
The Highly Productive Thermothelomyces heterothallica C1 Expression System as a Host for Rapid Development of Influenza Vaccines

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Expressed Influenza Antigens
2.2. Generation of Strains
2.3. The Upstream Process—Production of Influenza Antigens
2.4. The Downstream Process—Purification of Influenza Antigens
2.5. Control Proteins
2.6. Analytical Methods for the Characterization of the Expressed Proteins
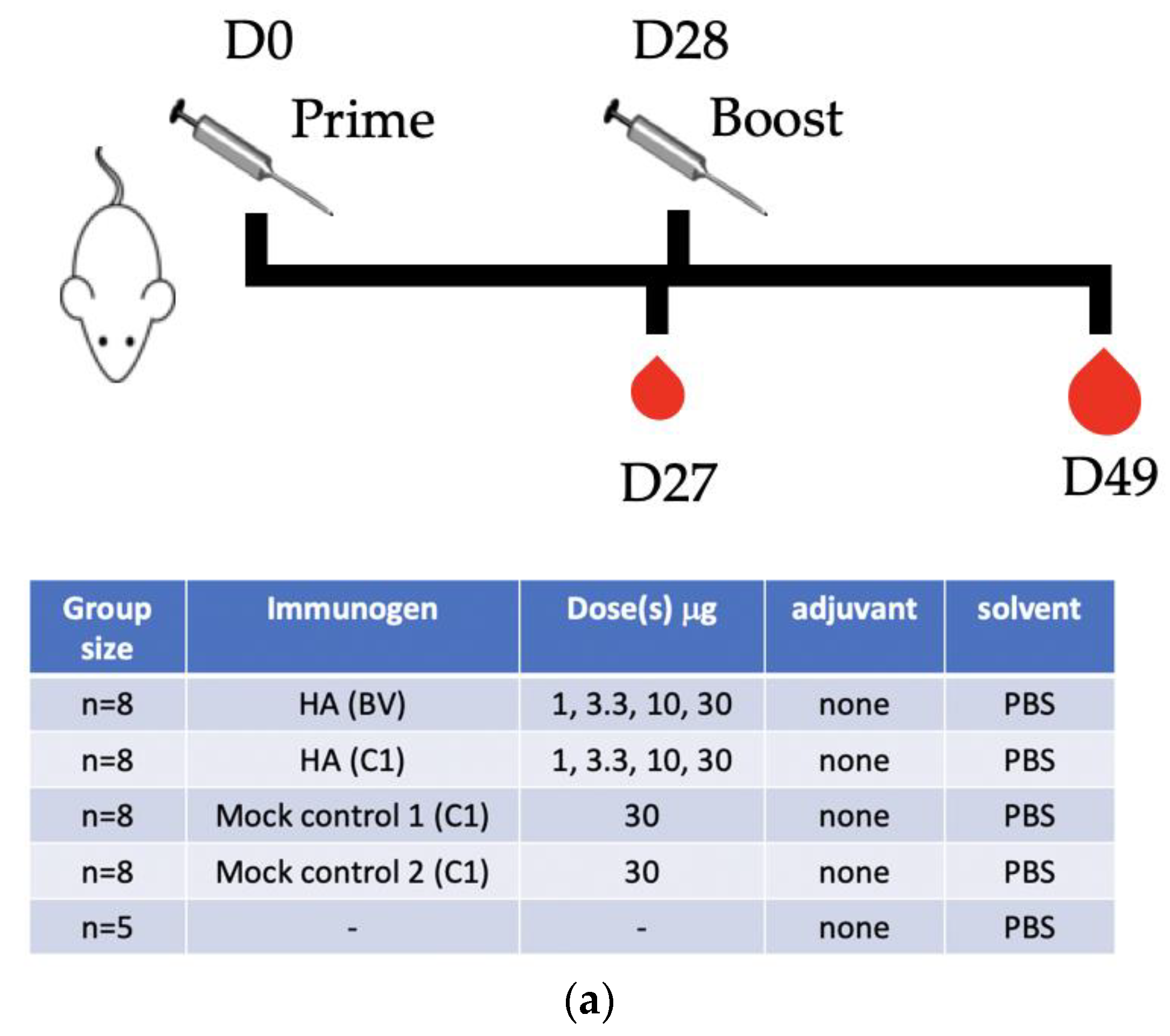
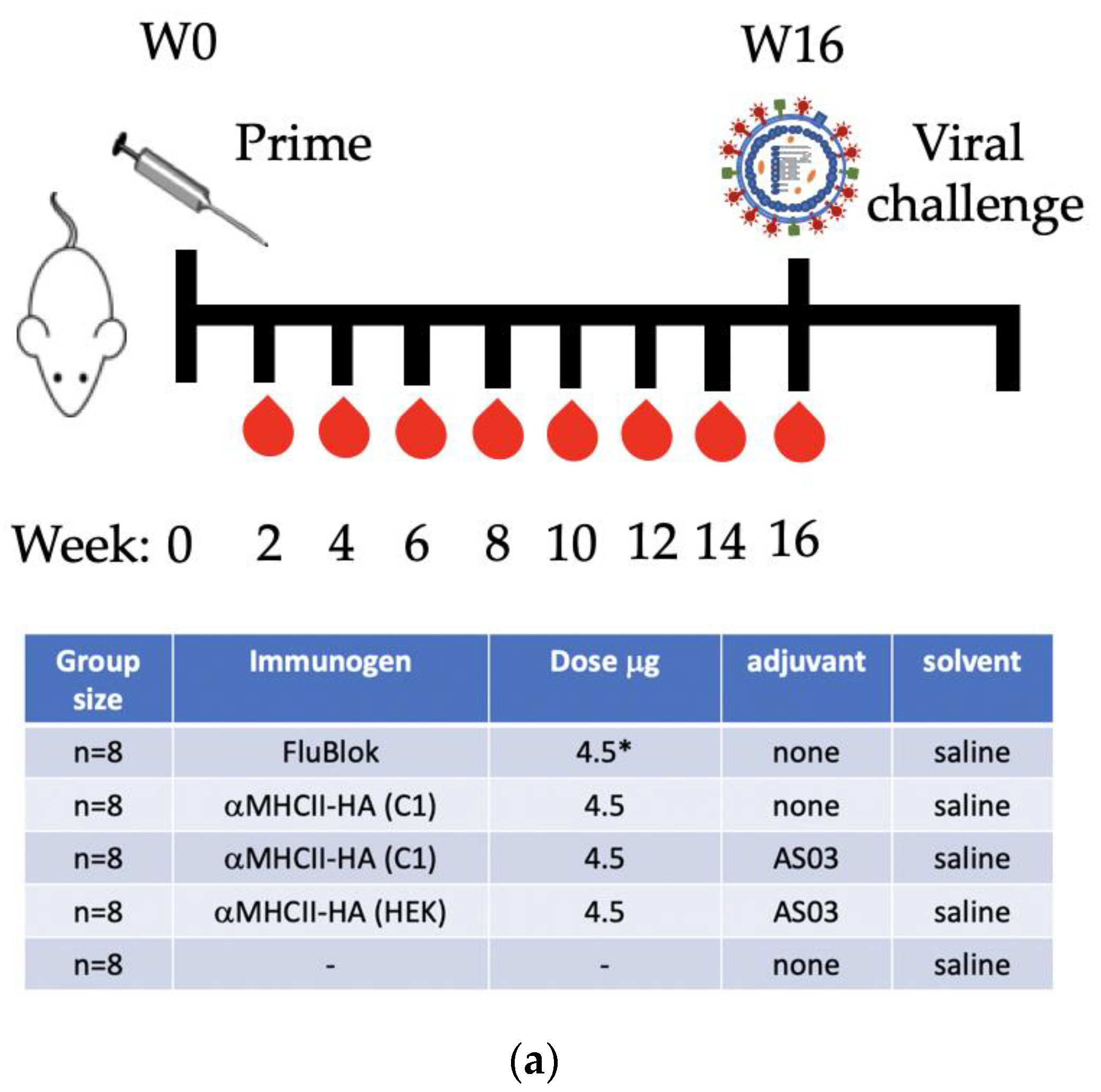
2.7. Animal Studies
2.8. Hemagglutination Inhibition (HI) Assay
2.9. Sandwich ELISA
3. Results
3.1. Influenza Hemagglutinin Produced in C1 (C1-HA (New Caledonia/20/99))
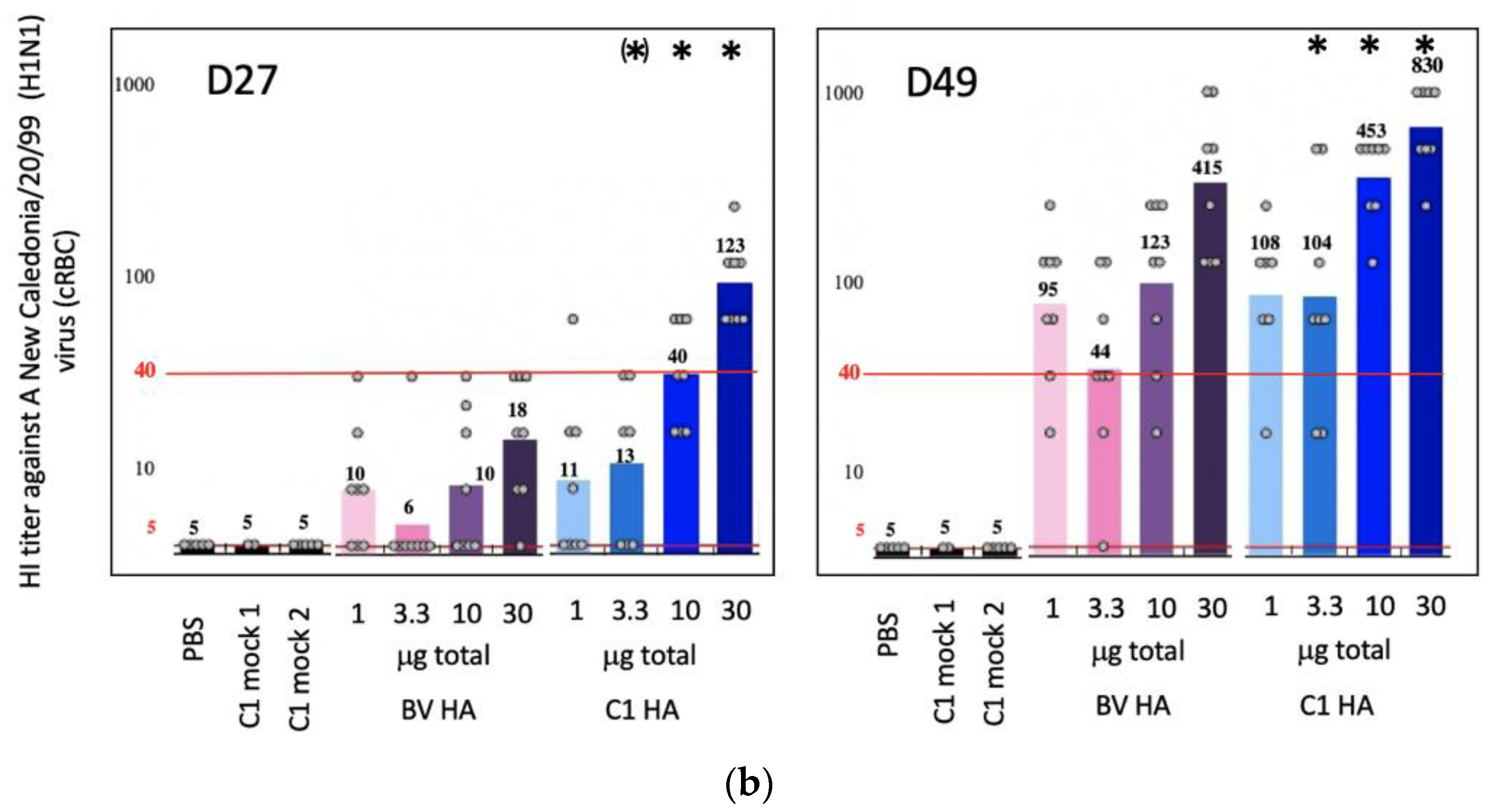
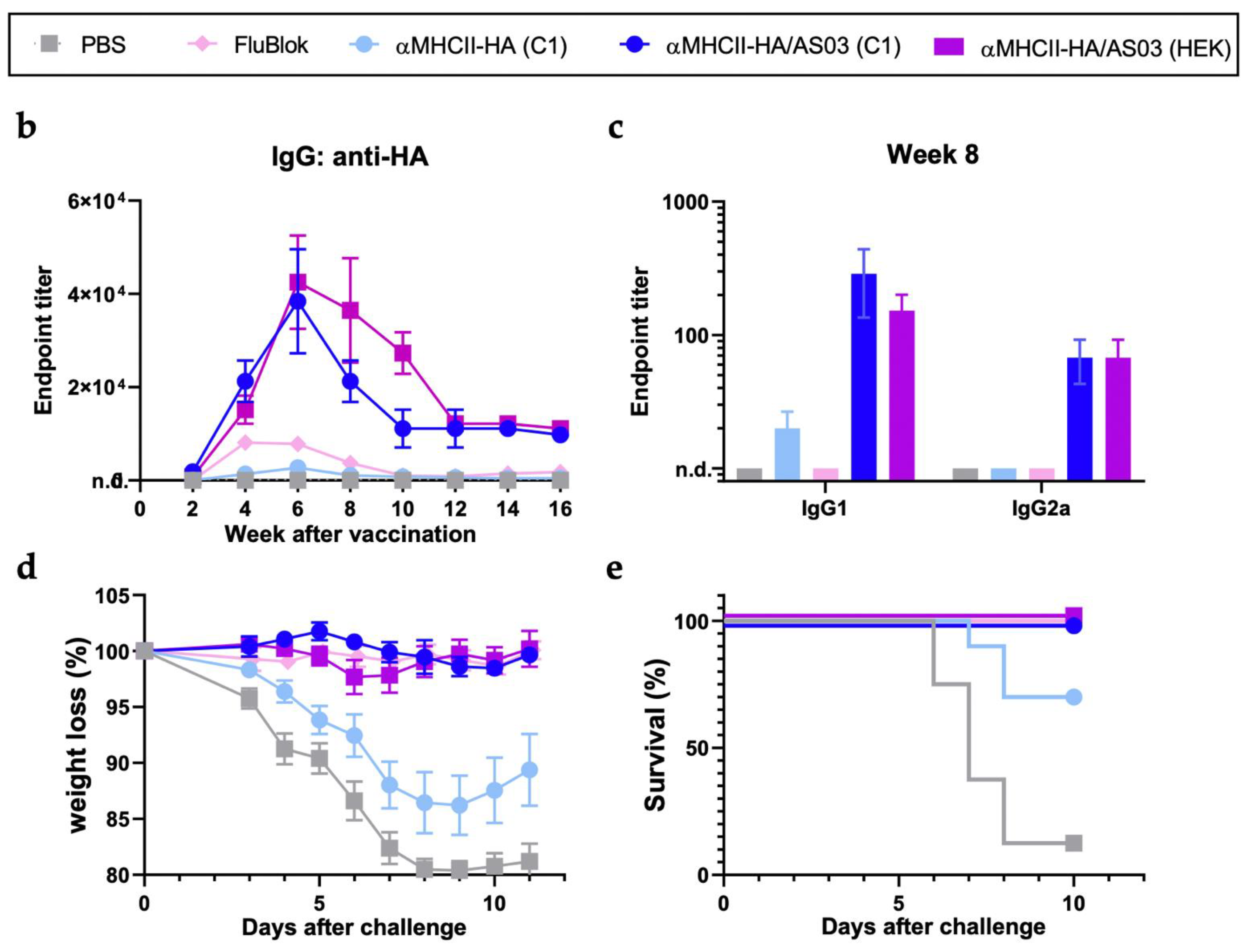
3.2. C1-HA (New Caledonia/20/99) as an Influenza Vaccine Candidate
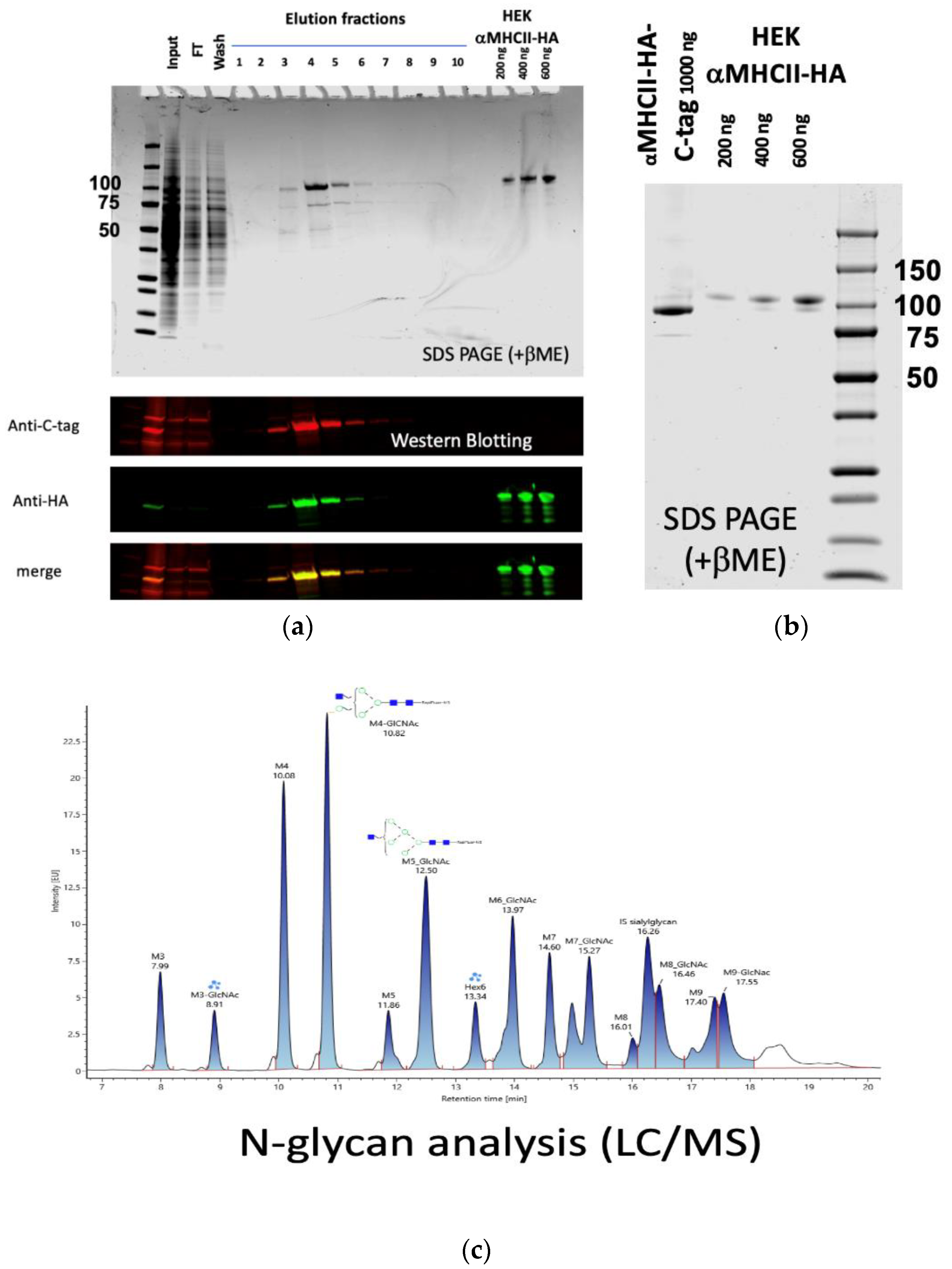
3.3. A Secreted MHCII-Targeted HA (California/7/2009) Produced in C1
3.4. C1 αMHCII-HA (California/7/2009)-C-Tag as an Influenza Vaccine Candidate
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Girard, M.P.; Cherian, T.; Pervikov, Y.; Kieny, M.P. A Review of Vaccine Research and Development: Human Acute Respiratory Infections. Vaccine 2005, 23, 5708–5724. [Google Scholar] [CrossRef]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of Global Seasonal Influenza-Associated Respiratory Mortality: A Modelling Study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Bullard, B.L.; Weaver, E.A. Strategies Targeting Hemagglutinin as a Universal Influenza Vaccine. Vaccines 2021, 9, 257. [Google Scholar] [CrossRef]
- Segaloff, H.; Melidou, A.; Adlhoch, C.; Pereyaslov, D.; Robesyn, E.; Penttinen, P.; Olsen, S.J. Who European Region And The European Influenza Surveillance Network Co-Circulation of Influenza A(H1N1)Pdm09 and Influenza A(H3N2) Viruses, World Health Organization (WHO) European Region, October 2018 to February 2019. Eurosurveillance 2019, 24, 1900125. [Google Scholar] [CrossRef] [Green Version]
- Bouvier, N.M.; Palese, P. The Biology of Influenza Viruses. Vaccine 2008, 26 (Suppl. S4), D49–D53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westgeest, K.B.; Russell, C.A.; Lin, X.; Spronken, M.I.J.; Bestebroer, T.M.; Bahl, J.; van Beek, R.; Skepner, E.; Halpin, R.A.; de Jong, J.C.; et al. Genomewide Analysis of Reassortment and Evolution of Human Influenza A(H3N2) Viruses Circulating between 1968 and 2011. J. Virol. 2014, 88, 2844–2857. [Google Scholar] [CrossRef] [Green Version]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and Ecology of Influenza A Viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef] [PubMed]
- Saunders-Hastings, P.R.; Krewski, D. Reviewing the History of Pandemic Influenza: Understanding Patterns of Emergence and Transmission. Pathogens 2016, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Rota, P.A.; Wallis, T.R.; Harmon, M.W.; Rota, J.S.; Kendal, A.P.; Nerome, K. Cocirculation of Two Distinct Evolutionary Lineages of Influenza Type B Virus since 1983. Virology 1990, 175, 59–68. [Google Scholar] [CrossRef]
- Goto, H.; Kawaoka, Y. A Novel Mechanism for the Acquisition of Virulence by a Human Influenza A Virus. Proc. Natl. Acad. Sci. USA 1998, 95, 10224–10228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doms, R.W.; Helenius, A. Quaternary Structure of Influenza Virus Hemagglutinin after Acid Treatment. J. Virol. 1986, 60, 833–839. [Google Scholar] [CrossRef] [Green Version]
- Skehel, J.J.; Wiley, D.C. Receptor Binding and Membrane Fusion in Virus Entry: The Influenza Hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, S.J.; Haire, L.F.; Russell, R.J.; Stevens, D.J.; Xiao, B.; Ha, Y.; Vasisht, N.; Steinhauer, D.A.; Daniels, R.S.; Elliot, A.; et al. The Structure and Receptor Binding Properties of the 1918 Influenza Hemagglutinin. Science 2004, 303, 1838–1842. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. The Human Antibody Response to Influenza A Virus Infection and Vaccination. Nat. Rev. Immunol. 2019, 19, 383–397. [Google Scholar] [CrossRef]
- Lee, S.; Ryu, J.-H. Influenza Viruses: Innate Immunity and MRNA Vaccines. Front. Immunol. 2021, 12, 710647. [Google Scholar] [CrossRef] [PubMed]
- Schild, G.C.; Oxford, J.S.; Dowdle, W.R.; Coleman, M.; Pereira, M.S.; Chakraverty, P. Antigenic Variation in Current Influenza A Viruses: Evidence for a High Frequency of Antigenic “drift” for the Hong Kong Virus. Bull. World Health Organ. 1974, 51, 1–11. [Google Scholar] [PubMed]
- Ray, R.; Dos Santos, G.; Buck, P.O.; Claeys, C.; Matias, G.; Innis, B.L.; Bekkat-Berkani, R. A Review of the Value of Quadrivalent Influenza Vaccines and Their Potential Contribution to Influenza Control. Hum. Vaccines Immunother. 2017, 13, 1640–1652. [Google Scholar] [CrossRef] [Green Version]
- WHO. Influenza (Seasonal). Available online: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 15 December 2021).
- Pérez Rubio, A.; Eiros, J.M. Cell Culture-Derived Flu Vaccine: Present and Future. Hum. Vaccines Immunother. 2018, 14, 1874–1882. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.M.J.; Izikson, R.; Post, P.; Dunkle, L. Safety, Efficacy, and Immunogenicity of Flublok in the Prevention of Seasonal Influenza in Adults. Ther. Adv. Vaccines 2015, 3, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A Novel Coronavirus Outbreak of Global Health Concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Chams, N.; Chams, S.; Badran, R.; Shams, A.; Araji, A.; Raad, M.; Mukhopadhyay, S.; Stroberg, E.; Duval, E.J.; Barton, L.M.; et al. COVID-19: A Multidisciplinary Review. Front. Public Health 2020, 8, 383. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Vu, M.N.; Kelly, H.G.; Kent, S.J.; Wheatley, A.K. Current and Future Nanoparticle Vaccines for COVID-19. EBioMedicine 2021, 74, 103699. [Google Scholar] [CrossRef]
- de Oliveira Daian e Silva, D.S.; da Fonseca, F.G. The Rise of Vectored Vaccines: A Legacy of the COVID-19 Global Crisis. Vaccines 2021, 9, 1101. [Google Scholar] [CrossRef] [PubMed]
- Singh, B. Myceliophthora Thermophila Syn. Sporotrichum Thermophile: A Thermophilic Mould of Biotechnological Potential. Crit. Rev. Biotechnol. 2016, 36, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Marin-Felix, Y.; Stchigel, A.M.; Miller, A.N.; Guarro, J.; Cano-Lira, J.F. A Re-Evaluation of the Genus Myceliophthora (Sordariales, Ascomycota): Its Segregation into Four Genera and Description of Corynascus Fumimontanus Sp. Nov. Mycologia 2015, 107, 619–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berka, R.M.; Grigoriev, I.V.; Otillar, R.; Salamov, A.; Grimwood, J.; Reid, I.; Ishmael, N.; John, T.; Darmond, C.; Moisan, M.-C.; et al. Comparative Genomic Analysis of the Thermophilic Biomass-Degrading Fungi Myceliophthora Thermophila and Thielavia Terrestris. Nat. Biotechnol. 2011, 29, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Visser, H.; Joosten, V.; Punt, P.J.; Gusakov, A.V.; Olson, P.T.; Joosten, R.; Bartels, J.; Visser, J.; Sinitsyn, A.P.; Emalfarb, M.A.; et al. RESEARCH: Development of a Mature Fungal Technology and Production Platform for Industrial Enzymes Based on a Myceliophthora Thermophila Isolate, Previously Known as Chrysosporium Lucknowense C1. Ind. Biotechnol. 2011, 7, 214–223. [Google Scholar] [CrossRef]
- Gusakov, A.V.; Salanovich, T.N.; Antonov, A.I.; Ustinov, B.B.; Okunev, O.N.; Burlingame, R.; Emalfarb, M.; Baez, M.; Sinitsyn, A.P. Design of Highly Efficient Cellulase Mixtures for Enzymatic Hydrolysis of Cellulose. Biotechnol. Bioeng. 2007, 97, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, L.; Sun, T.; Xu, J.; Ji, J.; Liu, Q.; Tian, C. Direct Production of Commodity Chemicals from Lignocellulose Using Myceliophthora Thermophila. Metab. Eng. 2020, 61, 416–426. [Google Scholar] [CrossRef]
- GRN, No. 292 Cellulase Enzyme Preparation Derived from a Genetically Modified Strain of Myceliophthora thermophila. Available online: https://www.cfsanappsexternal.fda.gov/scripts/fdcc/?set=GRASNotices&id=292&sort=GRN_No&order=DESC&startrow=1&type=basic&search=Myceliophthora (accessed on 15 December 2021).
- Emalfarb, M.; Verwoerd, T.C.; Alfenito, M.R.; Baer, M.; Legastelois, I.; Kazek, M.-P.; Bernard, M.-C.; Dubayle, J.; Kensinger, R. Production of Flu Vaccine in Myceliophthora thermophila. U.S. Patent Application No. 16/640,483, 2020. Available online: https://patents.google.com/patent/WO2019038623A1/en (accessed on 21 August 2020).
- Emalfarb, M.A.; Punt, P.J.; van Zeijl, C.M.J. Expression-regulating sequences and expression products in the field of filamentous fungi. U.S. Patent No 7,906,309, 15 March 2011. [Google Scholar]
- Manicassamy, B.; Medina, R.A.; Hai, R.; Tsibane, T.; Stertz, S.; Nistal-Villán, E.; Palese, P.; Basler, C.F.; García-Sastre, A. Protection of Mice against Lethal Challenge with 2009 H1N1 Influenza A Virus by 1918-like and Classical Swine H1N1 Based Vaccines. PLoS Pathog. 2010, 6, e1000745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, H.; Berzofsky, J.A. Enhancement of Antigenic Potency in Vitro and Immunogenicity In Vivo by Coupling the Antigen to Anti-Immunoglobulin. J. Immunol. 1986, 136, 58–65. [Google Scholar]
- Grodeland, G.; Fredriksen, A.B.; Løset, G.Å.; Vikse, E.; Fugger, L.; Bogen, B. Antigen Targeting to Human HLA Class II Molecules Increases Efficacy of DNA Vaccination. J. Immunol. 2016, 197, 3575–3585. [Google Scholar] [CrossRef]
- Biragyn, A.; Belyakov, I.M.; Chow, Y.-H.; Dimitrov, D.S.; Berzofsky, J.A.; Kwak, L.W. DNA Vaccines Encoding Human Immunodeficiency Virus-1 Glycoprotein 120 Fusions with Proinflammatory Chemoattractants Induce Systemic and Mucosal Immune Responses. Blood 2002, 100, 1153–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredriksen, A.B.; Sandlie, I.; Bogen, B. DNA Vaccines Increase Immunogenicity of Idiotypic Tumor Antigen by Targeting Novel Fusion Proteins to Antigen-Presenting Cells. Mol. Ther. 2006, 13, 776–785. [Google Scholar] [CrossRef]
- Carayanniotis, G.; Barber, B.H. Adjuvant-Free IgG Responses Induced with Antigen Coupled to Antibodies against Class II MHC. Nature 1987, 327, 59–61. [Google Scholar] [CrossRef]
- Grødeland, G.; Fossum, E.; Bogen, B. Polarizing T and B Cell Responses by APC-Targeted Subunit Vaccines. Front. Immunol. 2015, 6, 367. [Google Scholar] [CrossRef]
- Jin, J.; Hjerrild, K.A.; Silk, S.E.; Brown, R.E.; Labbé, G.M.; Marshall, J.M.; Wright, K.E.; Bezemer, S.; Clemmensen, S.B.; Biswas, S.; et al. Accelerating the Clinical Development of Protein-Based Vaccines for Malaria by Efficient Purification Using a Four Amino Acid C-Terminal ‘C-Tag’. Int. J. Parasitol. 2017, 47, 435–446. [Google Scholar] [CrossRef]
- van den Brink, J.; van Muiswinkel, G.C.J.; Theelen, B.; Hinz, S.W.A.; de Vries, R.P. Efficient Plant Biomass Degradation by Thermophilic Fungus Myceliophthora Heterothallica. Appl. Environ. Microbiol. 2013, 79, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.D.; Ross, T.M. H3N2 Influenza Viruses in Humans: Viral Mechanisms, Evolution, and Evaluation. Hum. Vaccines Immunother. 2018, 14, 1840–1847. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.; Zaia, J. Why Glycosylation Matters in Building a Better Flu Vaccine. Mol. Cell. Proteom. 2019, 18, 2348–2358. [Google Scholar] [CrossRef]
- Aebischer, A.; Wernike, K.; König, P.; Franzke, K.; Wichgers Schreur, P.J.; Kortekaas, J.; Vitikainen, M.; Wiebe, M.; Saloheimo, M.; Tchelet, R.; et al. Development of a Modular Vaccine Platform for Multimeric Antigen Display Using an Orthobunyavirus Model. Vaccines 2021, 9, 651. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, L.A.; Ramos, Y.; Andújar, I.; Torres, E.O.; Cabrera, G.; Martín, A.; Roche, D.; Chinea, G.; Becquet, M.; González, I.; et al. In-Solution Buffer-Free Digestion Allows Full-Sequence Coverage and Complete Characterization of Post-Translational Modifications of the Receptor-Binding Domain of SARS-CoV-2 in a Single ESI–MS Spectrum. Anal. Bioanal. Chem. 2021, 413, 7559–7585. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-F. Discovering Novel Zoonotic Viruses. WANG, Lin-Fa. Discovering novel zoonotic viruses. N. S. W. Public Health Bull. 2011, 22, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Zárate, S.; Taboada, B.; Yocupicio-Monroy, M.; Arias, C.F. Human Virome. Arch. Med. Res. 2017, 48, 701–716. [Google Scholar] [CrossRef]
- Meslin, F.X.; Stöhr, K.; Heymann, D. Public Health Implications of Emerging Zoonoses. Rev. Sci. Tech. 2000, 19, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-F.; Crameri, G. Emerging Zoonotic Viral Diseases. Rev. Sci. Tech. 2014, 33, 569–581. [Google Scholar] [CrossRef]
- Hassell, J.M.; Begon, M.; Ward, M.J.; Fèvre, E.M. Urbanization and Disease Emergence: Dynamics at the Wildlife-Livestock-Human Interface. Trends Ecol. Evol. 2017, 32, 55–67. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keresztes, G.; Baer, M.; Alfenito, M.R.; Verwoerd, T.C.; Kovalchuk, A.; Wiebe, M.G.; Andersen, T.K.; Saloheimo, M.; Tchelet, R.; Kensinger, R.; et al. The Highly Productive Thermothelomyces heterothallica C1 Expression System as a Host for Rapid Development of Influenza Vaccines. Vaccines 2022, 10, 148. https://doi.org/10.3390/vaccines10020148
Keresztes G, Baer M, Alfenito MR, Verwoerd TC, Kovalchuk A, Wiebe MG, Andersen TK, Saloheimo M, Tchelet R, Kensinger R, et al. The Highly Productive Thermothelomyces heterothallica C1 Expression System as a Host for Rapid Development of Influenza Vaccines. Vaccines. 2022; 10(2):148. https://doi.org/10.3390/vaccines10020148
Chicago/Turabian StyleKeresztes, Gabor, Mark Baer, Mark R. Alfenito, Theo C. Verwoerd, Andriy Kovalchuk, Marilyn G. Wiebe, Tor Kristian Andersen, Markku Saloheimo, Ronen Tchelet, Richard Kensinger, and et al. 2022. "The Highly Productive Thermothelomyces heterothallica C1 Expression System as a Host for Rapid Development of Influenza Vaccines" Vaccines 10, no. 2: 148. https://doi.org/10.3390/vaccines10020148
APA StyleKeresztes, G., Baer, M., Alfenito, M. R., Verwoerd, T. C., Kovalchuk, A., Wiebe, M. G., Andersen, T. K., Saloheimo, M., Tchelet, R., Kensinger, R., Grødeland, G., & Emalfarb, M. (2022). The Highly Productive Thermothelomyces heterothallica C1 Expression System as a Host for Rapid Development of Influenza Vaccines. Vaccines, 10(2), 148. https://doi.org/10.3390/vaccines10020148