
In Silico Designed Multi-Epitope Immunogen “Tpme-VAC/LGCM-2022” May Induce Both Cellular and Humoral Immunity against Treponema pallidum Infection


 , ,
, ,  ,
,  , , ,
, , ,  ,
,  and
and 
Abstract
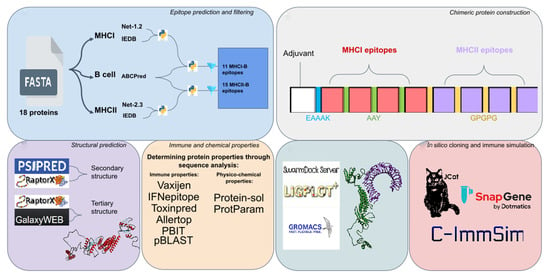
:
1. Introduction
2. Materials and Methods
2.1. Selection of Target Antigenic Proteins
2.2. Prediction of MHC-I Allele Binding CTL Epitopes
2.3. Prediction of MHC-II Allele Binding HTL Epitopes
2.4. Prediction of B-Cell Epitopes
2.5. Filtering Best Epitopes from Each Protein
2.6. Construction of Multi-Epitope Immunogen Sequence
2.7. Prediction of Antigenicity, IFN-γ Induction, Toxicity, and Allergenicity of the Multi-Epitope Immunogen
2.8. Physico-Chemical Properties and Host and Microbiota Homology Analyses
2.9. Secondary Structure Prediction
2.10. Tertiary Structure and Refinement
2.11. Prediction of Conformational B Cell Epitopes
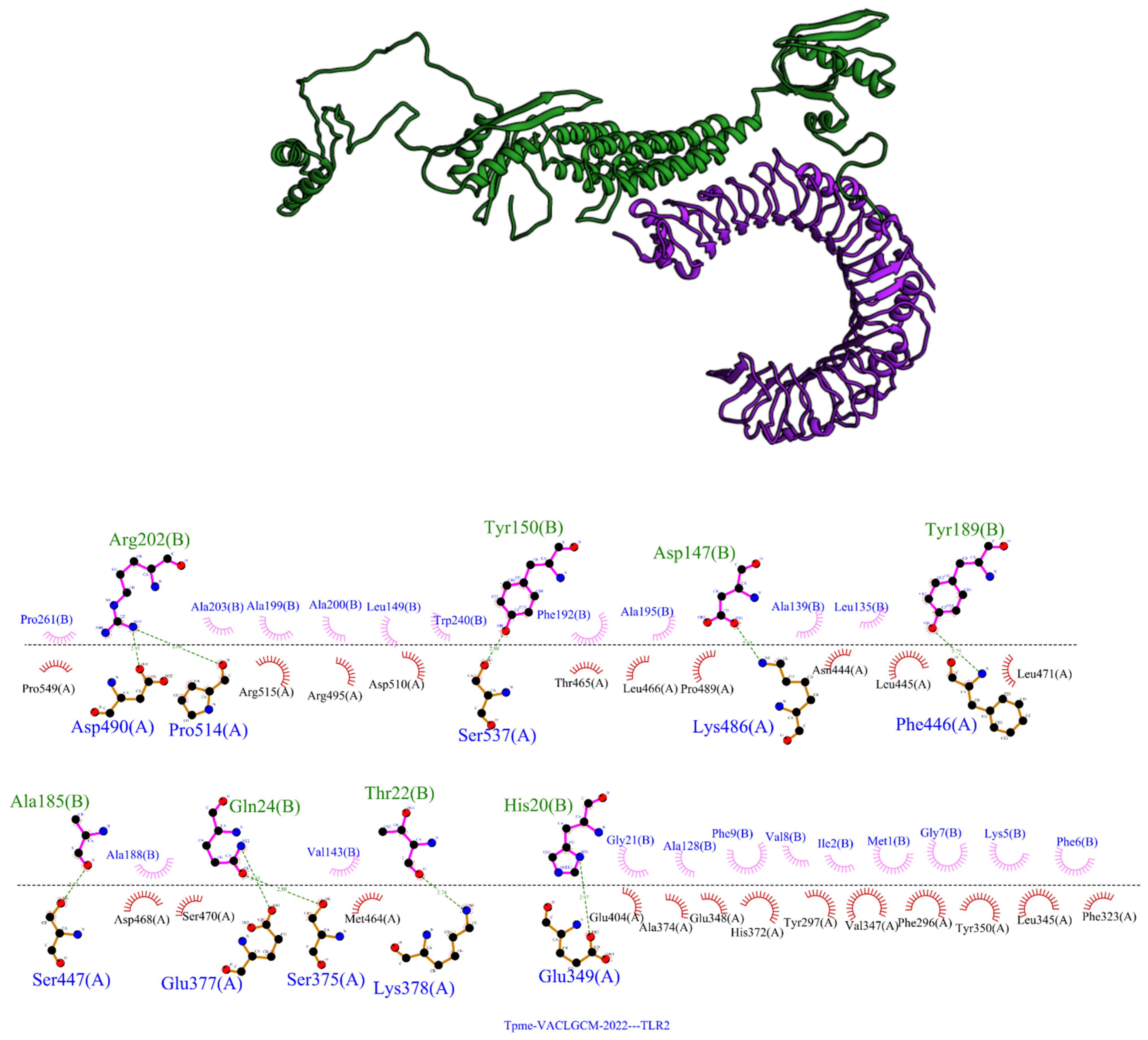
2.12. Molecular Docking between the Chimeric Protein and the TLR-2 Recepto
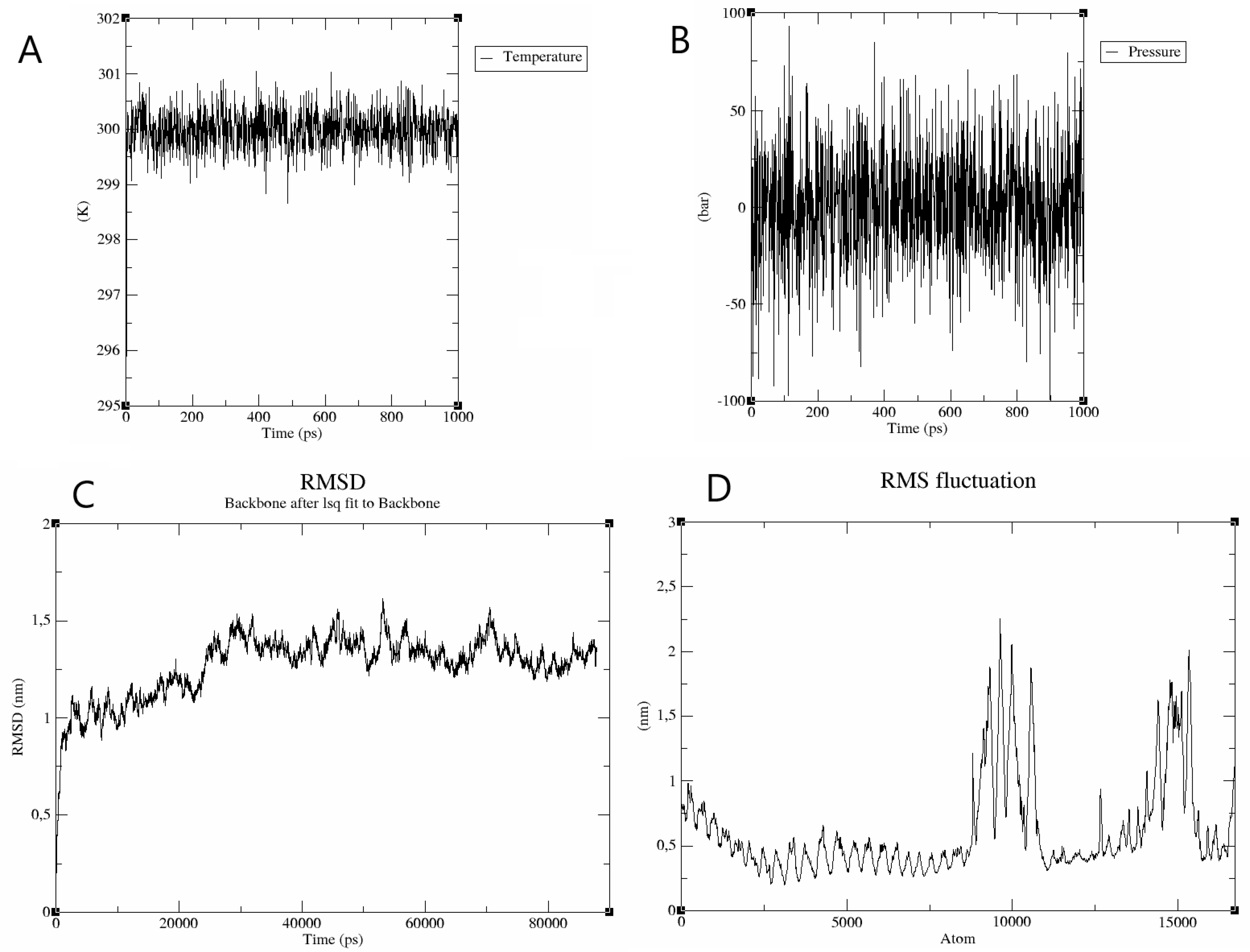
2.13. Molecular Dynamics Simulation of the Receptor-Ligand Complex
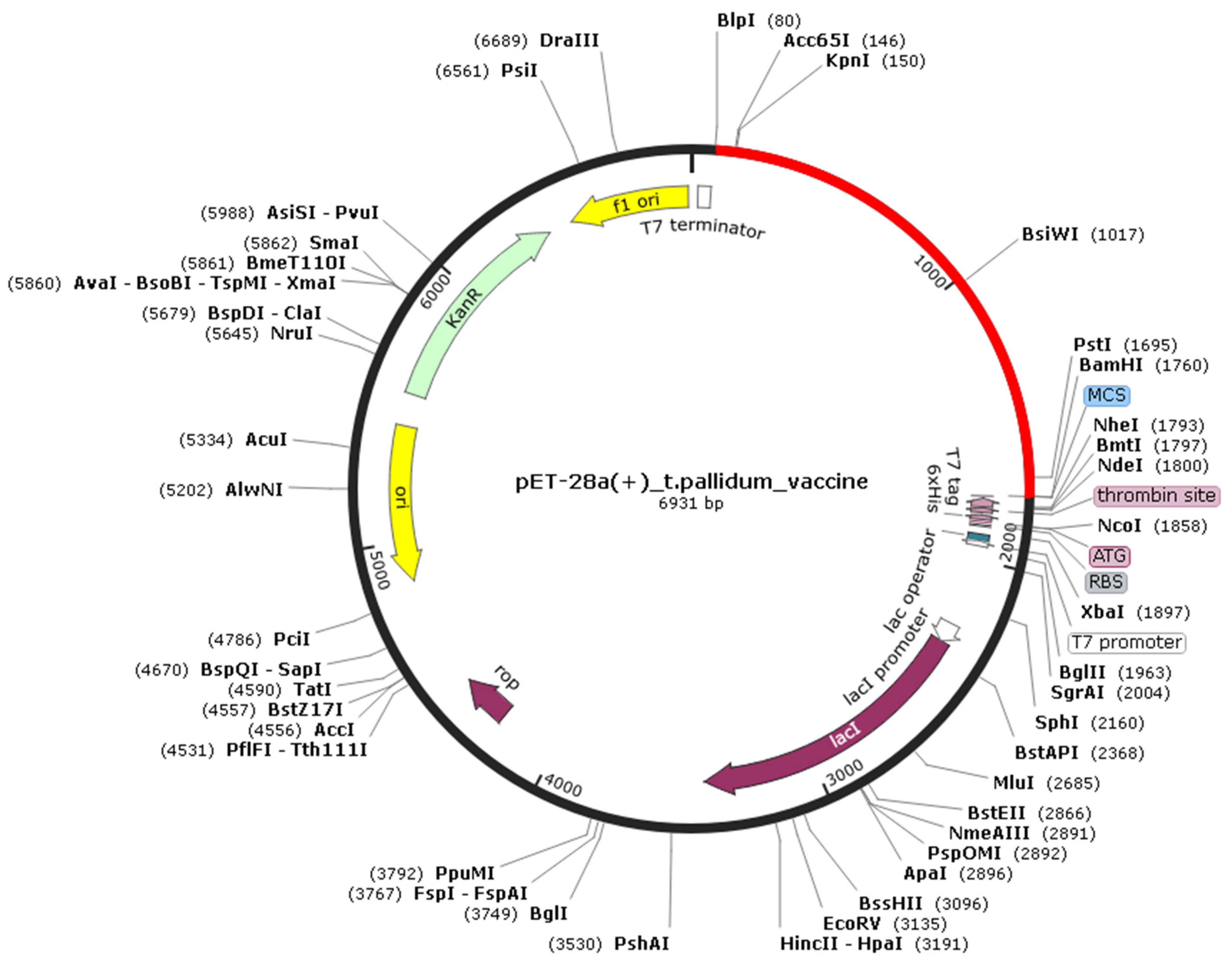
2.14. In Silico Cloning
2.15. Immune Simulation of Multi-Epitope Immunogen
3. Results
3.1. Predicted CTL Epitopes
3.2. Predicted HTL and B-Cell Epitopes
3.3. Overlapping Epitopes for Both Humoral and Cellular Responses
3.4. Constructed Multi-Epitope Vaccine Sequence (Tpme-VAC/LGCM-2022), and Host and Microbiota Homology
3.5. Secondary and Tertiary Structural Properties of Tpme-VAC/LGCM-2022
3.6. Antigenicity, IFN-γ Production, and Conformational B-Cell Epitopes in Tpme-VAC/LGCM-2022
3.7. Physico-Chemical Properties, Toxicity, and Allergenicity of Tpme-VAC/LGCM-2022
3.8. Tpme-VAC/LGCM-2022docks with the TLR2 Receptor
3.9. Tpme-VAC/LGCM-2022-TLR2 Complex Is Stable in Molecular Dynamics Simulation
3.10. Codon Adaptation and in silico Cloning of Tpme-VAC/LGCM-2022
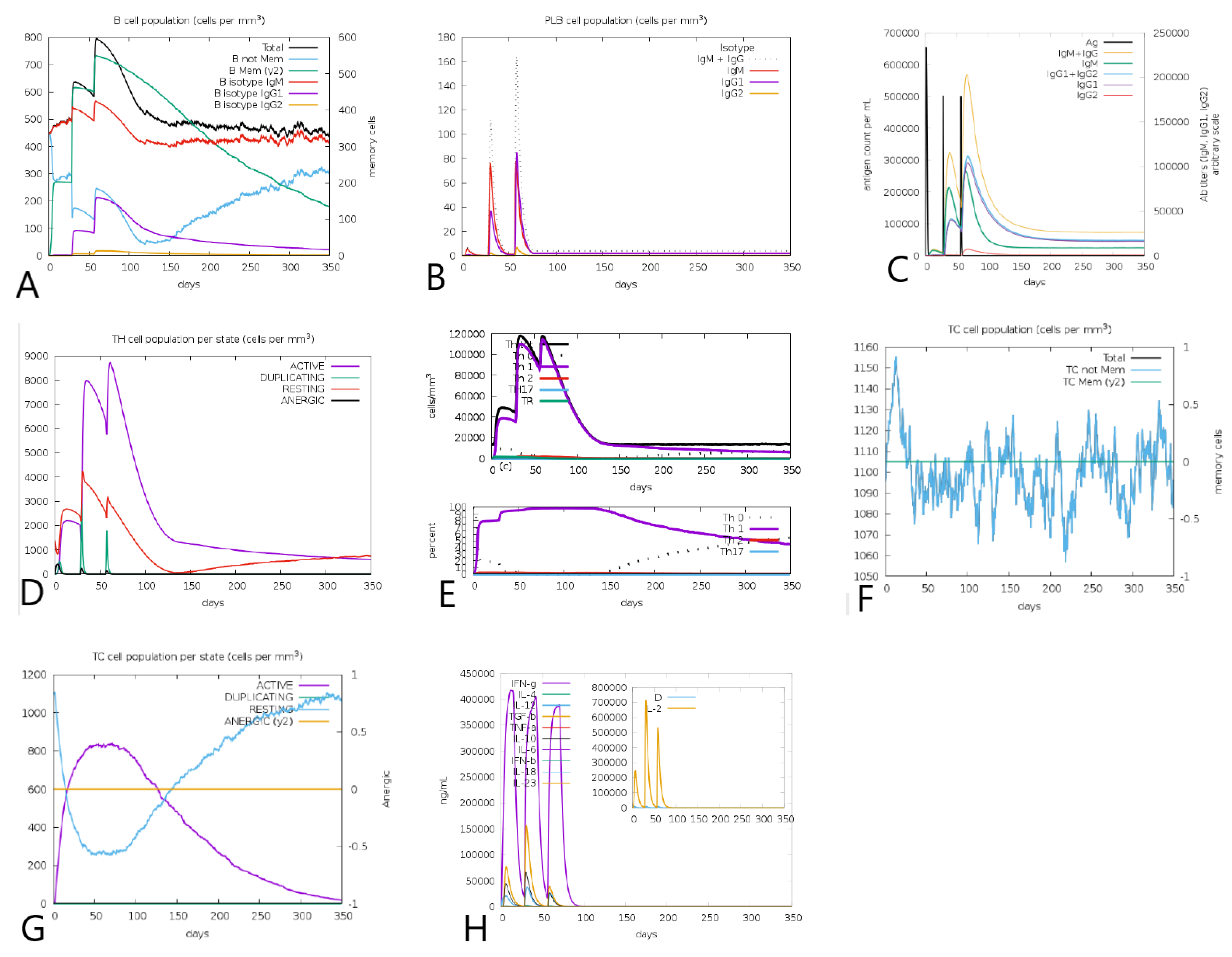
3.11. Tpme-VAC/LGCM-2022 Could Simulate Immune Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Radolf, J.; Deka, R.; Anand, A.; Šmajs, D.; Norgard, M.; Yang, X. Treponema Pallidum, the Syphilis Spirochete: Making a Living as a Stealth Pathogen. Nat. Rev. Microbiol. 2016, 14, 744–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmondson, D.G.; Hu, B.; Norris, S.J. Long-Term In Vitro Culture of the Syphilis Spirochete Treponema Pallidum Subsp. Pallidum. mBio 2018, 9, e01153-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, A.K.; Tiwari, S.; Jamal, S.B.; Oliveira, L.d.C.; Alves, L.G.; Azevedo, V.; Ghosh, P.; Oliveira, C.J.F.; Soares, S.C. The Pan-Genome of Treponema Pallidum Reveals Differences in Genome Plasticity between Subspecies Related to Venereal and Non-Venereal Syphilis. BMC Genom. 2020, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Spiteri, G.; Unemo, M.; Mårdh, O.; Amato-Gauci, A. The Resurgence of Syphilis in High-Income Countries in the 2000s: A Focus on Europe. Epidemiol. Infect. 2019, 147, e134. [Google Scholar] [CrossRef] [Green Version]
- WHO Data on Syphilis. Available online: https://www.who.int/data/gho/data/themes/topics/topic-details/GHO/data-on-syphilis (accessed on 21 July 2021).
- Li, Y.; Li, J.; Hu, W.; Luo, H.; Zhou, J.; Li, C.; Chen, C. Gene Subtype Analysis of Treponema Pallidum for Drug Resistance to Azithromycin. Exp. Ther. Med. 2018, 16, 1009–1013. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.; Otero, C.E.; Li, K.; Valencia, S.M.; Nelson, A.N.; Permar, S.R. Vaccines for Perinatal and Congenital Infections—How Close Are We? Front. Pediatrics 2020, 8, 569. [Google Scholar] [CrossRef]
- Hook, E.W. Syphilis. Lancet 2017, 389, 1550–1557. [Google Scholar] [CrossRef]
- Cameron, C.E.; Lukehart, S.A. Current Status of Syphilis Vaccine Development: Need, Challenges, Prospects. Vaccine 2014, 32, 1602. [Google Scholar] [CrossRef] [Green Version]
- Lithgow, K.V.; Cameron, C.E. Vaccine Development for Syphilis. Expert Rev. Vaccines 2016, 16, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, V.; Rungta, T.; Goyal, K.; Singh, M.P. Designing a Multi-Epitope Based Vaccine to Combat Kaposi Sarcoma Utilizing Immunoinformatics Approach. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Palatnik-de-Sousa, C.B.; Soares, I.d.S.; Rosa, D.S. Editorial: Epitope Discovery and Synthetic Vaccine Design. Front. Immunol. 2018, 9, 826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, D.D. In Silico Vaccine Design: A Tutorial in Immunoinformatics. Healthc. Anal. 2022, 2, 100044. [Google Scholar] [CrossRef]
- Jaiswal, A.; Tiwari, S.; Jamal, S.; Barh, D.; Azevedo, V.; Soares, S. An In Silico Identification of Common Putative Vaccine Candidates against Treponema Pallidum: A Reverse Vaccinology and Subtractive Genomics Based Approach. Int. J. Mol. Sci. 2017, 18, 402. [Google Scholar] [CrossRef]
- Haynes, A.M.; Godornes, C.; Ke, W.; Giacani, L. Evaluation of the Protective Ability of the Treponema Pallidum Subsp. Pallidum Tp0126 OmpW Homolog in the Rabbit Model of Syphilis. Infect. Immun. 2019, 87, e00323-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lithgow, K.V.; Hof, R.; Wetherell, C.; Phillips, D.; Houston, S.; Cameron, C.E. A Defined Syphilis Vaccine Candidate Inhibits Dissemination of Treponema Pallidum Subspecies Pallidum. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, C.; Lukehart, S.; van Voorhis, W. Protection against Syphilis Correlates with Specificity of Antibodies to the Variable Regions of Treponema Pallidum Repeat Protein K. Infect. Immun. 2003, 71, 5605–5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, S.; Nielsen, M.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource Program 2003-2018: Reflections and Outlook. Immunogenetics 2020, 72, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, J.; Sidney, J.; Chung, J.; Brander, C.; Peters, B.; Sette, A. Functional Classification of Class II Human Leukocyte Antigen (HLA) Molecules Reveals Seven Different Supertypes and a Surprising Degree of Repertoire Sharing across Supertypes. Immunogenetics 2011, 63, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-Scale Validation of Methods for Cytotoxic T-Lymphocyte Epitope Prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.H.; Zhang, G.L.; Tongchusak, S.; Reinherz, E.L.; Brusic, V. Evaluation of MHC-II Peptide Binding Prediction Servers: Applications for Vaccine Research. BMC Bioinform. 2008, 9, S22. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved Methods for Predicting Peptide Binding Affinity to MHC Class II Molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Galanis, K.A.; Nastou, K.C.; Papandreou, N.C.; Petichakis, G.N.; Pigis, D.G.; Iconomidou, V.A. Linear B-Cell Epitope Prediction for In Silico Vaccine Design: A Performance Review of Methods Available via Command-Line Interface. Int. J. Mol. Sci. 2021, 22, 3210. [Google Scholar] [CrossRef]
- Zhao, W.; Sher, X. Systematically Benchmarking Peptide-MHC Binding Predictors: From Synthetic to Naturally Processed Epitopes. PLoS Comput. Biol. 2018, 14, e1006457. [Google Scholar] [CrossRef] [PubMed]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting Population Coverage of T-Cell Epitope-Based Diagnostics and Vaccines. BMC Bioinform. 2006, 7, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosrati, M.; Hajizade, A.; Nazarian, S.; Amani, J.; Namvar Vansofla, A.; Tarverdizadeh, Y. Designing a Multi-Epitope Vaccine for Cross-Protection against Shigella Spp: An Immunoinformatics and Structural Vaccinology Study. Mol. Immunol. 2019, 116, 106–116. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of Interferon-Gamma Inducing MHC Class-II Binders. Biol. Direct 2013, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Bangov, I.; Flower, D.; Doytchinova, I. AllerTOP v.2—A Server for in Silico Prediction of Allergens. J. Mol. Model 2014, 20, 1–6. [Google Scholar] [CrossRef]
- Walker, J. The Proteomics Protocols Handbook; Springer Science and Business Media LLC: Totowa, NJ, USA, 2005. [Google Scholar] [CrossRef]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein–Sol: A Web Tool for Predicting Protein Solubility from Sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shende, G.; Haldankar, H.; Barai, R.; Bharmal, M.; Shetty, V.; Idicula-Thomas, S. PBIT: Pipeline Builder for Identification of Drug Targets for Infectious Diseases. Bioinformatics 2017, 33, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Jones, D. Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.; Mezulis, S.; Yates, C.; Wass, M.; Sternberg, M. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-Based Protein Structure Modeling Using the RaptorX Web Server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y. Protein Structure and Function Prediction Using I-TASSER. Curr. Protoc. Bioinform. 2015, 52, 5.8.1–5.8.15. [Google Scholar] [CrossRef]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB Server for Protein Structure Prediction and Refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Bui, H.; Li, W.; Fusseder, N.; Bourne, P.; Sette, A.; Peters, B. ElliPro: A New Structure-Based Tool for the Prediction of Antibody Epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [Green Version]
- Marra, C.M.; Sahi, S.K.; Tantalo, L.C.; Ho, E.L.; Dunaway, S.B.; Jones, T.; Hawn, T.R. Toll-like Receptor Polymorphisms Are Associated with Increased Neurosyphilis Risk. Sex Transm. Dis. 2014, 41, 440. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchala, M.; Moal, I.; Chaleil, R.; Fernandez-Recio, J.; Bates, P. SwarmDock: A Server for Flexible Protein-Protein Docking. Bioinformatics 2013, 29, 807–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.; Swindells, M. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.; Jahn, D. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to Its Potential Expression Host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]
- Moyle, P.; Toth, I. Modern Subunit Vaccines: Development, Components, and Research Opportunities. Chem. Med. Chem. 2013, 8, 360–376. [Google Scholar] [CrossRef]
- Jameie, F.; Dalimi, A.; Pirestani, M.; Mohebali, M. Development of a Multi-Epitope Recombinant Protein for the Diagnosis of Human Visceral Leishmaniasis. Iran. J. Parasitol. 2021, 16, 1–10. [Google Scholar] [CrossRef]
- Radolf, J.D.; Kumar, S. The Treponema Pallidum Outer Membrane. Curr. Top. Microbiol. Immunol. 2017, 415, 1–38. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE ID/NAME | MHC | EPITOPE | PERCENTILE RANK | |
|---|---|---|---|---|
| 1 | TP_0049 | I | HLRTFLAAV | 0.12 |
| 2 | II | CPSVCHLRTFLAAVR | 0.9 | |
| 3 | I | SVCGPDFLY | 0.22 | |
| 4 | TP_0323 | I | ASVALFYAY | 0.1 |
| 5 | II | VGMAVAASVALFYAY | 1.1 | |
| 6 | II | IELFSALPYALTVVV | 0.6 | |
| 7 | II | EGLMMFGAFSTATVT | 0.7 | |
| 8 | TP_0335 | I | AAAVTEYAF | 0.14 |
| 9 | II | VLHAAAAVTEYAFVL | 0.8 | |
| 10 | I | AVHALWNAY | 0.05 | |
| 11 | I | HALWNAYAI | 0.21 | |
| 12 | II | VHALWNAYAIAAAAR | 0.25 | |
| 13 | I | TLFAGAAGA | 0.07 | |
| 14 | II | RPAGSATLFAGAAGA | 0.9 | |
| 15 | TP_0430/ntpK | I | AAAAGADAL | 0.59 |
| 16 | II | GRAAAAGADALAETG | 0.25 | |
| 17 | I | GMFGAAAVL | 0.15 | |
| 18 | II | GMFGAAAVLGISAVG | 0.4 | |
| 19 | TP_0435/nlpE | I | YMGAPGAGK | 0.11 |
| 20 | TP_0557 | I | RAVRTLLII | 0.72 |
| 21 | II | KRMWRAVRTLLIICA | 0.5 | |
| 22 | TP_0733 | II | GGGGFHLGYEYFFTK | 0.3 |
| 23 | TP_0972/ftr1 | II | VGVFVAIRFLSVRLP | 0.12 |
| 24 | TP_0326/BamA | II | GIVSFDFFFDAAMVY | 0.12 |
| 25 | II | GQKWTYELYLEILQK | 0.03 | |
| 26 | tprK | II | DYAQARAPAAGAKVS | 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, L.G.R.; Rodrigues, T.C.V.; Jaiswal, A.K.; Santos, R.G.; Kato, R.B.; Barh, D.; Alzahrani, K.J.; Banjer, H.J.; Soares, S.d.C.; Azevedo, V.; et al. In Silico Designed Multi-Epitope Immunogen “Tpme-VAC/LGCM-2022” May Induce Both Cellular and Humoral Immunity against Treponema pallidum Infection. Vaccines 2022, 10, 1019. https://doi.org/10.3390/vaccines10071019
Gomes LGR, Rodrigues TCV, Jaiswal AK, Santos RG, Kato RB, Barh D, Alzahrani KJ, Banjer HJ, Soares SdC, Azevedo V, et al. In Silico Designed Multi-Epitope Immunogen “Tpme-VAC/LGCM-2022” May Induce Both Cellular and Humoral Immunity against Treponema pallidum Infection. Vaccines. 2022; 10(7):1019. https://doi.org/10.3390/vaccines10071019
Chicago/Turabian StyleGomes, Lucas Gabriel Rodrigues, Thaís Cristina Vilela Rodrigues, Arun Kumar Jaiswal, Roselane Gonçalves Santos, Rodrigo Bentes Kato, Debmalya Barh, Khalid J. Alzahrani, Hamsa Jameel Banjer, Siomar de Castro Soares, Vasco Azevedo, and et al. 2022. "In Silico Designed Multi-Epitope Immunogen “Tpme-VAC/LGCM-2022” May Induce Both Cellular and Humoral Immunity against Treponema pallidum Infection" Vaccines 10, no. 7: 1019. https://doi.org/10.3390/vaccines10071019
APA StyleGomes, L. G. R., Rodrigues, T. C. V., Jaiswal, A. K., Santos, R. G., Kato, R. B., Barh, D., Alzahrani, K. J., Banjer, H. J., Soares, S. d. C., Azevedo, V., & Tiwari, S. (2022). In Silico Designed Multi-Epitope Immunogen “Tpme-VAC/LGCM-2022” May Induce Both Cellular and Humoral Immunity against Treponema pallidum Infection. Vaccines, 10(7), 1019. https://doi.org/10.3390/vaccines10071019