De Klerk S, Deshpande G, Bester D, Marnewick JL, Sieck G, Laher I, Essop MF. The Impact of Sugar-Sweetened Beverage Consumption on the Liver: A Proteomics-Based Analysis. Antioxidants. 2020; 9(7):569. https://doi.org/10.3390/antiox9070569
Chicago/Turabian StyleBenade, Janina, Lucien Sher, Sheneez De Klerk, Gaurang Deshpande, Dirk Bester, Jeanine L. Marnewick, Gary Sieck, Ismail Laher, and M. Faadiel Essop. 2020. "The Impact of Sugar-Sweetened Beverage Consumption on the Liver: A Proteomics-Based Analysis" Antioxidants 9, no. 7: 569. https://doi.org/10.3390/antiox9070569
APA StyleBenade, J., Sher, L., De Klerk, S., Deshpande, G., Bester, D., Marnewick, J. L., Sieck, G., Laher, I., & Essop, M. F. (2020). The Impact of Sugar-Sweetened Beverage Consumption on the Liver: A Proteomics-Based Analysis. Antioxidants, 9(7), 569. https://doi.org/10.3390/antiox9070569