Gene Expression Driven by a Strong Viral Promoter in MVA Increases Vaccination Efficiency by Enhancing Antibody Responses and Unmasking CD8+ T Cell Epitopes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1. Ag, Peptides and Media
2.2. Mice
2.3. Plasmid Construction
2.4. Viruses
2.5. Generation of Recombinant Viruses
2.6. Western Blot Analysis
2.7. Bone Marrow Derived DC Preparation, in Vitro Stimulation and Injection
2.8. Flow Cytometric Analysis
2.9. Proliferation Assay
2.10. Staining for Intracellular Cytokines
2.11. In Vitro Ag Presentation Assay
2.12. Immunization Studies
2.13. Detection of Ag Specific Abs
2.14. Detection of IFN-γ-Producing Cells by ELISPOT
2.15. Detection of IFN-γ-Producing CD4+ T Cells by Flow Cytometry
2.16. Statistical Analysis
3. Results
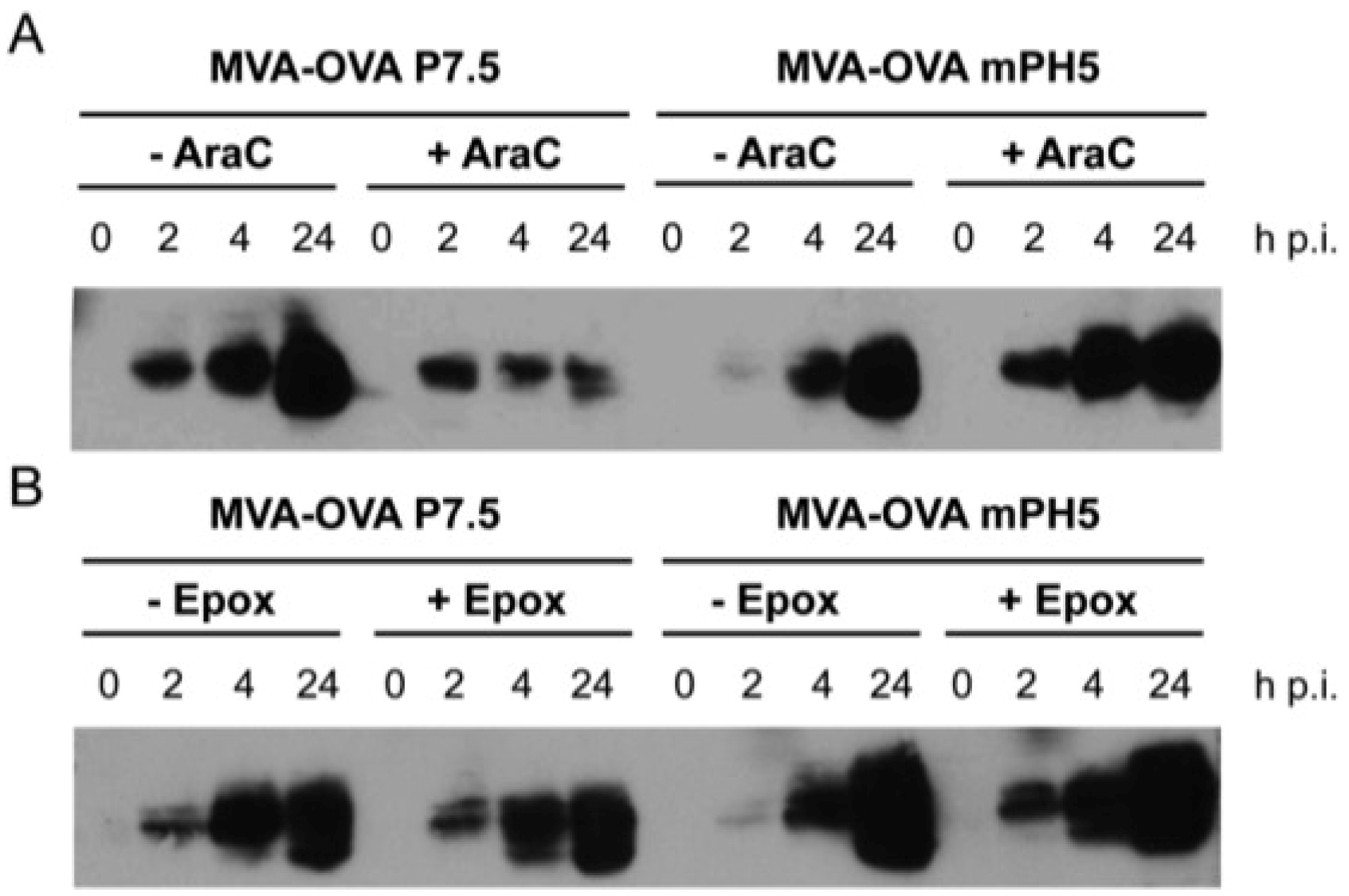
3.1. The Use of Different Promoters Results in Changes in the Profile and Turnover of Proteins Expressed by rMVA in Infected Cells
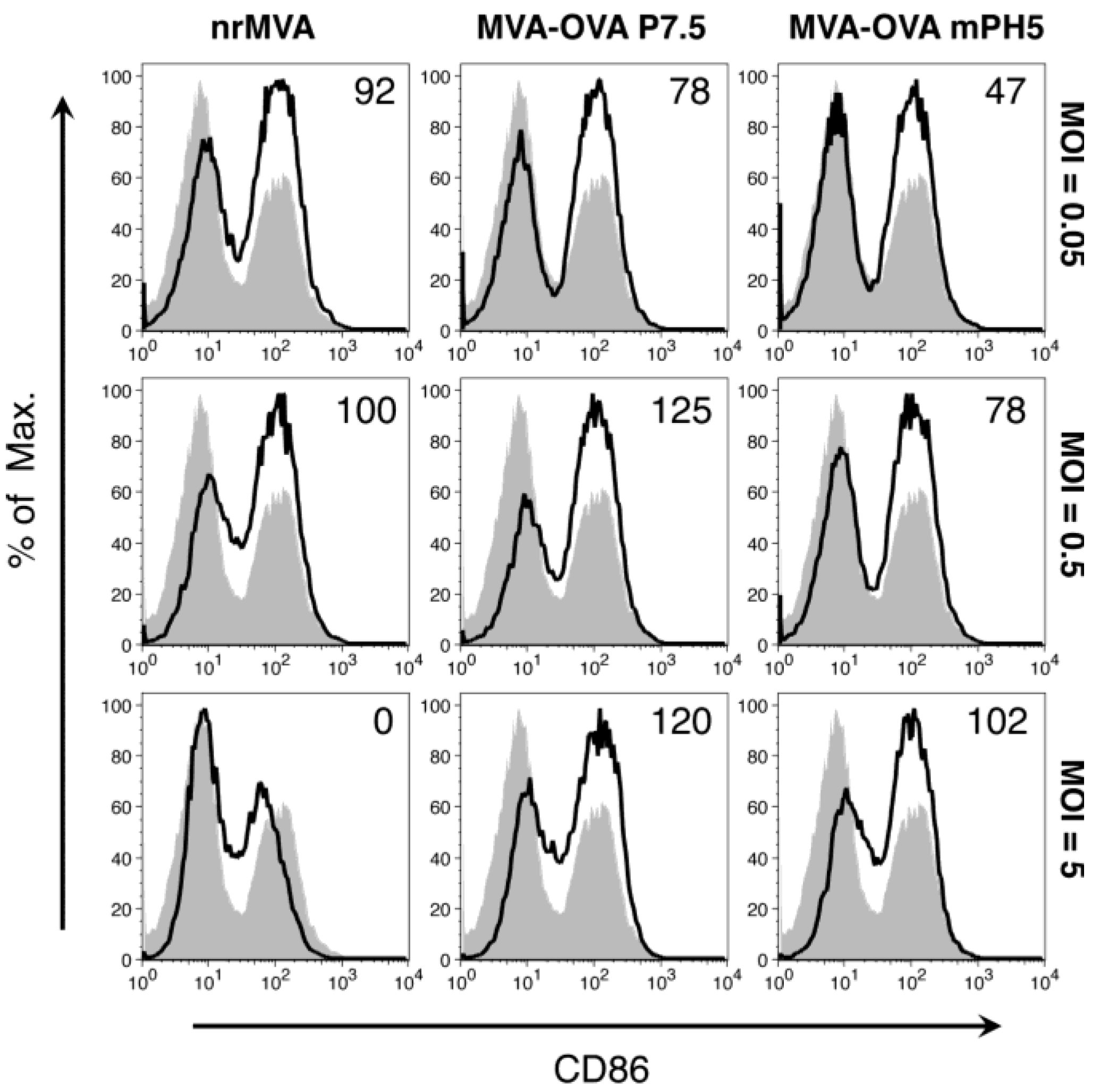
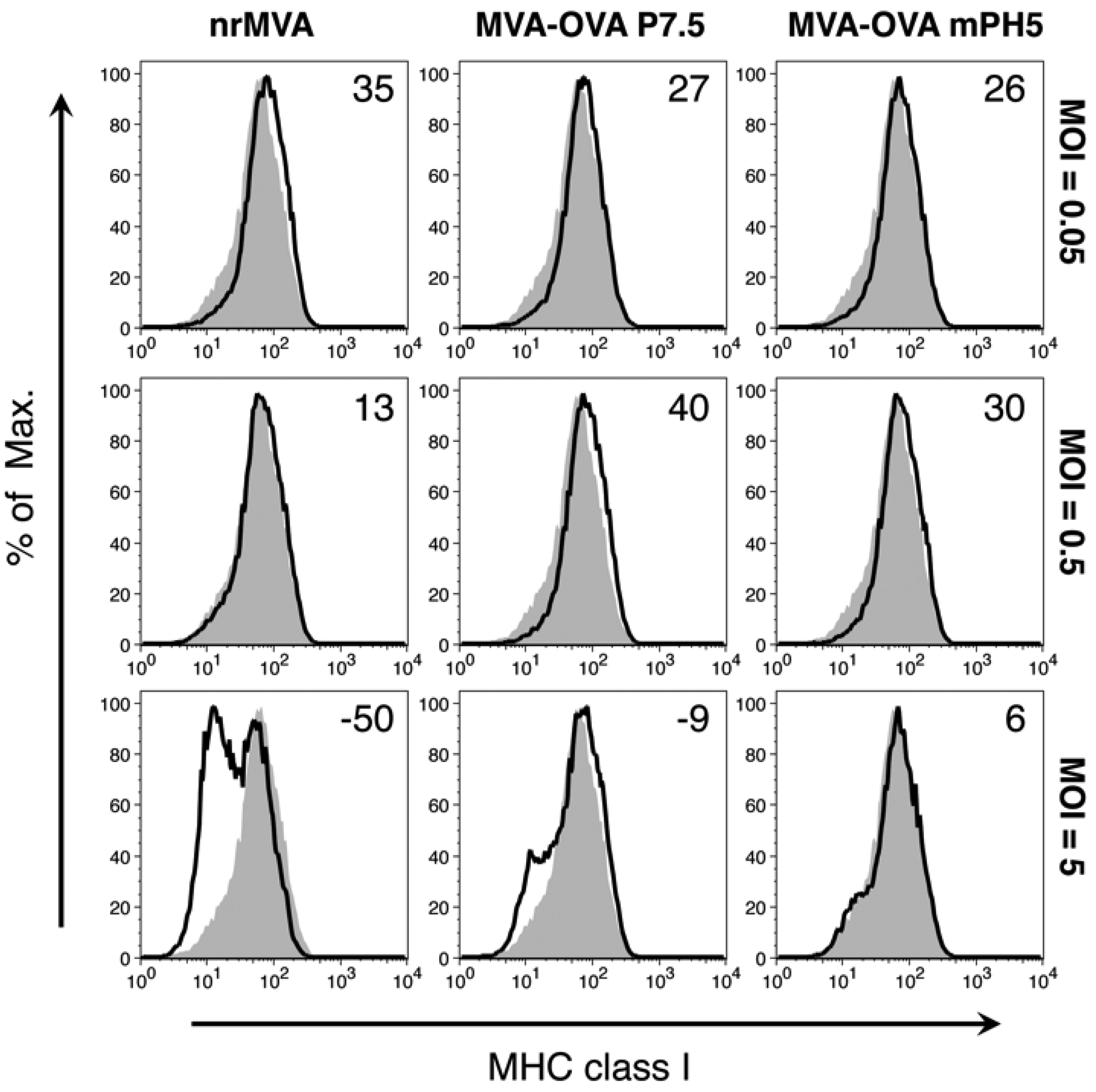
3.2. Activation of Murine DCs Is Primarily Dependent on the MOI of rMVA, but It Is Also Affected by the Promoter Selected To Drive Ag Expression



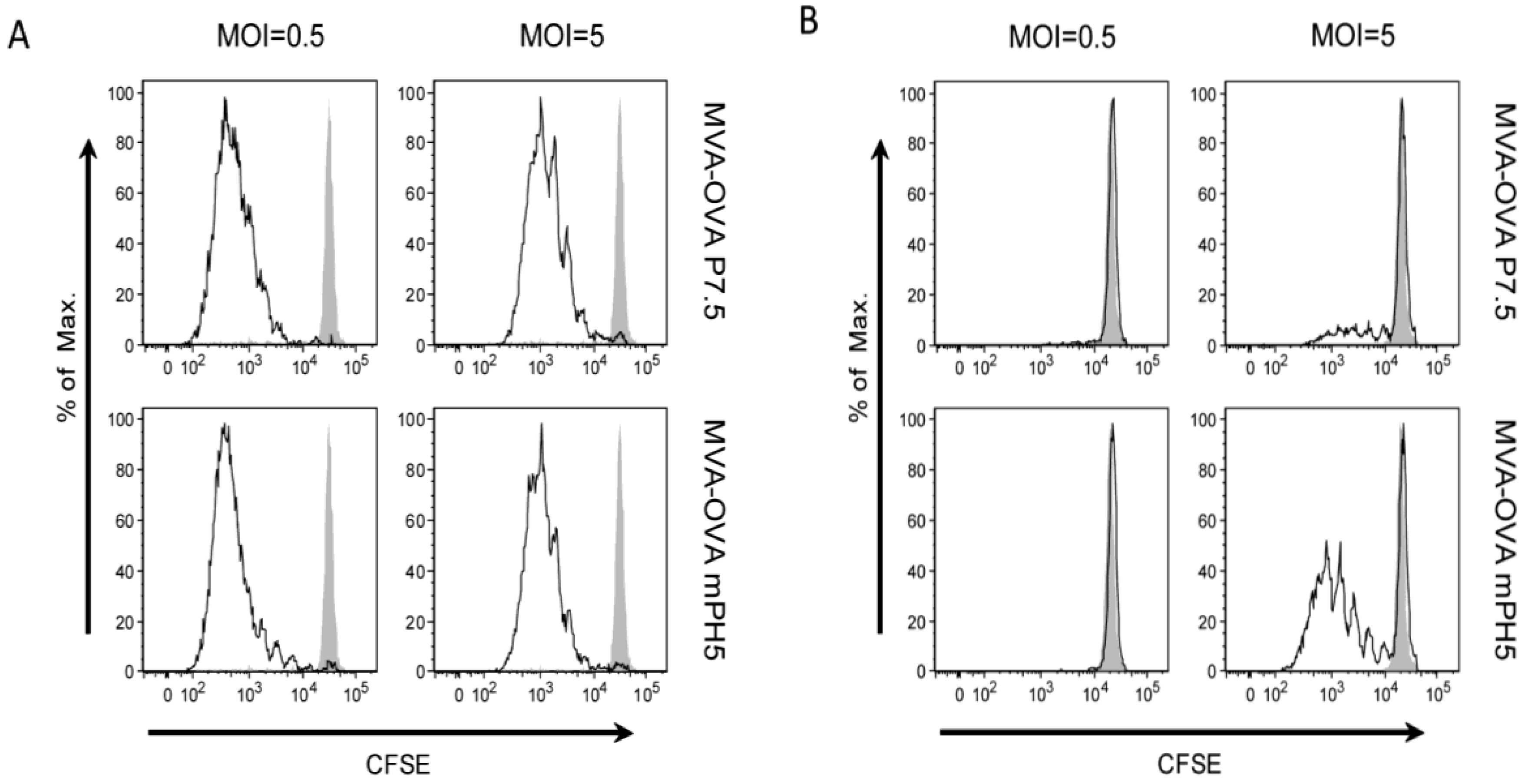
3.3. The Strength of Viral Ag Expression during the Early Phase Shapes Ag Presentation


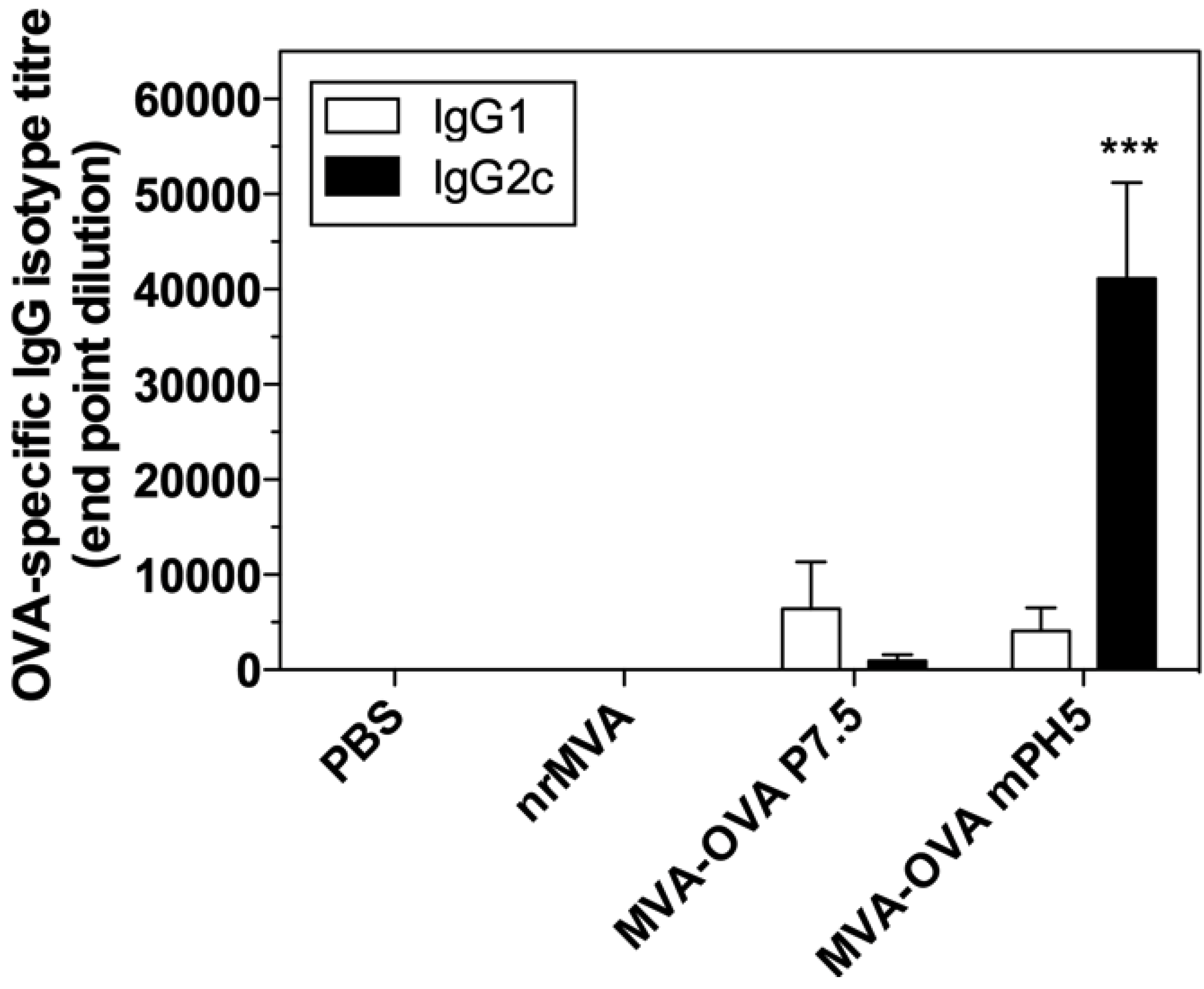
3.4. Immunization with rMVA Expressing OVA Driven by Different Promoters Induced Different Ab Responses

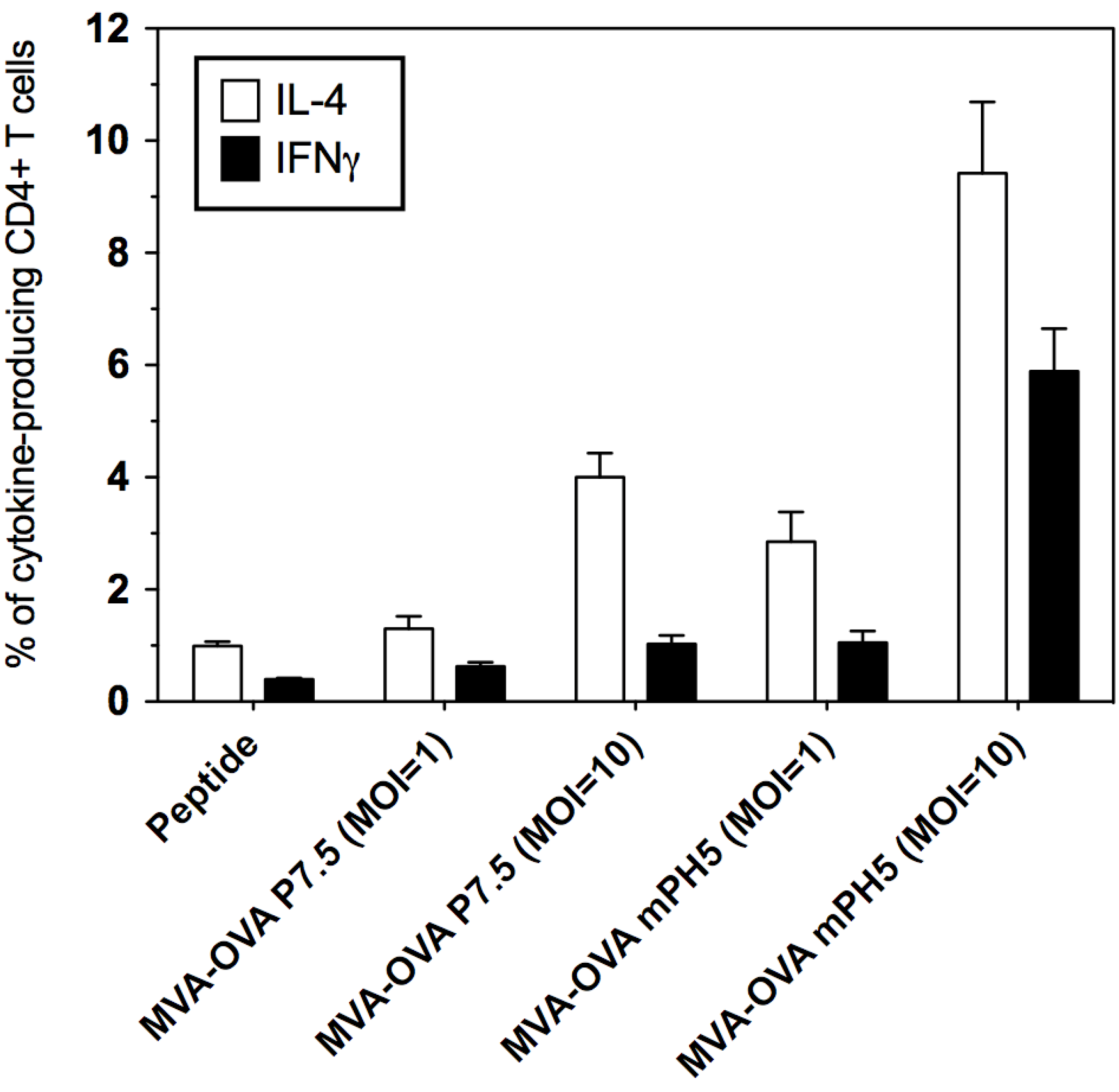
3.5. Immunization with rMVA Expressing OVA Driven by Different Promoters Shape the CD8+ T Cell Immune Response

4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carroll, M.W.; Moss, B. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: Propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 1997, 238, 198–211. [Google Scholar] [CrossRef]
- Drexler, I.; Heller, K.; Wahren, B.; Erfle, V.; Sutter, G. Highly attenuated modified vaccinia virus ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J. Gen. Virol. 1998, 79, 347–352. [Google Scholar]
- Sutter, G.; Moss, B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. USA 1992, 89, 10847–10851. [Google Scholar] [CrossRef]
- Blanchard, T.J.; Alcami, A.; Andrea, P.; Smith, G.L. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: Implications for use as a human vaccine. J. Gen. Virol. 1998, 79, 1159–1167. [Google Scholar]
- Sancho, M.C.; Schleich, S.; Griffiths, G.; Krijnse-Locker, J. The block in assembly of modified vaccinia virus Ankara in hela cells reveals new insights into vaccinia virus morphogenesis. J. Virol. 2002, 76, 8318–8334. [Google Scholar] [CrossRef]
- Mayr, A.; Stickl, H.; Muller, H.K.; Danner, K.; Singer, H. The smallpox vaccination strain MVA: Marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism (author’s transl). Zent. Bakteriol. B 1978, 167, 375–390. [Google Scholar]
- Slifka, M.K. The future of smallpox vaccination: Is MVA the key? Med. Immunol. 2005, 4. [Google Scholar] [CrossRef] [Green Version]
- Stickl, H.; Hochstein-Mintzel, V.; Mayr, A.; Huber, H.C.; Schafer, H.; Holzner, A. MVA vaccination against smallpox: Clinical tests with an attenuated live vaccinia virus strain (MVA). Dtsch. Med. Wochenschr. 1974, 99, 2386–2392. [Google Scholar] [CrossRef]
- Cosma, A.; Nagaraj, R.; Buhler, S.; Hinkula, J.; Busch, D.H.; Sutter, G.; Goebel, F.D.; Erfle, V. Therapeutic vaccination with MVA-HIV-1 nef elicits Nef-specific T-helper cell responses in chronically HIV-1 infected individuals. Vaccine 2003, 22, 21–29. [Google Scholar] [CrossRef]
- Bejon, P.; Mwacharo, J.; Kai, O.K.; Todryk, S.; Keating, S.; Lang, T.; Gilbert, S.C.; Peshu, N.; Marsh, K.; Hill, A.V.; et al. Immunogenicity of the candidate malaria vaccines FP9 and modified vaccinia virus Ankara encoding the pre-erythrocytic antigen ME-TRAP in 1–6 year old children in a malaria endemic area. Vaccine 2006, 24, 4709–4715. [Google Scholar] [CrossRef]
- Burgers, W.A.; Shephard, E.; Monroe, J.E.; Greenhalgh, T.; Binder, A.; Hurter, E.; van Harmelen, J.H.; Williamson, C.; Williamson, A.L. Construction, characterization, and immunogenicity of a multigene modified vaccinia Ankara (MVA) vaccine based on HIV type 1 subtype C. AIDS Res. Hum. Retrovir. 2008, 24, 195–206. [Google Scholar] [CrossRef]
- Dorrell, L.; Williams, P.; Suttill, A.; Brown, D.; Roberts, J.; Conlon, C.; Hanke, T.; McMichael, A. Safety and tolerability of recombinant modified vaccinia virus ankara expressing an HIV-1 gag/multiepitope immunogen (MVA.HIVA) in HIV-1-infected persons receiving combination antiretroviral therapy. Vaccine 2007, 25, 3277–3283. [Google Scholar] [CrossRef]
- Dorrell, L.; Yang, H.; Ondondo, B.; Dong, T.; di Gleria, K.; Suttill, A.; Conlon, C.; Brown, D.; Williams, P.; Bowness, P.; et al. Expansion and diversification of virus-specific T cells following immunization of human immunodeficiency virus type 1 (HIV-1)-infected individuals with a recombinant modified vaccinia virus Ankara/HIV-1 gag vaccine. J. Virol. 2006, 80, 4705–4716. [Google Scholar] [CrossRef]
- Meyer, R.G.; Britten, C.M.; Siepmann, U.; Petzold, B.; Sagban, T.A.; Lehr, H.A.; Weigle, B.; Schmitz, M.; Mateo, L.; Schmidt, B.; et al. A phase I vaccination study with tyrosinase in patients with stage II melanoma using recombinant modified vaccinia virus ankara (MVA-HTYR). Cancer Immunol. Immunother. 2005, 54, 453–467. [Google Scholar] [CrossRef]
- Moorthy, V.S.; Pinder, M.; Reece, W.H.; Watkins, K.; Atabani, S.; Hannan, C.; Bojang, K.; McAdam, K.P.; Schneider, J.; Gilbert, S.; et al. Safety and immunogenicity of DNA/modified vaccinia virus Ankara malaria vaccination in African adults. J. Infect. Dis. 2003, 188, 1239–1244. [Google Scholar] [CrossRef]
- Webster, D.P.; Dunachie, S.; McConkey, S.; Poulton, I.; Moore, A.C.; Walther, M.; Laidlaw, S.M.; Peto, T.; Skinner, M.A.; Gilbert, S.C.; et al. Safety of recombinant fowlpox strain FP9 and modified vaccinia virus Ankara vaccines against liver-stage P. falciparum malaria in non-immune volunteers. Vaccine 2006, 24, 3026–3034. [Google Scholar] [CrossRef]
- Gilbert, S.C. Clinical development of modified vaccinia virus ankara vaccines. Vaccine 2013, 31, 4241–4246. [Google Scholar] [CrossRef]
- Hui, E.P.; Taylor, G.S.; Jia, H.; Ma, B.B.; Chan, S.L.; Ho, R.; Wong, W.L.; Wilson, S.; Johnson, B.F.; Edwards, C.; et al. Phase I trial of recombinant modified vaccinia Ankara encoding Epstein-Barr viral tumor antigens in nasopharyngeal carcinoma patients. Cancer Res. 2013, 73, 1676–1688. [Google Scholar] [CrossRef]
- Hayes, P.; Gilmour, J.; von Lieven, A.; Gill, D.; Clark, L.; Kopycinski, J.; Cheeseman, H.; Chung, A.; Alter, G.; Dally, L.; et al. Safety and immunogenicity of DNA prime and modified vaccinia Ankara virus-HIV subtype c vaccine boost in healthy adults. Clin. Vaccine Immunol. CVI 2013, 20, 397–408. [Google Scholar] [CrossRef]
- Di Nicola, M.; Siena, S.; Bregni, M.; Longoni, P.; Magni, M.; Milanesi, M.; Matteucci, P.; Mortarini, R.; Anichini, A.; Parmiani, G.; et al. Gene transfer into human dendritic antigen-presenting cells by vaccinia virus and adenovirus vectors. Cancer Gene Ther. 1998, 5, 350–356. [Google Scholar]
- Kastenmuller, W.; Drexler, I.; Ludwig, H.; Erfle, V.; Peschel, C.; Bernhard, H.; Sutter, G. Infection of human dendritic cells with recombinant vaccinia virus MVA reveals general persistence of viral early transcription but distinct maturation-dependent cytopathogenicity. Virology 2006, 350, 276–288. [Google Scholar] [CrossRef]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Engelmayer, J.; Larsson, M.; Subklewe, M.; Chahroudi, A.; Cox, W.I.; Steinman, R.M.; Bhardwaj, N. Vaccinia virus inhibits the maturation of human dendritic cells: A novel mechanism of immune evasion. J. Immunol. 1999, 163, 6762–6768. [Google Scholar]
- Gasteiger, G.; Kastenmuller, W.; Ljapoci, R.; Sutter, G.; Drexler, I. Cross-priming of cytotoxic T cells dictates antigen requisites for modified vaccinia virus Ankara vector vaccines. J. Virol. 2007, 81, 11925–11936. [Google Scholar]
- Liu, L.; Chavan, R.; Feinberg, M.B. Dendritic cells are preferentially targeted among hematolymphocytes by modified vaccinia virus ankara and play a key role in the induction of virus-specific T cell responses in vivo. BMC Immunol. 2008, 9. [Google Scholar] [CrossRef]
- Shortman, K. Dendritic cells: Multiple subtypes, multiple origins, multiple functions. Immunol. Cell Biol. 2000, 78, 161–165. [Google Scholar] [CrossRef]
- Kastenmuller, W.; Gasteiger, G.; Gronau, J.H.; Baier, R.; Ljapoci, R.; Busch, D.H.; Drexler, I. Cross-competition of CD8+ T cells shapes the immunodominance hierarchy during boost vaccination. J. Exp. Med. 2007, 204, 2187–2198. [Google Scholar] [CrossRef]
- Wyatt, L.S.; Shors, S.T.; Murphy, B.R.; Moss, B. Development of a replication-deficient recombinant vaccinia virus vaccine effective against parainfluenza virus 3 infection in an animal model. Vaccine 1996, 14, 1451–1458. [Google Scholar] [CrossRef]
- Norder, M.; Becker, P.D.; Drexler, I.; Link, C.; Erfle, V.; Guzman, C.A. Modified vaccinia virus Ankara exerts potent immune modulatory activities in a murine model. PLoS One 2010, 5, e11400. [Google Scholar]
- Catipovic, B.; dal Porto, J.; Mage, M.; Johansen, T.E.; Schneck, J.P. Major histocompatibility complex conformational epitopes are peptide specific. J. Exp. Med. 1992, 176, 1611–1618. [Google Scholar] [CrossRef]
- Lipford, G.B.; Hoffman, M.; Wagner, H.; Heeg, K. Primary in vivo responses to ovalbumin. Probing the predictive value of the Kb binding motif. J. Immunol. 1993, 150, 1212–1222. [Google Scholar]
- Mo, A.X.; van Lelyveld, S.F.; Craiu, A.; Rock, K.L. Sequences that flank subdominant and cryptic epitopes influence the proteolytic generation of MHC class I-presented peptides. J. Immunol. 2000, 164, 4003–4010. [Google Scholar] [CrossRef]
- Sadegh-Nasseri, S.; McConnell, H.M. A kinetic intermediate in the reaction of an antigenic peptide and I-Ek. Nature 1989, 337, 274–276. [Google Scholar] [CrossRef]
- Clarke, S.R.; Barnden, M.; Kurts, C.; Carbone, F.R.; Miller, J.F.; Heath, W.R. Characterization of the ovalbumin-specific TCR transgenic line OT-I: Mhc elements for positive and negative selection. Immunol. Cell Biol. 2000, 78, 110–117. [Google Scholar] [CrossRef]
- Kelly, J.M.; Sterry, S.J.; Cose, S.; Turner, S.J.; Fecondo, J.; Rodda, S.; Fink, P.J.; Carbone, F.R. Identification of conserved T cell receptor CDR 3 residues contacting known exposed peptide side chains from a major histocompatibility complex class I-bound determinant. Eur. J. Immunol. 1993, 23, 3318–3326. [Google Scholar] [CrossRef]
- Robertson, J.M.; Jensen, P.E.; Evavold, B.D. Do11.10 and OT-II T cells recognize a C-terminal ovalbumin 323–339 epitope. J. Immunol. 2000, 164, 4706–4712. [Google Scholar] [CrossRef]
- Staib, C.; Drexler, I.; Sutter, G. Construction and isolation of recombinant MVA. Methods Mol. Biol. 2004, 269, 77–100. [Google Scholar]
- Staib, C.; Lowel, M.; Erfle, V.; Sutter, G. Improved host range selection for recombinant modified vaccinia virus ankara. Biotechniques 2003, 34, 694–696, 698, 700. [Google Scholar]
- Wyatt, L.S.; Carroll, M.W.; Czerny, C.P.; Merchlinsky, M.; Sisler, J.R.; Moss, B. Marker rescue of the host range restriction defects of modified vaccinia virus Ankara. Virology 1998, 251, 334–342. [Google Scholar] [CrossRef]
- Cochran, M.A.; Puckett, C.; Moss, B. In vitro mutagenesis of the promoter region for a vaccinia virus gene: Evidence for tandem early and late regulatory signals. J. Virol. 1985, 54, 30–37. [Google Scholar]
- Inaba, K.; Inaba, M.; Romani, N.; Aya, H.; Deguchi, M.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992, 176, 1693–1702. [Google Scholar] [CrossRef]
- Lutz, M.B.; Kukutsch, N.; Ogilvie, A.L.; Rossner, S.; Koch, F.; Romani, N.; Schuler, G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 1999, 223, 77–92. [Google Scholar] [CrossRef]
- FlowJo software, version 9.6.4. TreeStar Inc.: Ashland, OR, USA, 2013.
- Becker, P.D.; Norder, M.; Guzman, C.A.; Grinstein, S. Immune modulator adamantylamide dipeptide stimulates efficient major histocompatibility complex class I-restricted responses in mice. Clin. Vaccine Immunol. CVI 2007, 14, 538–543. [Google Scholar] [CrossRef]
- GraphPad Prism software for Windows, version 5.02. GraphPad Software, Inc.: La Jolla, CA, USA, 2013.
- Chakrabarti, S.; Sisler, J.R.; Moss, B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques 1997, 23, 1094–1097. [Google Scholar]
- Satheshkumar, P.S.; Anton, L.C.; Sanz, P.; Moss, B. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 2009, 83, 2469–2479. [Google Scholar] [CrossRef]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef]
- Princiotta, M.F.; Finzi, D.; Qian, S.B.; Gibbs, J.; Schuchmann, S.; Buttgereit, F.; Bennink, J.R.; Yewdell, J.W. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 2003, 18, 343–354. [Google Scholar] [CrossRef]
- Zhang, X.; Cassis-Ghavami, F.; Eller, M.; Currier, J.; Slike, B.M.; Chen, X.; Tartaglia, J.; Marovich, M.; Spearman, P. Direct comparison of antigen production and induction of apoptosis by canarypox virus- and modified vaccinia virus ankara-human immunodeficiency virus vaccine vectors. J. Virol. 2007, 81, 7022–7033. [Google Scholar] [CrossRef]
- Smith, G.L.; Moss, B. Infectious poxvirus vectors have capacity for at least 25,000 base pairs of foreign DNA. Gene 1983, 25, 21–28. [Google Scholar] [CrossRef]
- Schlosser, E.; Otero, C.; Wuensch, C.; Kessler, B.; Edelmann, M.; Brunisholz, R.; Drexler, I.; Legler, D.F.; Groettrup, M. A novel cytosolic class I antigen-processing pathway for endoplasmic-reticulum-targeted proteins. EMBO Rep. 2007, 8, 945–951. [Google Scholar] [CrossRef]
- Pascutti, M.F.; Rodriguez, A.M.; Falivene, J.; Giavedoni, L.; Drexler, I.; Gherardi, M.M. Interplay between modified vaccinia virus ankara and dendritic cells: Phenotypic and functional maturation of bystander dendritic cells. J. Virol. 2011, 85, 5532–5545. [Google Scholar] [CrossRef]
- Brandler, S.; Lepelley, A.; Desdouits, M.; Guivel-Benhassine, F.; Ceccaldi, P.E.; Levy, Y.; Schwartz, O.; Moris, A. Preclinical studies of a modified vaccinia virus Ankara-based HIV candidate vaccine: Antigen presentation and antiviral effect. J. Virol. 2010, 84, 5314–5328. [Google Scholar] [CrossRef]
- Climent, N.; Guerra, S.; Garcia, F.; Rovira, C.; Miralles, L.; Gomez, C.E.; Pique, N.; Gil, C.; Gatell, J.M.; Esteban, M.; et al. Dendritic cells exposed to MVA-based HIV-1 vaccine induce highly functional HIV-1-specific CD8+ T cell responses in HIV-1-infected individuals. PLoS One 2011, 6, e19644. [Google Scholar] [CrossRef]
- Dolan, B.P.; Sharma, A.A.; Gibbs, J.S.; Cunningham, T.J.; Bennink, J.R.; Yewdell, J.W. MHC class I antigen processing distinguishes endogenous antigens based on their translation from cellular vs. Viral mrna. Proc. Natl. Acad. Sci. USA 2012, 109, 7025–7030. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Becker, P.D.; Nörder, M.; Weissmann, S.; Ljapoci, R.; Erfle, V.; Drexler, I.; Guzmán, C.A. Gene Expression Driven by a Strong Viral Promoter in MVA Increases Vaccination Efficiency by Enhancing Antibody Responses and Unmasking CD8+ T Cell Epitopes. Vaccines 2014, 2, 581-600. https://doi.org/10.3390/vaccines2030581
Becker PD, Nörder M, Weissmann S, Ljapoci R, Erfle V, Drexler I, Guzmán CA. Gene Expression Driven by a Strong Viral Promoter in MVA Increases Vaccination Efficiency by Enhancing Antibody Responses and Unmasking CD8+ T Cell Epitopes. Vaccines. 2014; 2(3):581-600. https://doi.org/10.3390/vaccines2030581
Chicago/Turabian StyleBecker, Pablo D., Miriam Nörder, Sebastian Weissmann, Ronny Ljapoci, Volker Erfle, Ingo Drexler, and Carlos A. Guzmán. 2014. "Gene Expression Driven by a Strong Viral Promoter in MVA Increases Vaccination Efficiency by Enhancing Antibody Responses and Unmasking CD8+ T Cell Epitopes" Vaccines 2, no. 3: 581-600. https://doi.org/10.3390/vaccines2030581
APA StyleBecker, P. D., Nörder, M., Weissmann, S., Ljapoci, R., Erfle, V., Drexler, I., & Guzmán, C. A. (2014). Gene Expression Driven by a Strong Viral Promoter in MVA Increases Vaccination Efficiency by Enhancing Antibody Responses and Unmasking CD8+ T Cell Epitopes. Vaccines, 2(3), 581-600. https://doi.org/10.3390/vaccines2030581