
Designing of a Novel Multi-Antigenic Epitope-Based Vaccine against E. hormaechei: An Intergraded Reverse Vaccinology and Immunoinformatics Approach

 ,
,  , , and
, , and 
Abstract
:1. Introduction
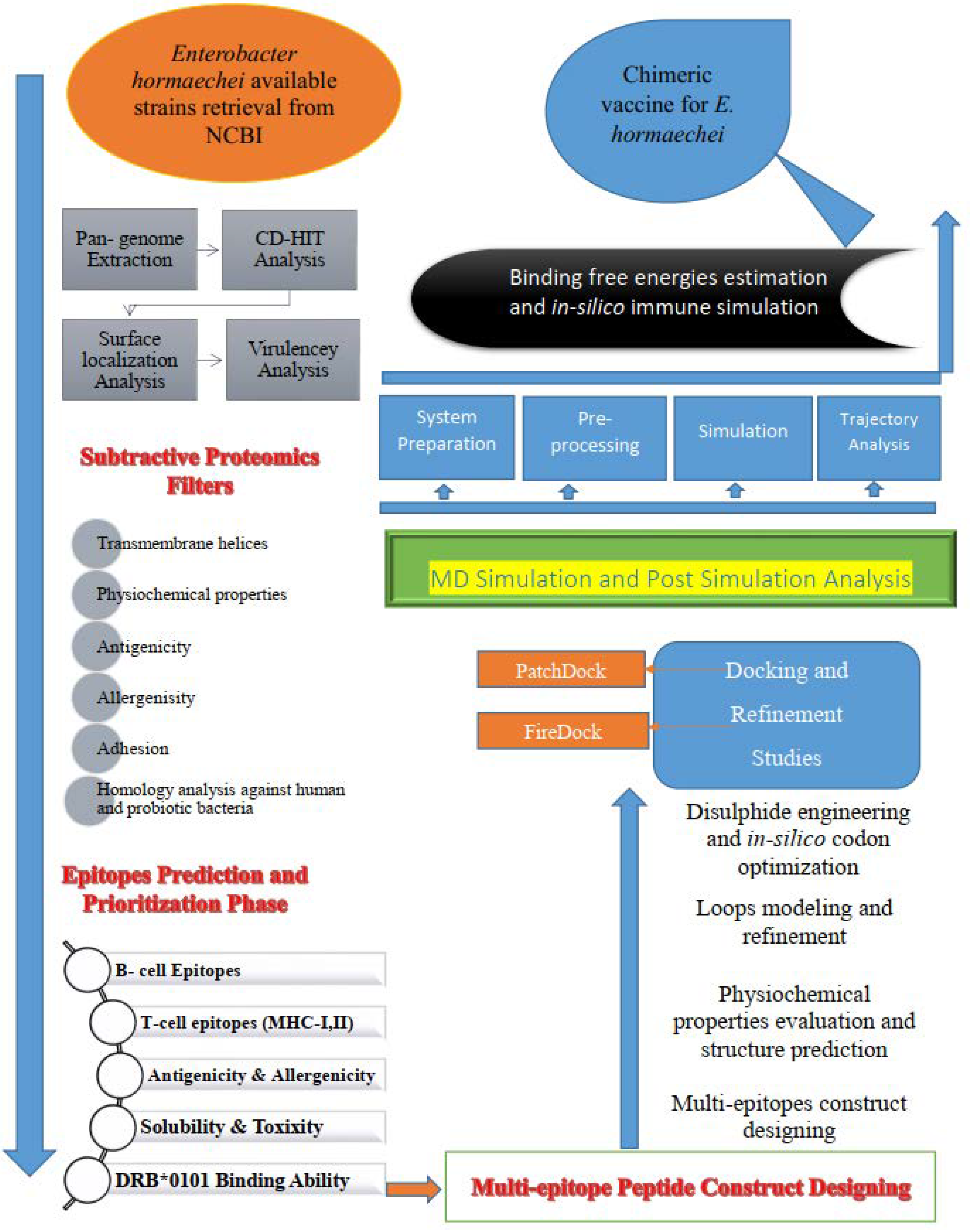
2. Research Methodology
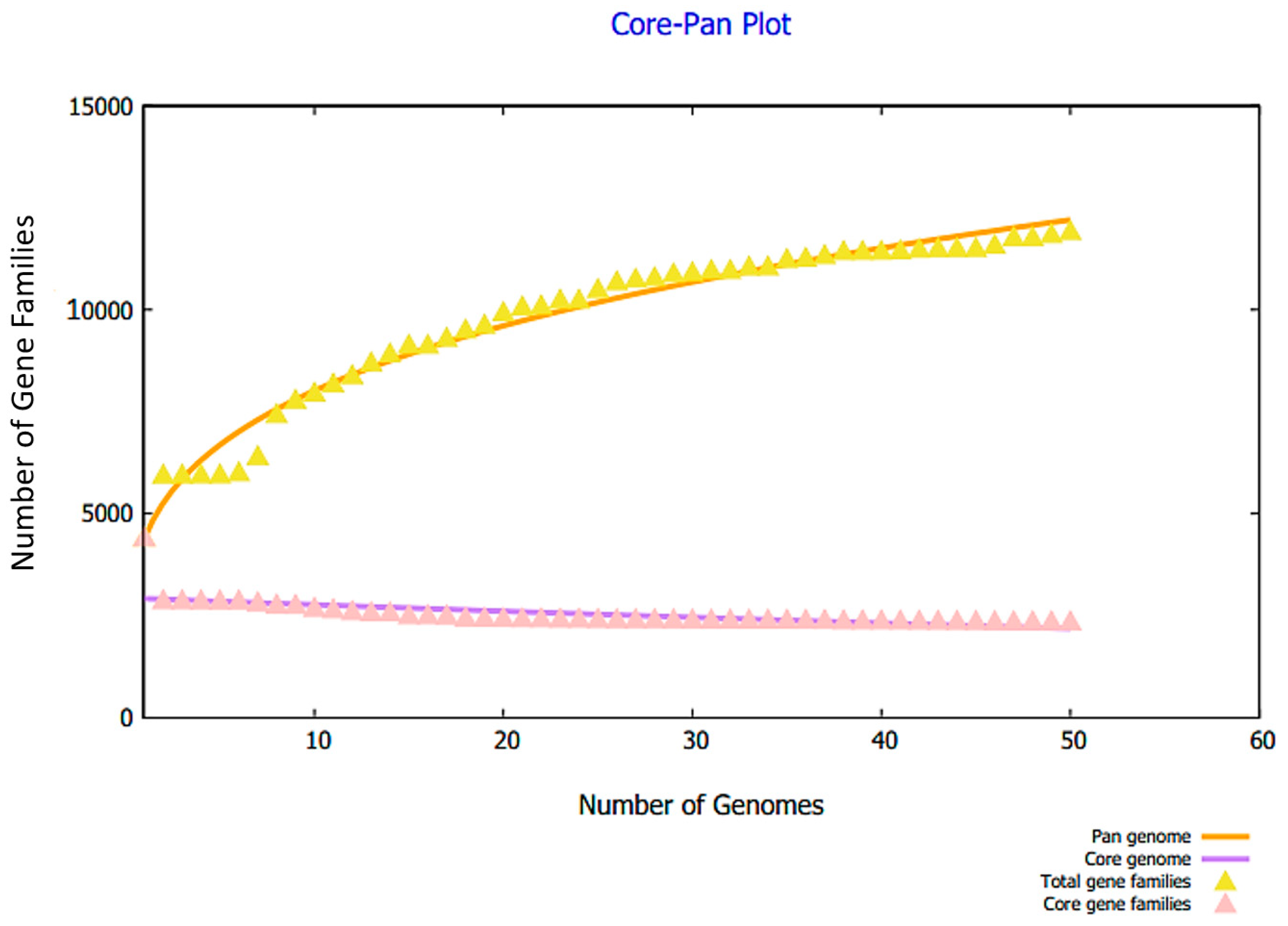
2.1. Retrieval of Complete Proteome and Core Proteome Identification of E. hormaechei
2.2. B- and T-Cell Epitope Mapping
2.3. Construction and Processing of the Multi-Epitope Vaccine Model
2.4. Molecular Interaction Analysis Study
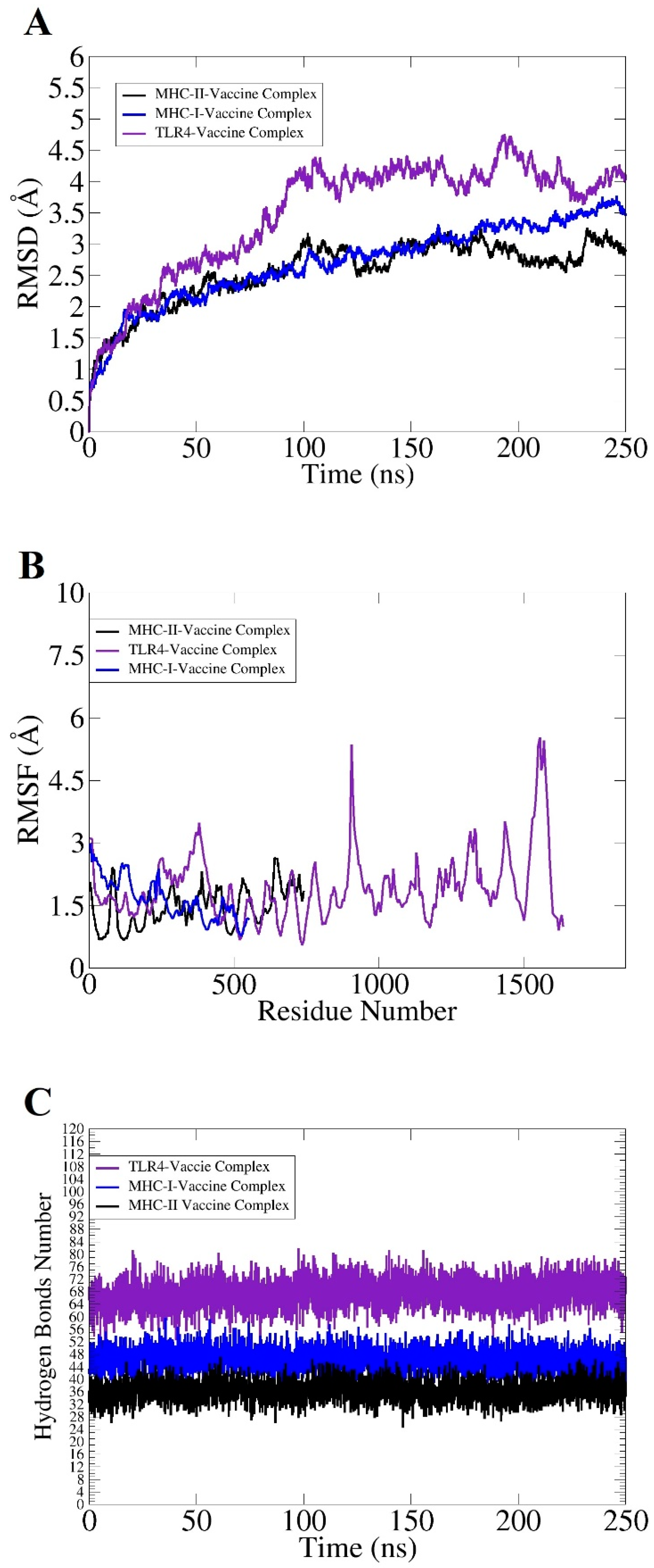
2.5. Molecular Dynamic (MD) Simulation and Binding Free Energy Calculation
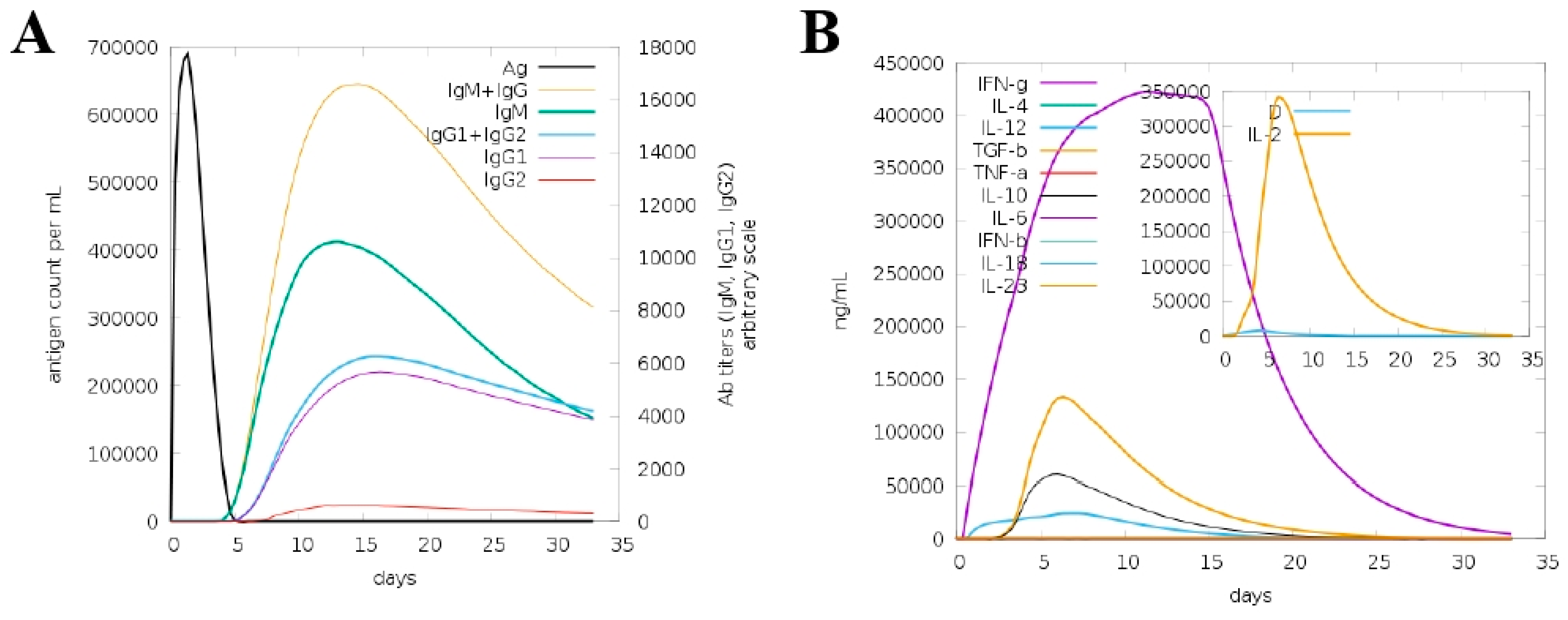
2.6. Immune Simulation
3. Results and Discussion
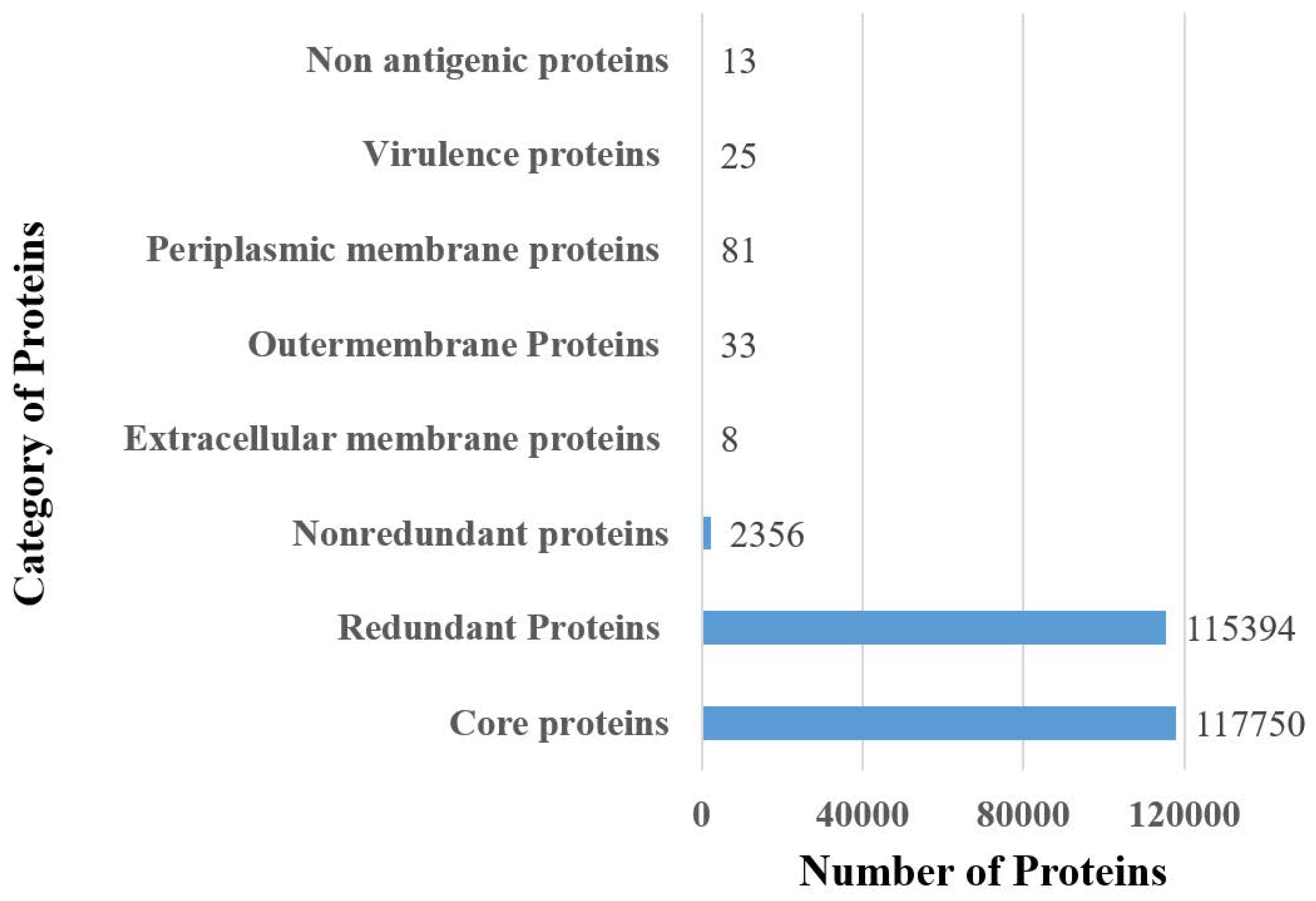
3.1. Complete Proteome Retrieval and Identification of Core, Non-Redundant, Surface-Localized, Virulent Proteins
3.2. Physiochemical Properties, Transmembrane Helices, Allergenicity and Homology Analysis
3.3. Prioritization of Potential B- and T-Cell (MHC-I, MHC-II) Epitopes
3.4. Multi-Epitope-Based Vaccine Design and Processing
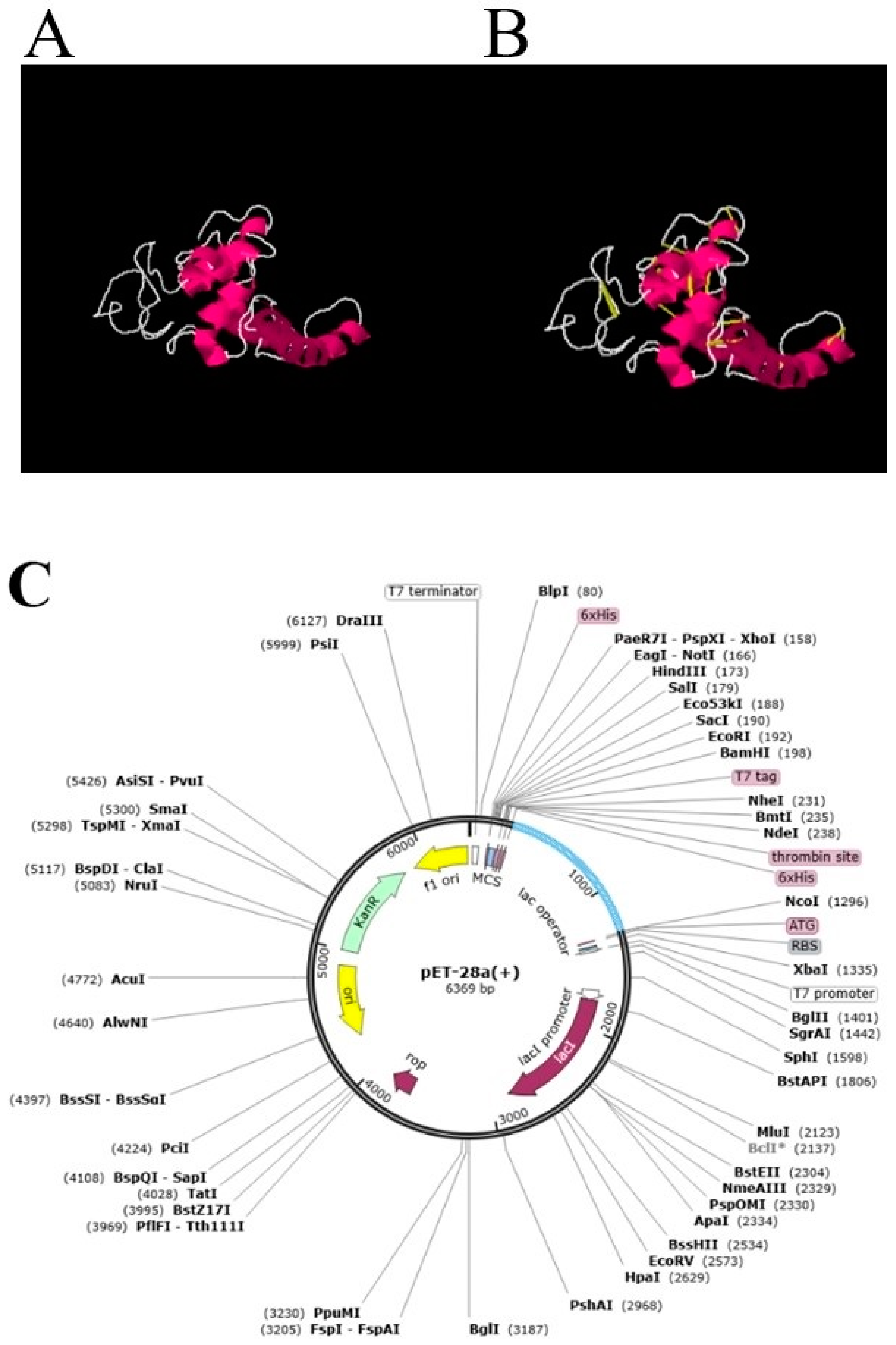
3.5. Loop Modeling, Refinement, Disulfide Engineering and In Silico Codon Optimization
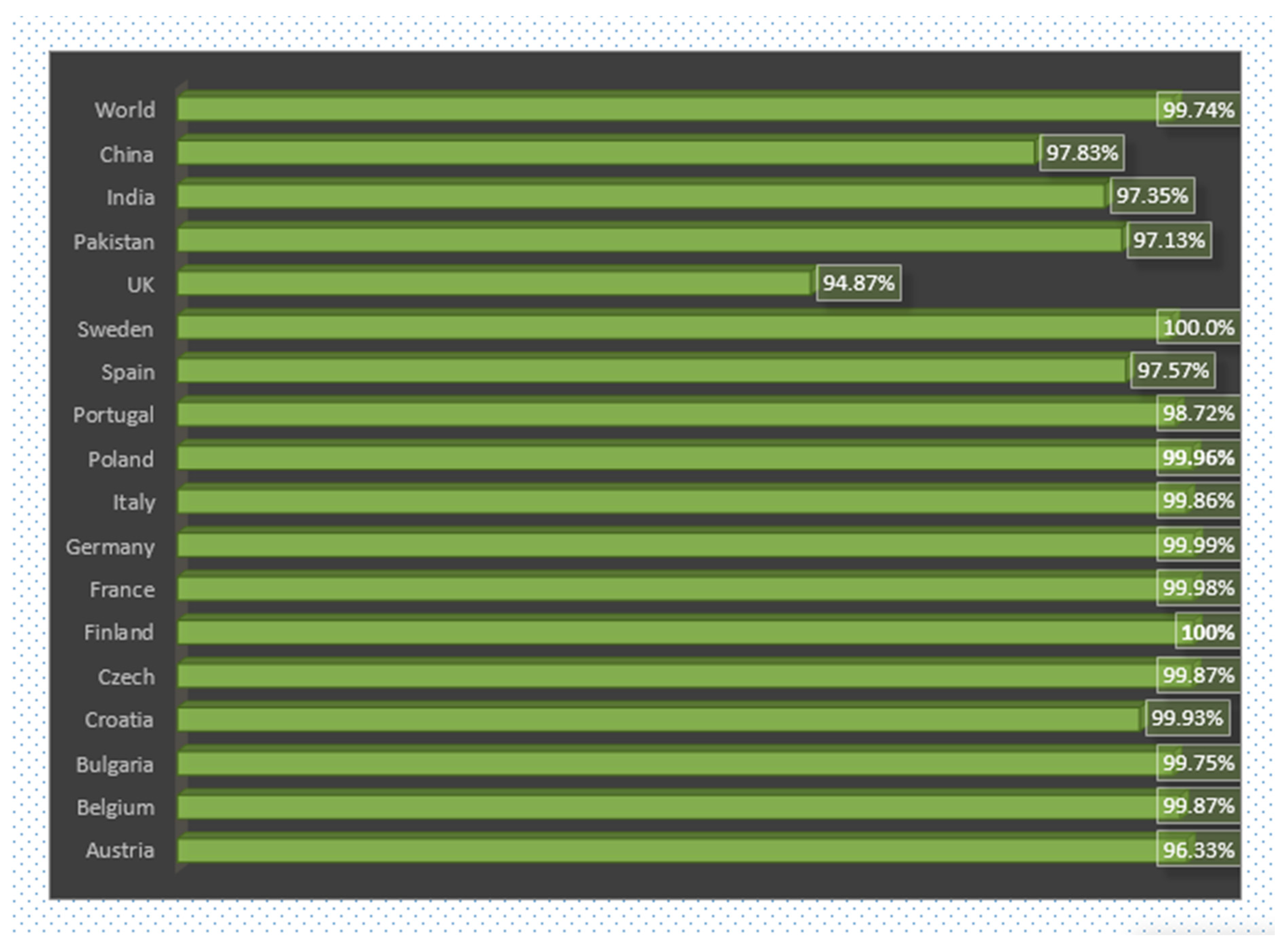
3.6. World and Country Wise Population Coverage Analysis of Vaccine
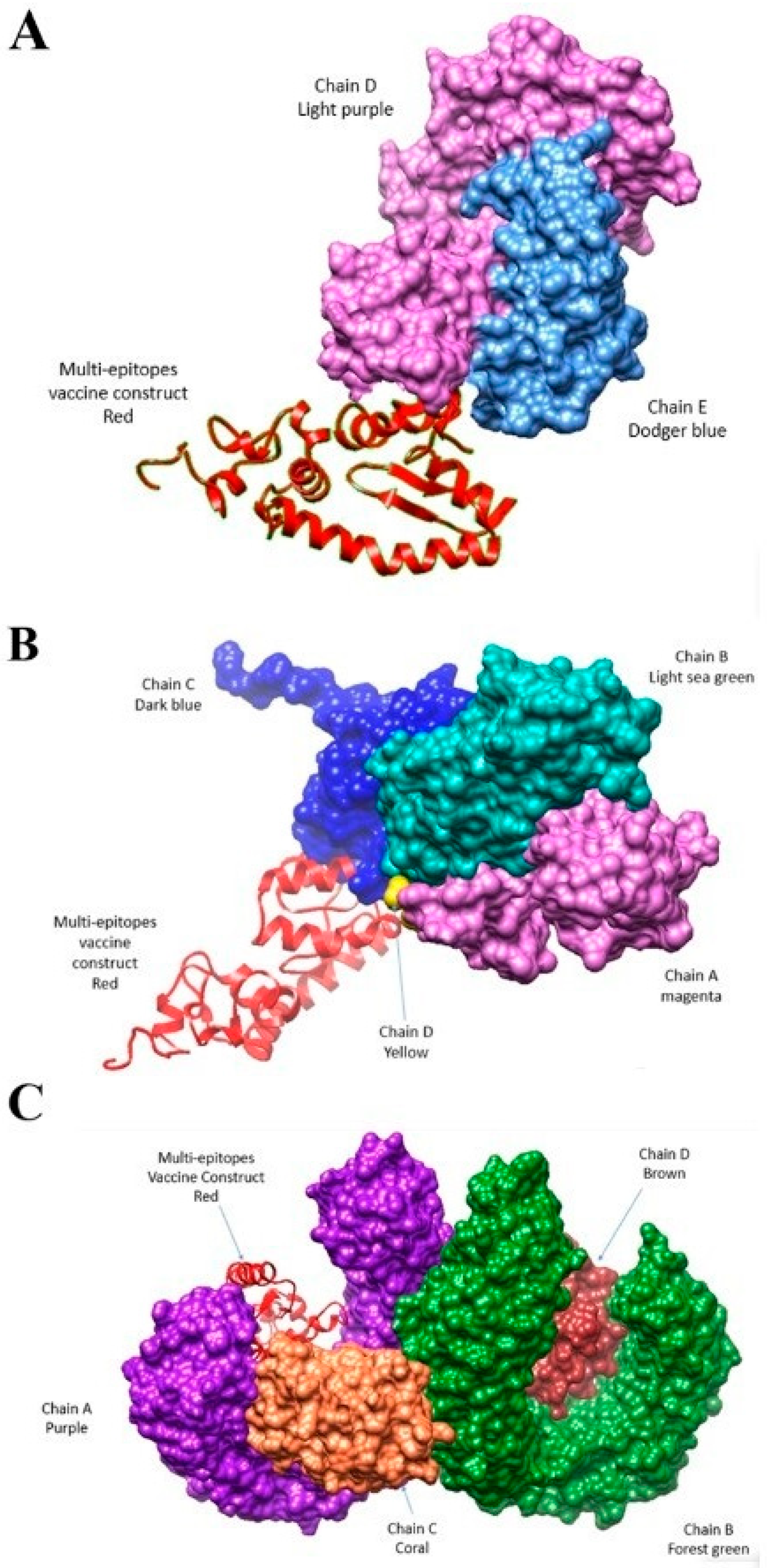
3.7. Docking and Simulation Analysis
3.8. Free Binding Energy Estimation of Docked Molecules
3.9. Interactive Residues of Vaccine-MHC-I, Vaccine MHC-II and Vaccine TLR-4
3.10. In Silico Host Immune Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Feye, K.M.; Shi, Z.; Pavlidis, H.O.; Kogut, M.; Ashworth, A.J.; Ricke, S.C. A Historical Review on Antibiotic Resistance of Foodborne Campylobacter. Front. Microbiol. 2019, 10, 1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, A.H.; Moore, L.S.P.; Sundsfjord, A.; Steinbakk, M.; Regmi, S.; Karkey, A.; Guerin, P.J.; Piddock, L.J. Understanding the Mechanisms and Drivers of Antimicrobial Resistance. Lancet 2016, 387, 176–187. [Google Scholar] [CrossRef]
- Golkar, Z.; Bagasra, O.; Pace, D.G. Bacteriophage Therapy: A Potential Solution for the Antibiotic Resistance Crisis. J. Infect. Dev. Ctries. 2014, 8, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903. [Google Scholar] [CrossRef] [Green Version]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; Salamat, M.K.F.; et al. Antibiotic Resistance: A Rundown of a Global Crisis. Infect. Drug Resist. 2018, 11, 1645. [Google Scholar] [CrossRef] [Green Version]
- van Duin, D.; Doi, Y. The Global Epidemiology of Carbapenemase-Producing Enterobacteriaceae. Virulence 2017, 8, 460–469. [Google Scholar] [CrossRef]
- Woerther, P.-L.; Lepeule, R.; Burdet, C.; Decousser, J.-W.; Ruppé, É.; Barbier, F. Carbapenems and Alternative β-Lactams for the Treatment of Infections Due to Extended-Spectrum β-Lactamase-Producing Enterobacteriaceae: What Impact on Intestinal Colonisation Resistance? Int. J. Antimicrob. Agents 2018, 52, 762–770. [Google Scholar] [CrossRef]
- Khalifa, H.O.; Soliman, A.M.; Ahmed, A.M.; Shimamoto, T.; Hara, T.; Ikeda, M.; Kuroo, Y.; Kayama, S.; Sugai, M.; Shimamoto, T. High Carbapenem Resistance in Clinical Gram-Negative Pathogens Isolated in Egypt. Microb. Drug Resist. 2017, 23, 838–844. [Google Scholar] [CrossRef]
- Wang, Z.; Duan, L.; Liu, F.; Hu, Y.; Leng, C.; Kan, Y.; Yao, L.; Shi, H. First Report of Enterobacter Hormaechei with Respiratory Disease in Calves. BMC Vet. Res. 2020, 16, 1. [Google Scholar] [CrossRef]
- Ai, W.; Zhou, Y.; Wang, B.; Zhan, Q.; Hu, L.; Xu, Y.; Guo, Y.; Wang, L.; Yu, F.; Li, X. First Report of Coexistence of BlaSFO-1 and BlaNDM-1 β-Lactamase Genes as Well as Colistin Resistance Gene Mcr-9 in a Transferrable Plasmid of a Clinical Isolate of Enterobacter hormaechei. Front. Microbiol. 2021, 12, 676113. [Google Scholar] [CrossRef]
- Daurel, C.; Fiant, A.-L.; Brémont, S.; Courvalin, P.; Leclercq, R. Emergence of an Enterobacter Hormaechei Strain with Reduced Susceptibility to Tigecycline under Tigecycline Therapy. Antimicrob. Agents Chemother. 2009, 53, 4953–4954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, A.S.; Eisenstadt, R.; Morrison, J.O. Enhanced in Vitro Bactericidal Activity of Amikacin or Gentamicin Combined with Three New Extended-Spectrum Cephalosporins against Cephalothin-Resistant Members of the Family Enterobacteriaceae. Antimicrob. Agents Chemother. 1984, 25, 725–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, K.B.; Alsolami, A.; Khalifa, A.M.; Khalil, N.A.; Moursi, S.; Osman, A.; Fahad, D.; Rakha, E.; Rashidi, M.; Moussa, S.; et al. A Multi-Point Surveillance for Antimicrobial Resistance Profiles among Clinical Isolates of Gram-Negative Bacteria Recovered from Major Ha’il Hospitals, Saudi Arabia. Microorganisms 2021, 9, 2024. [Google Scholar] [CrossRef] [PubMed]
- Masignani, V.; Pizza, M.; Moxon, E.R. The Development of a Vaccine against Meningococcus B Using Reverse Vaccinology. Front. Immunol. 2019, 10, 751. [Google Scholar] [CrossRef] [PubMed]
- Seib, K.L.; Zhao, X.; Rappuoli, R. Developing Vaccines in the Era of Genomics: A Decade of Reverse Vaccinology. Clin. Microbiol. Infect. 2012, 18, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leow, C.Y.; Chuah, C.; Abdul Majeed, A.B.; Mohd Nor, N.; Leow, C.H. Application of Reverse Vaccinology and Immunoinformatic Strategies for the Identification of Vaccine Candidates Against Shigella Flexneri. In Bacterial Vaccines; Springer: Berlin, Germany, 2022; pp. 17–35. [Google Scholar]
- Sette, A.; Rappuoli, R. Reverse Vaccinology: Developing Vaccines in the Era of Genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Seib, K.L.; Dougan, G.; Rappuoli, R. The Key Role of Genomics in Modern Vaccine and Drug Design for Emerging Infectious Diseases. PLoS Genet. 2009, 5, e1000612. [Google Scholar] [CrossRef] [Green Version]
- Del Tordello, E.; Rappuoli, R.; Delany, I. Reverse Vaccinology: Exploiting Genomes for Vaccine Design. In Human Vaccines; Elsevier: Amsterdam, The Netherlands, 2017; pp. 65–86. [Google Scholar]
- NCBI Resource Coordinators. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an Ultra-Fast Pan-Genome Analysis Pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved Protein Subcellular Localization Prediction with Refined Localization Subcategories and Predictive Capabilities for All Prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Sanober, G.; Ahmad, S.; Azam, S.S. Identification of Plausible Drug Targets by Investigating the Druggable Genome of MDR Staphylococcus Epidermidis. Gene Rep. 2017, 7, 147–153. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Lu, C.; Hwang, J. Prediction of Protein Subcellular Localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of Putative Vaccine Candidates against Helicobacter Pylori Exploiting Exoproteome and Secretome: A Reverse Vaccinology Based Approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Ismail, M.; Sajid, Z.; Ali, A.; Wu, X.; Muhammad, S.A.; Shaikh, R.S. Prediction of Prophylactic Peptide Vaccine Candidates for Human Papillomavirus (HPV): Immunoinformatics and Reverse Vaccinology Approaches. Curr. Proteom. 2021, 18, 178–192. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, P.; Luo, J.; Jiang, Y. Secreted Protein Prediction System Combining CJ-SPHMM, TMHMM, and PSORT. Mamm. Genome 2003, 14, 859–865. [Google Scholar] [CrossRef]
- Ismail, S.; Shahid, F.; Khan, A.; Bhatti, S.; Ahmad, S.; Naz, A.; Almatroudi, A.; ul Qamar, M.T. Pan-Vaccinomics Approach Towards a Universal Vaccine Candidate Against WHO Priority Pathogens to Address Growing Global Antibiotic Resistance. Comput. Biol. Med. 2021, 136, 104705. [Google Scholar] [CrossRef]
- Blast, N. Basic Local Alignment Search Tool. Natl. Libr. Med. Natl. Cent. Biotechnol. Inf. 2015, 43, D6–D17. [Google Scholar]
- Nosrati, M.; Hajizade, A.; Nazarian, S.; Amani, J.; Vansofla, A.N.; Tarverdizadeh, Y. Designing a Multi-Epitope Vaccine for Cross-Protection against Shigella Spp: An Immunoinformatics and Structural Vaccinology Study. Mol. Immunol. 2019, 116, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Furrie, E.; Macfarlane, S.; Cummings, J.H.; Macfarlane, G.T. Systemic Antibodies towards Mucosal Bacteria in Ulcerative Colitis and Crohn’s Disease Differentially Activate the Innate Immune Response. Gut 2004, 53, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P.; et al. IEDB-AR: Immune Epitope Database—Analysis Resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The Immune Epitope Database (IEDB) 3. 0. Nucleic Acids Res. 2014, 43, D405–D412. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing Multi-Epitope Vaccine against Staphylococcus Aureus by Employing Subtractive Proteomics, Reverse Vaccinology and Immuno-Informatics Approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef]
- Chand, Y.; Singh, S. Prioritization of Potential Vaccine Candidates and Designing a Multiepitope-Based Subunit Vaccine against Multidrug-Resistant Salmonella Typhi Str. CT18: A Subtractive Proteomics and Immunoinformatics Approach. Microb. Pathog. 2021, 159, 105150. [Google Scholar] [CrossRef]
- Ismail, S.; Ahmad, S.; Azam, S.S. Vaccinomics to Design a Novel Single Chimeric Subunit Vaccine for Broad-Spectrum Immunological Applications Targeting Nosocomial Enterobacteriaceae Pathogens. Eur. J. Pharm. Sci. 2020, 146, 105258. [Google Scholar] [CrossRef]
- Sun, J.B.; Czerkinsky, C.; Holmgren, J. Mucosally Induced Immunological Tolerance, Regulatory T Cells and the Adjuvant Effect by Cholera Toxin B Subunit. Scand. J. Immunol. 2010, 71, 1–11. [Google Scholar] [CrossRef]
- ProtParam, E. ExPASy-ProtParam Tool; SIB: Lausanne, Switzerland, 2017. [Google Scholar]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A Protein Structure and Structural Feature Prediction Server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [Green Version]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate Sequence-Based Prediction of Protein Solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB Server for Protein Structure Prediction and Refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef] [PubMed]
- Bower, M.; Hedberg, J.; Kuswara, A. Conceptualising Web 2.0 Enabled Learning Designs. Same Places Differ. Spaces. Proc. Ascilite Auckl. 2009, 2009, 1153–1162. [Google Scholar]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to Its Potential Expression Host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Sathiamoorthy, S.; Shin, J.A. Boundaries of the Origin of Replication: Creation of a PET-28a-Derived Vector with P15A Copy Control Allowing Compatible Coexistence with PET Vectors. PLoS ONE 2012, 7, e47259. [Google Scholar] [CrossRef] [Green Version]
- Dorosti, H.; Eslami, M.; Negahdaripour, M.; Ghoshoon, M.B.; Gholami, A.; Heidari, R.; Dehshahri, A.; Erfani, N.; Nezafat, N.; Ghasemi, Y. Vaccinomics Approach for Developing Multi-Epitope Peptide Pneumococcal Vaccine. J. Biomol. Struct. Dyn. 2019, 37, 3524–3535. [Google Scholar] [CrossRef]
- Bibi, S.; Ullah, I.; Zhu, B.; Adnan, M.; Liaqat, R.; Kong, W.-B.; Niu, S. In Silico Analysis of Epitope-Based Vaccine Candidate against Tuberculosis Using Reverse Vaccinology. Sci. Rep. 2021, 11, 1249. [Google Scholar] [CrossRef]
- Rakhmetov, A.; Lee, S.P.; Grebinyk, D.; Ostapchenko, L.; Chae, H.Z. Simulation of Peroxiredoxin II and Brain-Type Creatine Kinase Protein-Protein Interaction Using the on-Line Docking Server ClusPro 2.0. J. Appl. Pharm. Sci. 2015, 5, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of Three-Dimensional Structural Information of Biological Macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Vanga, S.K.; Orsat, V.; Raghavan, V. Application of Molecular Dynamic Simulation to Study Food Proteins: A Review. Crit. Rev. Food Sci. Nutr. 2018, 58, 2779–2789. [Google Scholar] [CrossRef] [PubMed]
- Kalita, P.; Lyngdoh, D.L.; Padhi, A.K.; Shukla, H.; Tripathi, T. Development of Multi-Epitope Driven Subunit Vaccine against Fasciola Gigantica Using Immunoinformatics Approach. Int. J. Biol. Macromol. 2019, 138, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. The FF14SB Force Field. Amber 2014, 14, 29–31. [Google Scholar]
- Kerrigan, J.E. AMBER 10.0 Introductory Tutorial. USA. 2009. Available online: https://ambermd.org/tutorials/ (accessed on 10 January 2022).
- Allemailem, K.S. A Comprehensive Computer Aided Vaccine Design Approach to Propose a Multi-Epitopes Subunit Vaccine against Genus Klebsiella Using Pan-Genomics, Reverse Vaccinology, and Biophysical Techniques. Vaccines 2021, 9, 1087. [Google Scholar] [CrossRef]
- Butt, A.M.; Tahir, S.; Nasrullah, I.; Idrees, M.; Lu, J.; Tong, Y. Mycoplasma Genitalium: A Comparative Genomics Study of Metabolic Pathways for the Identification of Drug and Vaccine Targets. Infect. Genet. Evol. 2012, 12, 53–62. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Chukwudozie, O.S.; Gray, C.M.; Fagbayi, T.A.; Chukwuanukwu, R.C.; Oyebanji, V.O.; Bankole, T.T.; Adewole, R.A.; Daniel, E.M. Immuno-Informatics Design of a Multimeric Epitope Peptide Based Vaccine Targeting SARS-CoV-2 Spike Glycoprotein. PLoS ONE 2021, 16, e0248061. [Google Scholar] [CrossRef]
- Pereira, B.A.S.; Silva, F.S.; Rebello, K.M.; Marín-Villa, M.; Traub-Cseko, Y.M.; Andrade, T.C.B.; Bertho, Á.L.; Caffarena, E.R.; Alves, C.R. In Silico Predicted Epitopes from the COOH-Terminal Extension of Cysteine Proteinase B Inducing Distinct Immune Responses during Leishmania (Leishmania) Amazonensis Experimental Murine Infection. BMC Immunol. 2011, 12, 44. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Sood, D.; Sharma, N.; Chandra, R. Multiepitope Subunit Vaccine to Evoke Immune Response against Acute Encephalitis. J. Chem. Inf. Model. 2019, 60, 421–433. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Saba Ismail, S.A.; Mirza, M.U.; Abbasi, S.W.; Ashfaq, U.A.; Chen, L.-L. Development of a Novel Multi-Epitope Vaccine against Crimean-Congo Hemorrhagic Fever Virus: An Integrated Reverse Vaccinology, Vaccine Informatics and Biophysics Approach. Front. Immunol. 2021, 12, 669812. [Google Scholar] [CrossRef]
- Ashfaq, U.A.; Saleem, S.; Masoud, M.S.; Ahmad, M.; Nahid, N.; Bhatti, R.; Almatroudi, A.; Khurshid, M. Rational Design of Multi Epitope-Based Subunit Vaccine by Exploring MERS-COV Proteome: Reverse Vaccinology and Molecular Docking Approach. PLoS ONE 2021, 16, e0245072. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Shahid, F.; Tahir ul Qamar, M.; Abbasi, S.W.; Sajjad, W.; Ismail, S.; Alrumaihi, F.; Allemailem, K.S.; Almatroudi, A.; Ullah Saeed, H.F. Immuno-Informatics Analysis of Pakistan-Based HCV Subtype-3a for Chimeric Polypeptide Vaccine Design. Vaccines 2021, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- ul Qamar, M.T.; Shahid, F.; Aslam, S.; Ashfaq, U.A.; Aslam, S.; Fatima, I.; Fareed, M.M.; Zohaib, A.; Chen, L.-L. Reverse Vaccinology Assisted Designing of Multiepitope-Based Subunit Vaccine against SARS-CoV-2. Infect. Dis. Poverty 2020, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name/Accession Number | Predicted Epitopes |
|---|---|
| >core/8236/1/Org1_Gene1515 (curlin minor subunit (CsgB)) | AAAGYDLANSEYNFAVNELSKSSFN |
| >core/5396/1/Org1_Gene1549 (flagellar basal-body rod protein (FlgF)) | HPVVGEAGPIAVPEGAEITIA |
| GSEVQRGDDDIFRLSAESQATRGPVLQADPT | |
| >core/2987/11/Org11_Gene4825 (flagellar basal body P-ring protein (FlgI)) | AGAQAGGSRVQVNQLNGG |
| GTGDQTMQAPF | |
| NNVVSQPDTPLGGGQTVVVPQTDISVRDRGGSLQSVRSSTD |
| Model | Root Mean Square Deviation (RMSD) | MolProbity | Clash Score | Poor Rotamers | Rama Favored | GALAXY Energy |
|---|---|---|---|---|---|---|
| Initial | 0.000 | 3.493 | 97.9 | 3.0 | 84.1 | 9003.96 |
| MODEL 1 | 0.922 | 1.471 | 2.1 | 0.0 | 92.1 | −3333.41 |
| MODEL 2 | 0.880 | 1.517 | 2.5 | 0.8 | 92.1 | −3327.95 |
| MODEL 3 | 0.882 | 1.419 | 1.8 | 0.8 | 92.1 | −3326.39 |
| MODEL 4 | 0.786 | 1.494 | 2.5 | 0.0 | 92.7 | −3318.02 |
| MODEL 5 | 0.768 | 1.494 | 2.5 | 0.0 | 92.7 | −3316.79 |
| MODEL 6 | 0.804 | 1.517 | 2.5 | 0.0 | 92.1 | −3315.93 |
| MODEL 7 | 0.829 | 1.578 | 2.9 | 0.0 | 91.5 | −3315.06 |
| MODEL 8 | 0.874 | 1.572 | 3.2 | 0.0 | 92.7 | −3312.58 |
| MODEL 9 | 0.667 | 1.396 | 1.8 | 0.0 | 92.7 | −3312.28 |
| MODEL 10 | 0.831 | 1.517 | 2.5 | 0.8 | 92.1 | −3310.59 |
| Pairs of Amino Acid Residues | Chi3 Angle | Energy (kcal/mol) |
|---|---|---|
| LEU4-TYR-33 | −74.84 | 3.02 |
| LYS5-VAL-8 | 104.11 | 4.61 |
| LEU13-LEU-29 | 118.91 | 3.55 |
| ALA17-ASN-25 | 89.76 | 2.21 |
| GLY21-ASN-25 | 91.77 | 4.63 |
| PRO23-THR-40 | −68.85 | 1.64 |
| ILE26-ILE-38 | 115.25 | 6.87 |
| LEU29-THR-36 | −108.75 | 6.36 |
| CYS30-THR-30 | −67.54 | 2.88 |
| ASN42-ILE-42 | 75.73 | 6.86 |
| PHE46-ALA-59 | 123.39 | 7.39 |
| GLU57-GLN-70 | 109.96 | 3.47 |
| GLY75-HIS-78 | 122.15 | 4.19 |
| LEU98-VAL-103 | −80.03 | 1.94 |
| ILE120-ALA-127 | 108.49 | 3.1 |
| ALA127-MET-134 | 74.1 | 6.81 |
| LYS133-TYR-136 | 105.58 | 2.38 |
| GLY141-ALA-145 | −85.33 | 6.8 |
| ALA145-THR-148 | 114.28 | 2.59 |
| Energy Parameter | TLR-4-Vaccine Complex | Standard Deviation | MHC-I-Vaccine Complex | Standard Deviation | MHC-II-Vaccine Complex | Standard Deviation |
|---|---|---|---|---|---|---|
| MM-GBSA | ||||||
| VDWAALS | −162.00 | 6.70 | −184.87 | 7.36 | −174.32 | 5.66 |
| EEL | −71.36 | 2.67 | −62.00 | 1.07 | −49.52 | 2.08 |
| Delta G gas | −233.36 | 7.25 | −246.87 | 5.41 | −223.84 | 6.43 |
| Delta G solv | 25.63 | 1.25 | 37.87 | 1.96 | 32.10 | 1.24 |
| Delta Total | −258.99 | 8.36 | −284.74 | 3.98 | −255.94 | 7.93 |
| MM-PBSA | ||||||
| VDWAALS | −162.00 | 6.70 | −184.87 | 7.36 | −174.32 | 5.66 |
| EEL | −71.36 | 2.67 | −62.00 | 1.07 | −49.52 | 2.08 |
| Delta G gas | −233.36 | 7.25 | −246.87 | 5.41 | −223.84 | 6.43 |
| Delta G solv | 27.57 | 0.65 | 33.10 | 2.08 | 37.02 | 3.01 |
| Delta Total | −260.93 | 7.64 | −279.97 | 5.37 | −260.86 | 9.31 |
| Vaccine-Complexes | Interactive Residues |
|---|---|
| Vaccine-MHC-I | Ala31, Arg88, Asn65, Asn35, Ile45, Lys84, Glu32, Phe69, Met1, Tyr48, His34, Lys12, Lys3,Tyr29ser4 7, Pro140, Glu480 |
| Vaccine-MHC-II | Asp28, Asp91, Arg97, Ala16, Phe46, Lys44, Tyr39, His115, Tyr97, Gly121, Lys83, Gln37, Thr22, Tyr18, Lys64, Lys3, Hhis34, Glu34, Thr27, Glu32 |
| Vaccine-TLR-4 | Asn526, Ala479, Asp379, Arg382, Lys477, Tyr451, Gln430, Ser381, Lys158, Lys420, Val338, Glu336, His334, Lys109, Lys477 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albekairi, T.H.; Alshammari, A.; Alharbi, M.; Alshammary, A.F.; Tahir ul Qamar, M.; Ullah, A.; Irfan, M.; Ahmad, S. Designing of a Novel Multi-Antigenic Epitope-Based Vaccine against E. hormaechei: An Intergraded Reverse Vaccinology and Immunoinformatics Approach. Vaccines 2022, 10, 665. https://doi.org/10.3390/vaccines10050665
Albekairi TH, Alshammari A, Alharbi M, Alshammary AF, Tahir ul Qamar M, Ullah A, Irfan M, Ahmad S. Designing of a Novel Multi-Antigenic Epitope-Based Vaccine against E. hormaechei: An Intergraded Reverse Vaccinology and Immunoinformatics Approach. Vaccines. 2022; 10(5):665. https://doi.org/10.3390/vaccines10050665
Chicago/Turabian StyleAlbekairi, Thamer H., Abdulrahman Alshammari, Metab Alharbi, Amal F. Alshammary, Muhammad Tahir ul Qamar, Asad Ullah, Muhammad Irfan, and Sajjad Ahmad. 2022. "Designing of a Novel Multi-Antigenic Epitope-Based Vaccine against E. hormaechei: An Intergraded Reverse Vaccinology and Immunoinformatics Approach" Vaccines 10, no. 5: 665. https://doi.org/10.3390/vaccines10050665
APA StyleAlbekairi, T. H., Alshammari, A., Alharbi, M., Alshammary, A. F., Tahir ul Qamar, M., Ullah, A., Irfan, M., & Ahmad, S. (2022). Designing of a Novel Multi-Antigenic Epitope-Based Vaccine against E. hormaechei: An Intergraded Reverse Vaccinology and Immunoinformatics Approach. Vaccines, 10(5), 665. https://doi.org/10.3390/vaccines10050665