
A Nanoparticle Based Sp17 Peptide Vaccine Exposes New Immuno-Dominant and Species Cross-reactive B Cell Epitopes

Abstract
:1. Introduction
2. Material and Methods
2.1. Peptides and Recombinant Sp17 Protein

2.2. Vaccine Formulations
2.3. Immunisations, Immunogenicity and Immune-Therapy
2.4. ELISPOT and ELISA Assays
2.5. Statistical Analysis
3. Results and Discussion
3.1. PSNPs-hSp17 Peptides Formulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conjugation conditions | hSp171–32-PSNPs | hSp1723–54-PSNPs | hSp1745–76-PSNPs | hSp1767–98-PSNPs | hSp1789–120-PSNPs | hSp17111–142-PSNPs |
|---|---|---|---|---|---|---|
| Conjugation buffer * | MES | MES | PBS | PBS | PBS | MES |
| Conjugation pH # | 5.5 | 5.5 | 7.5 | 6.5 | 6 | 6.2 |
| Conjugation efficiency § (%) | 94.5 ± 2.2 | 87.9 ± 2.9 | 95.2 ± 1.2 | 77.1 ± 3.7 | 69.8 ± 3.6 | 95.8 ± 1.1 |
| Size (nm) | 72.3 ± 4.8 | 52.5 ± 0.2 | 49.1 ± 1.4 | 48.8 ± 0.8 | 45.9 ± 0.5 | 55.9 ± 0.1 |
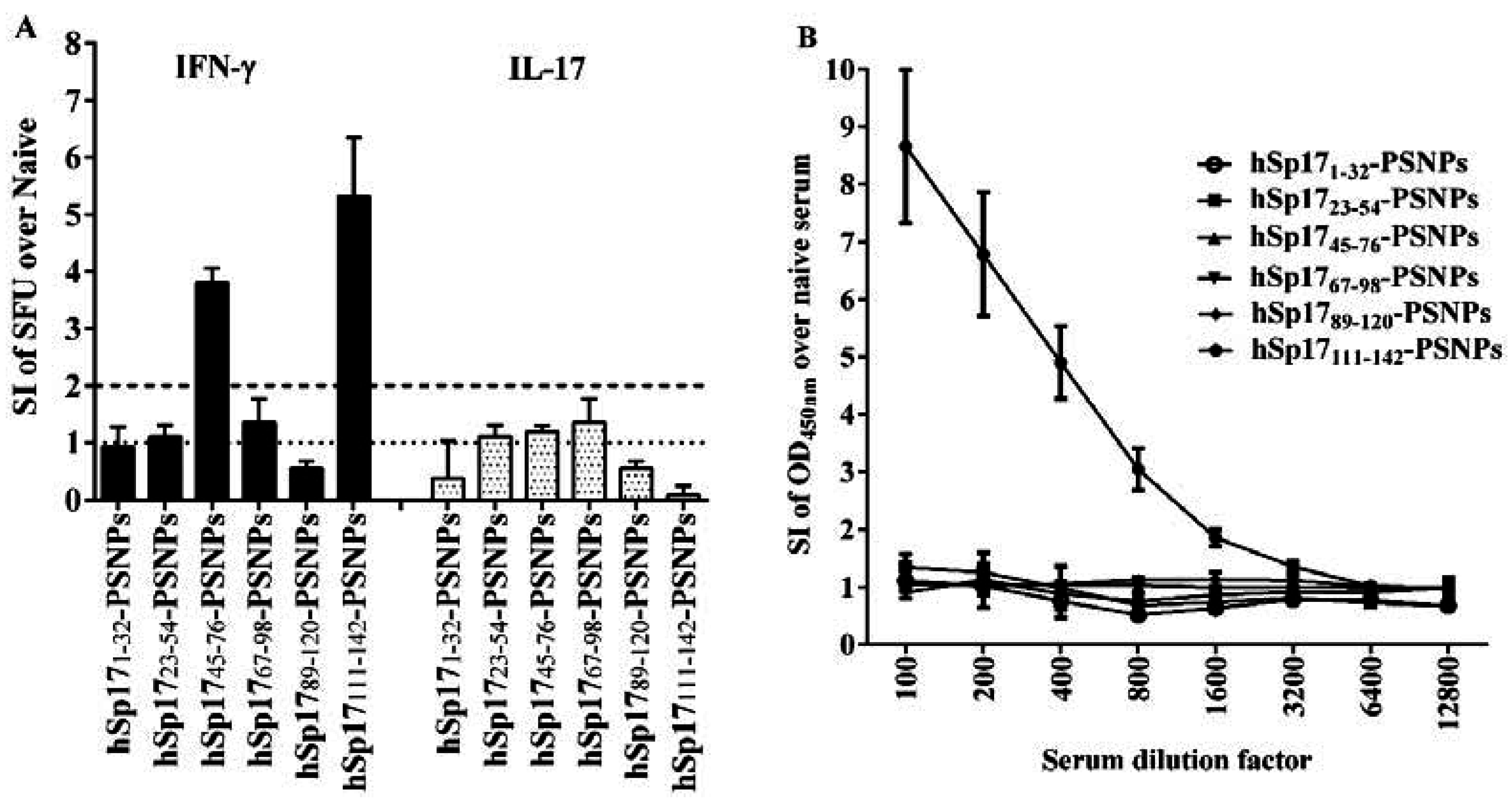
3.2. Immune Responses Induced by hSp17peptides-PSNPs Vaccine Formulations

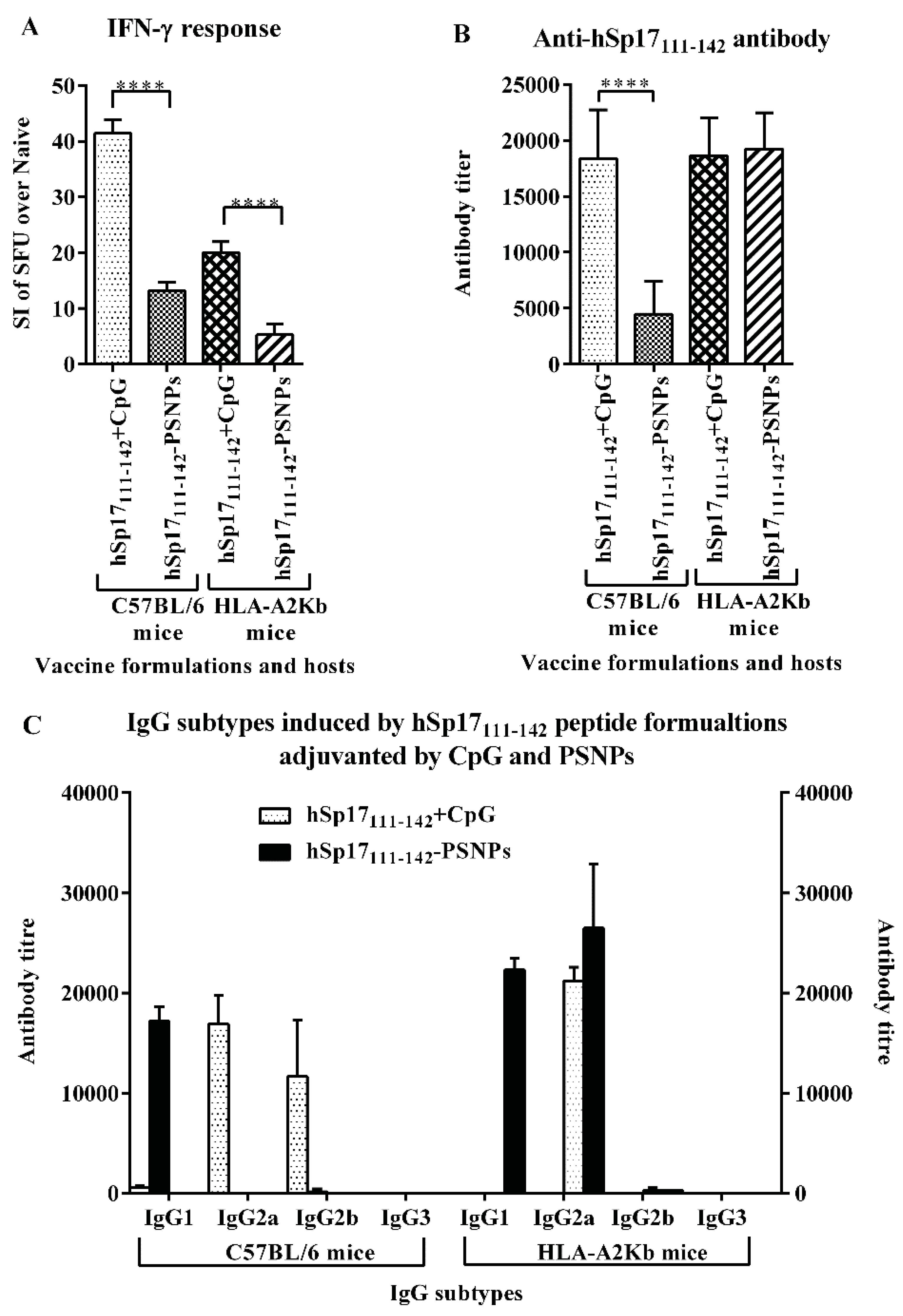
3.3. The Immunogenicity of hSp17111–142 Peptide may be Hampered by the Conjugation Process to PSNPs
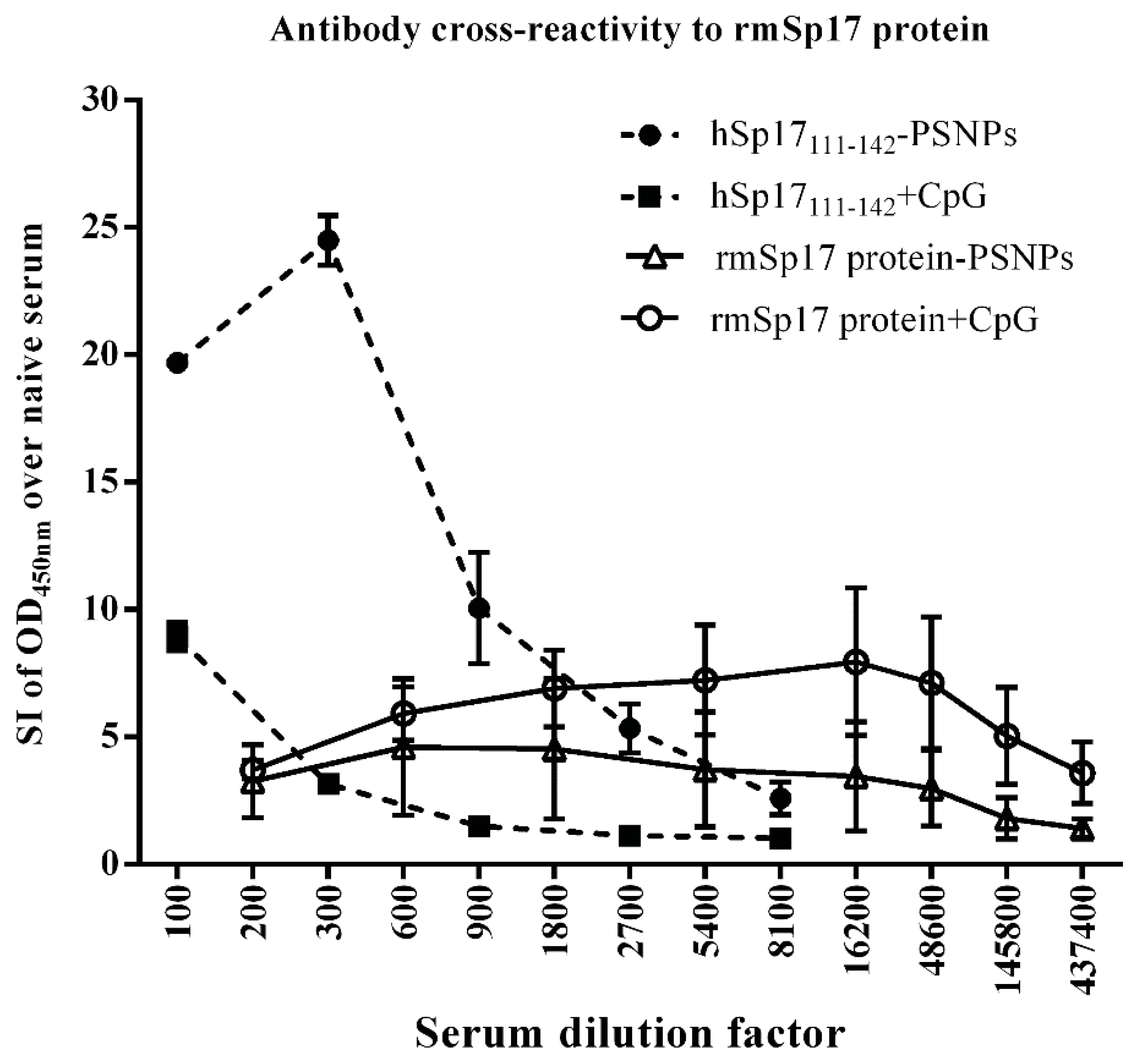
3.4. Strong Antibody Cross-reactivity to rmSp17 Protein


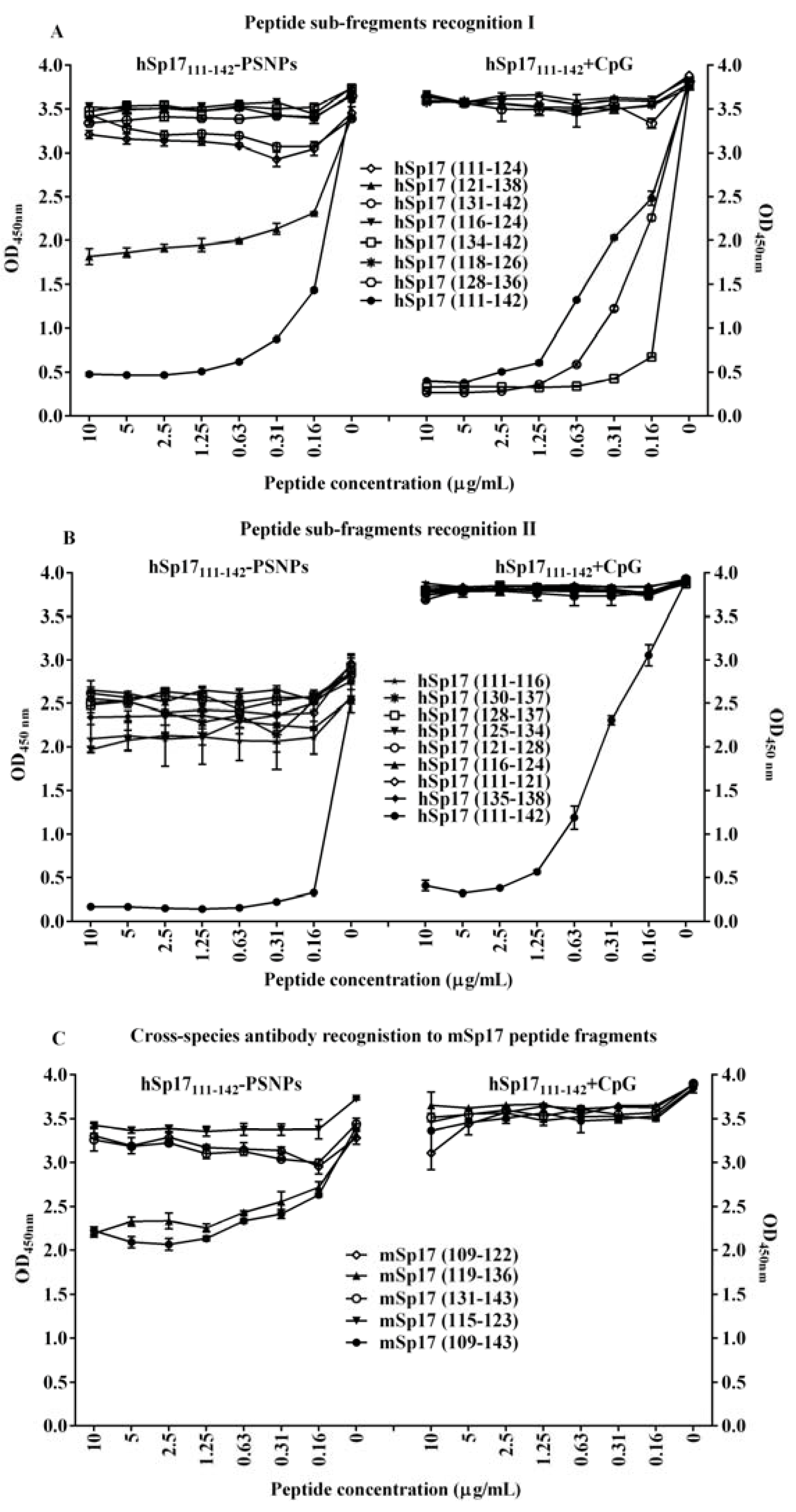
3.5. Changing the Dynamic of Epitope Recognitions after the Conjugation Process to PSNPs
| Amino Acid Position | Aligned Sequences |
|---|---|
| hSp17 | |
| 111–142 | KEKEEVAAVKIQAAFRGHIAREEAKKMKTNSL |
| 111–116 | KEKEEV |
| 111–124 | KEKEEVAAVKIQAA |
| 113–121 | KEEVAAVKI |
| 116–124 | VAAVKIQAA |
| 118–126 | AVKIQAAFR |
| 121–138 | IQAAFRGHIAREEAKKMK |
| 121–128 | IQAAFRGH |
| 125–134 | FRGHIAREEA |
| 128–136 | IAREEAKK |
| 128–137 | HIAREEAKKM |
| 130–137 | AREEAKKM |
| 131–142 | REEAKKMKTNSL |
| 134–142 | AKKMKTNSL |
| 135–138 | KKMK |
| mSp17 | |
| 109–143 | REQEEAAALKIQSLFRGHVAREEVKKMKSDKNENL |
| 109–122 | REQEEAAALKIQSL |
| 115–123 | AALKIQSLF |
| 119–136 | IQSLFRGHVAREEVKKMK |
| 131–143 | EVKKMKSDKNENL |

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix
| Position | Sequence | Sequence Homology between Human and Mouse | rmSp17 Reactivity |
|---|---|---|---|
| 2–10 | SIPFSNTHY | 100% | − |
| 11–19 | RIPQGFGNL | 100% | − |
| 12–19 | IPQGFGNL | 100% | − |
| 12–20 | IPQGFGNLL | 100% | − |
| 18–26 | NLLEGLTRE | 100% | − |
| 19–27 | LLEGLTREI | 100% | − |
| 22–30 | GLTREILRE | 100% | − |
| 27–35 | ILREQPDNI | 100% | − |
| 30–38 | EQPDNIPAF | 100% | − |
| 34–42 | NIPAFAAAY | 100% | − |
| 38–46 | FAAAYFESL | 89% | − |
| 39–46 | AAAYFENL | 89% | − |
| 39–47 | AAAYFESLL | 89% | − |
| 40–47 | AAYFENLL | 89% | − |
| 45–53 | SLLEKREKT | 89% | − |
| 66–74 | DRFYNNHAF | 100% | − |
| 91–99 | QISGKEEET | 11% | − |
| 111–119 | KEKEEVAAV | 56% § | − |
| 115–123 | AALKIQSLF | 56% § | − |
| 116–124 | VAAVKIQAA | 56% § | − |
| 134–142 | AKKMKTNSL | 44% § | − |
| 136–143 | KSDKNENL | 22% | − |
| 136–144 | KSDKNENLK | 22% | − |
| rmSp17 | recombinant murine Sp17 protein | ++++ ξ |
| Position | Sequence | Immunogen | |||||
|---|---|---|---|---|---|---|---|
| hSp171–32 | hSp1723–54 | hSp1745–76 | hSp1767–98 | hSp1789–120 | hSp17111–142 | ||
| 11–19 | RIPQGFGNL | - | |||||
| 12–20 | IPQGFGNLL | - | |||||
| 18–26 | NLLEGLTRE | - | |||||
| 19–27 | LLEGLTREI | - | |||||
| 22–30 | GLTREILRE | - | - | ||||
| 27–35 | ILREQPDNI | - | |||||
| 45–53 | SLLEKREKT | - | |||||
| 91–99 | QISGKEEET | - | |||||
| 111–119 | KEKEEVAAV | - | - | ||||
| 116–124 | VAAVKIQAA | - | - | ||||
| 134–142 | AKKMKTNSL | - | |||||
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Baldwin, L.A.; Huang, B.; Miller, R.W.; Tucker, T.; Goodrich, S.T.; Podzielinski, I.; DeSimone, C.P.; Ueland, F.R.; van Nagell, J.R.; Seamon, L.G. Ten-year relative survival for epithelial ovarian cancer. Obstet. Gynecol. 2012, 120, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Bali, A.; Reynolds, K. The current management of primary ovarian cancer: A review. Cancer Ther. 2004, 2, 305–316. [Google Scholar]
- Bookman, M.A.; Brady, M.F.; McGuire, W.P.; Harper, P.G.; Alberts, D.S.; Friedlander, M.; Colombo, N.; Fowler, J.M.; Argenta, P.A.; de Geest, K.; et al. Evaluation of new platinum-based treatment regimens in advanced-stage ovarian cancer: A phase III trial of the gynecologic cancer intergroup. J. Clin. Oncol. 2009, 27, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Hwu, P.; Freedman, R.S. The immunotherapy of patients with ovarian cancer. J. Immunother. 2002, 25, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Boyer, J.; Schullery, D.; Gimotty, P.; Gamerman, V.; Bender, J.; Levine, B.; Coukos, G.; Rubin, S.; Morgan, M.; et al. Phase I/II randomized trial of dendritic cell vaccination with or without cyclophosphamide for consolidation therapy of advanced ovarian cancer in first or second remission. Cancer Immunol. Immunother. 2012, 61, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, C.S.; Gnjatic, S.; Sabbatini, P.; Aghajanian, C.; Hensley, M.L.; Spriggs, D.R.; Iasonos, A.; Lee, H.; Dupont, B.; Pezzulli, S.; et al. Safety and immunogenicity study of NY-ESO-1B peptide and montanide ISA-51 vaccination of patients with epithelial ovarian cancer in high-risk first remission. Clin Cancer Res 2008, 14, 2740–2748. [Google Scholar] [CrossRef] [PubMed]
- Loveland, B.E.; Zhao, A.; White, S.; Gan, H.; Hamilton, K.; Xing, P.X.; Pietersz, G.A.; Apostolopoulos, V.; Vaughan, H.; Karanikas, V.; et al. Mannan-MUC1-pulsed dendritic cell immunotherapy: A phase I trial in patients with adenocarcinoma. Clin. Cancer Res. 2006, 12, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.L.; Quinn, M.A.; Grant, P.T.; Allen, D.G.; Jobling, T.W.; White, S.C.; Zhao, A.; Karanikas, V.; Vaughan, H.; Pietersz, G.; et al. A phase 2, single-arm study of an autologous dendritic cell treatment against mucin 1 in patients with advanced epithelial ovarian cancer. J. Immunother. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, P.; Tsuji, T.; Ferran, L.; Ritter, E.; Sedrak, C.; Tuballes, K.; Jungbluth, A.A.; Ritter, G.; Aghajanian, C.; Bell-McGuinn, K.; et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-iclc shows rapid induction of integrated immune response in ovarian cancer patients. Clin. Cancer Res. 2012, 18, 6497–6508. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Grabstein, K.H.; Sleath, P.R.; Cheever, M.A. Generation of immunity to the her-2/neu oncogenic protein in patients with breast and ovarian cancer using a peptide-based vaccine. Clin. Cancer Res. 1999, 5, 1289–1297. [Google Scholar] [PubMed]
- Straughn, J.M., Jr.; Shaw, D.R.; Guerrero, A.; Bhoola, S.M.; Racelis, A.; Wang, Z.; Chiriva-Internati, M.; Grizzle, W.E.; Alvarez, R.D.; Lim, S.H.; et al. Expression of sperm protein 17 (Sp17) in ovarian cancer. Int. J. Cancer 2004, 108, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Lea, I.A.; Adoyo, P.; O’Rand, M.G. Autoimmunogenicity of the human sperm protein Sp17 in vasectomized men and identification of linear B cell epitopes. Fertil. Steril. 1997, 67, 355–361. [Google Scholar] [CrossRef]
- Chiriva-Internati, M.; Wang, Z.; Salati, E.; Bumm, K.; Barlogie, B.; Lim, S.H. Sperm protein 17 (Sp17) is a suitable target for immunotherapy of multiple myeloma. Blood 2002, 100, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Chiriva-Internati, M.; Weidanz, J.A.; Yu, Y.; Frezza, E.E.; Jenkins, M.R.; Kennedy, R.C.; Cobos, E.; Kast, W.M. Sperm protein 17 is a suitable target for adoptive T-cell-based immunotherapy in human ovarian cancer. J. Immunother. 2008, 31, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Chiriva-Internati, M.; Yu, Y.; Mirandola, L.; Jenkins, M.R.; Chapman, C.; Cannon, M.; Cobos, E.; Kast, W.M. Cancer testis antigen vaccination affords long-term protection in a murine model of ovarian cancer. PLoS ONE 2010, 5, e10471. [Google Scholar] [CrossRef] [PubMed]
- Dadabayev, A.R.; Wang, Z.; Zhang, Y.; Zhang, J.; Robinson, W.R.; Lim, S.H. Cancer immunotherapy targeting Sp17: When should the laboratory findings be translated to the clinics? Am. J. Hematol. 2005, 80, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Voskens, C.J.; Strome, S.E.; Sewell, D.A. Synthetic peptide-based cancer vaccines: Lessons learned and hurdles to overcome. Curr. Mol. Med. 2009, 9, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Gao, Q.; Wilson, K.L.; Heyerick, A.; Plebanski, M. Mapping T and B cell epitopes in sperm protein 17 to support the development of an ovarian cancer vaccine. Vaccine 2015. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Richardson, R.T.; Widgren, E.E.; O’Rand, M.G. Characterization of Sp17: A ubiquitous three domain protein that binds heparin. Biochem. J. 2001, 357, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Fuchsberger, M.; Hochrein, H.; O’Keeffe, M. Activation of plasmacytoid dendritic cells. Immunol. Cell Biol. 2005, 83, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Mittag, D.; Proietto, A.I.; Loudovaris, T.; Mannering, S.I.; Vremec, D.; Shortman, K.; Wu, L.; Harrison, L.C. Human dendritic cell subsets from spleen and blood are similar in phenotype and function but modified by donor health status. J. Immunol. 2011, 186, 6207–6217. [Google Scholar] [CrossRef] [PubMed]
- Klinman, D.M.; Yi, A.K.; Beaucage, S.L.; Conover, J.; Krieg, A.M. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc. Natl. Acad. Sci. USA 1996, 93, 2879–2883. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.G.; Marshall, J.S. CpG-containing oligodeoxynucleotides induce TNF-alpha and IL-6 production but not degranulation from murine bone marrow-derived mast cells. J. Leukoc. Biol. 2001, 69, 253–262. [Google Scholar] [PubMed]
- Coward, J.; Kulbe, H.; Chakravarty, P.; Leader, D.; Vassileva, V.; Leinster, D.A.; Thompson, R.; Schioppa, T.; Nemeth, J.; Vermeulen, J.; et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin. Cancer Res. 2011, 17, 6083–6096. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.W.; Chen, M.W.; Hsiao, M.; Wang, S.; Chen, C.A.; Hsiao, S.M.; Chang, J.S.; Lai, T.C.; Rose-John, S.; Kuo, M.L.; et al. IL-6 trans-signaling in formation and progression of malignant ascites in ovarian cancer. Cancer Res. 2011, 71, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Zeskind, B. IL-6 and ovarian cancer––Letter. Clin. Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Pinciroli, P.; Alberti, C.; Sensi, M.; Canevari, S.; Tomassetti, A. An IL6-correlated signature in serous epithelial ovarian cancer associates with growth factor response. BMC Genomics 2013. [Google Scholar] [CrossRef] [PubMed]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [PubMed]
- Fifis, T.; Mottram, P.; Bogdanoska, V.; Hanley, J.; Plebanski, M. Short peptide sequences containing MHC class I and/or class II epitopes linked to nano-beads induce strong immunity and inhibition of growth of antigen-specific tumour challenge in mice. Vaccine 2004, 23, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Mottram, P.L.; Leong, D.; Crimeen-Irwin, B.; Gloster, S.; Xiang, S.D.; Meanger, J.; Ghildyal, R.; Vardaxis, N.; Plebanski, M. Type 1 and 2 immunity following vaccination is influenced by nanoparticle size: Formulation of a model vaccine for respiratory syncytial virus. Mol. Pharm. 2007, 4, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Kalkanidis, M.; Pietersz, G.A.; Mottram, P.L.; Crimeen-Irwin, B.; Ardipradja, K.; Plebanski, M. Methods for nano-particle based vaccine formulation and evaluation of their immunogenicity. Methods 2006, 40, 20–29. [Google Scholar]
- Scheerlinck, J.P.; Gloster, S.; Gamvrellis, A.; Mottram, P.L.; Plebanski, M. Systemic immune responses in sheep, induced by a novel nano-bead adjuvant. Vaccine 2006, 24, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Wilson, K.; Day, S.; Fuchsberger, M.; Plebanski, M. Methods of effective conjugation of antigens to nanoparticles as non-inflammatory vaccine carriers. Methods 2013, 60, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Karlson Tde, L.; Kong, Y.Y.; Hardy, C.L.; Xiang, S.D.; Plebanski, M. The signalling imprints of nanoparticle uptake by bone marrow derived dendritic cells. Methods 2013, 60, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Kong, Y.Y.; Hanley, J.; Fuchsberger, M.; Crimeen-Irwin, B.; Plebanski, M. Nanoparticles modify dendritic cell homeostasis and induce non-specific effects on immunity to malaria. Trans. R. Soc. Trop. Med. Hyg. 2015, 109, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Chiriva-Internati, M.; Wang, Z.; Pochopien, S.; Salati, E.; Lim, S.H. Identification of a sperm protein 17 CTL epitope restricted by HLA-A1. Int. J. Cancer 2003, 107, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Chiriva-Internati, M.; Wang, Z.; Salati, E.; Timmins, P.; Lim, S.H. Tumor vaccine for ovarian carcinoma targeting sperm protein 17. Cancer 2002, 94, 2447–2453. [Google Scholar] [CrossRef] [PubMed]
- Maletto, B.; Rópolo, A.; Morón, V.; Pistoresi-Palencia, M.C. CpG-DNA stimulates cellular and humoral immunity and promotes TH1 differentiation in aged BALB/C mice. J. Leukoc. Biol. 2002, 72, 447–454. [Google Scholar] [PubMed]
- Visciano, M.; Tagliamonte, M.; Tornesello, M.; Buonaguro, F.; Buonaguro, L. Effects of adjuvants on igg subclasses elicited by virus-like particles. J. Transl. Med. 2012, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Gao, Q.; Plebanski, M.; Department of Immunology and Pathology, Central Clinical School, Faculty of Medicine, Nursing and Health Sciences, Monash University, Melbourne, VIC, Australia. Unpublished data. 2015.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, S.D.; Gao, Q.; Wilson, K.L.; Heyerick, A.; Plebanski, M. A Nanoparticle Based Sp17 Peptide Vaccine Exposes New Immuno-Dominant and Species Cross-reactive B Cell Epitopes. Vaccines 2015, 3, 875-893. https://doi.org/10.3390/vaccines3040875
Xiang SD, Gao Q, Wilson KL, Heyerick A, Plebanski M. A Nanoparticle Based Sp17 Peptide Vaccine Exposes New Immuno-Dominant and Species Cross-reactive B Cell Epitopes. Vaccines. 2015; 3(4):875-893. https://doi.org/10.3390/vaccines3040875
Chicago/Turabian StyleXiang, Sue D., Qian Gao, Kirsty L. Wilson, Arne Heyerick, and Magdalena Plebanski. 2015. "A Nanoparticle Based Sp17 Peptide Vaccine Exposes New Immuno-Dominant and Species Cross-reactive B Cell Epitopes" Vaccines 3, no. 4: 875-893. https://doi.org/10.3390/vaccines3040875
APA StyleXiang, S. D., Gao, Q., Wilson, K. L., Heyerick, A., & Plebanski, M. (2015). A Nanoparticle Based Sp17 Peptide Vaccine Exposes New Immuno-Dominant and Species Cross-reactive B Cell Epitopes. Vaccines, 3(4), 875-893. https://doi.org/10.3390/vaccines3040875