Recent Insight into SARS-CoV2 Immunopathology and Rationale for Potential Treatment and Preventive Strategies in COVID-19



Abstract
:1. Introduction
2. What We Know about Immune Response to Coronaviruses
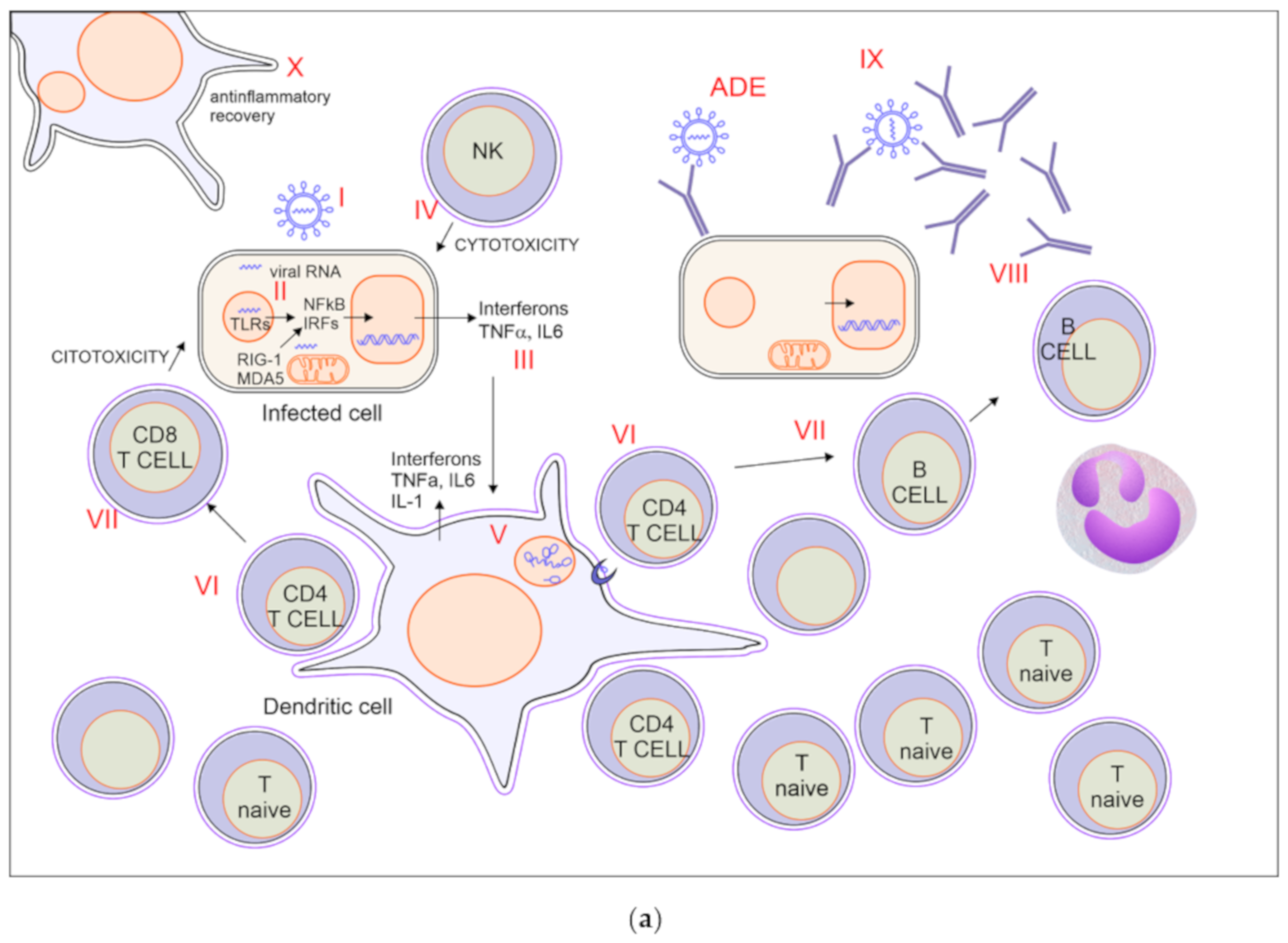
2.1. A Possible Scenario of Covid-19 Immune Response with Effective Recovery
2.1.1. Viral Entry via ACE2 Binding (Figure 1a I)
2.1.2. Innate Immune Response (Figure 1a II)
2.1.3. Production of Interferons (Figure 1a III)
2.1.4. Activation of NK-Mediated Killing of Infected Cells (Figure 1a IV)
2.1.5. Activation of Dendritic Cells, Macrophages, and Neutrophils (Figure 1a V)
2.1.6. Recruitment and Activation of Virus Specific CD4 T Cells (Figure 1a VI)
2.1.7. Activation of CD8 Mediated Cytotoxicity and B Cell (Figure 1a VII)
2.1.8. Production of Antibodies (Figure 1a VIII)
2.1.9. Block of Viral Spreading (Figure 1a IX)
2.1.10. Anti-Inflammatory Recovery (Figure 1a X)
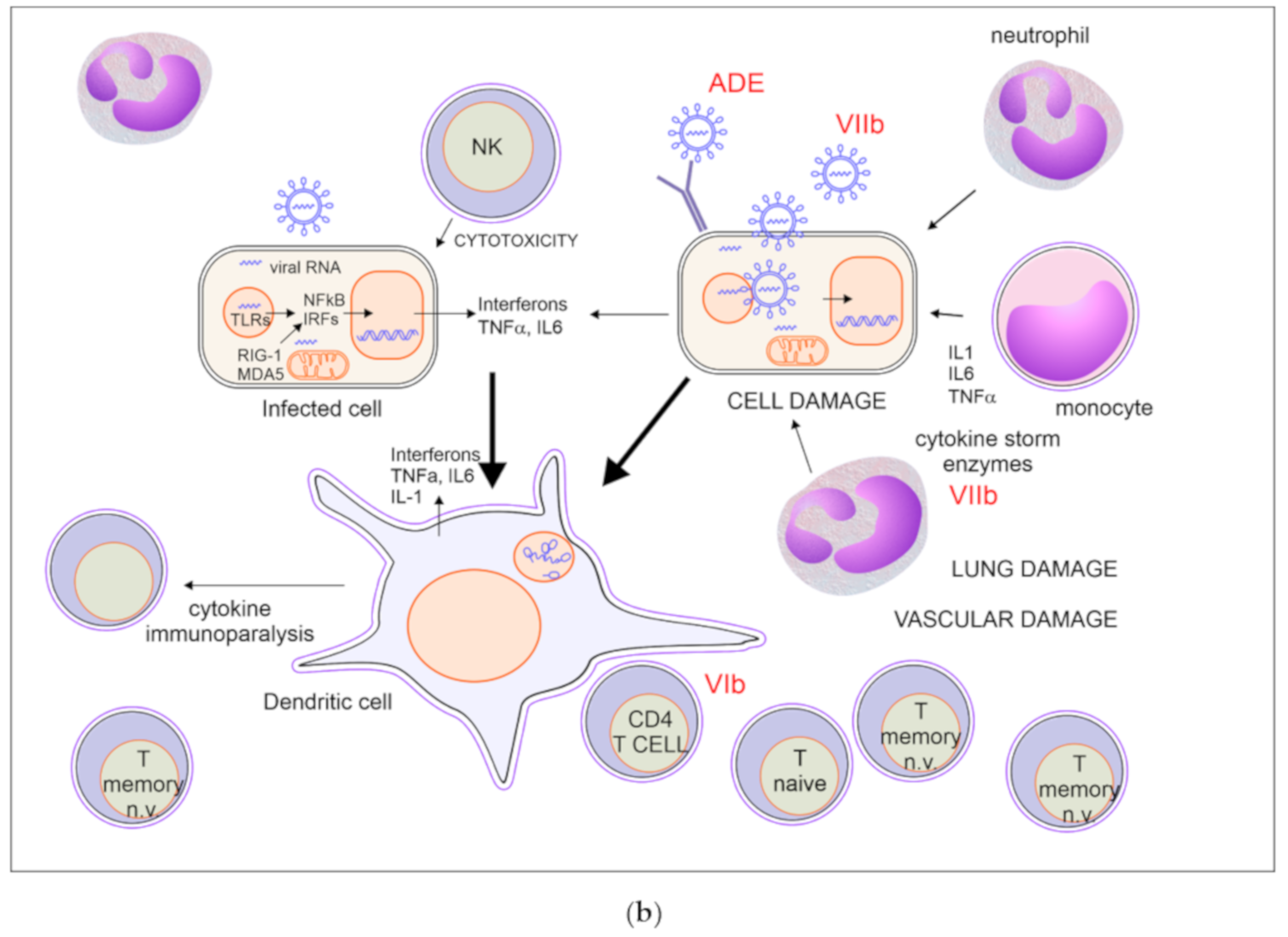
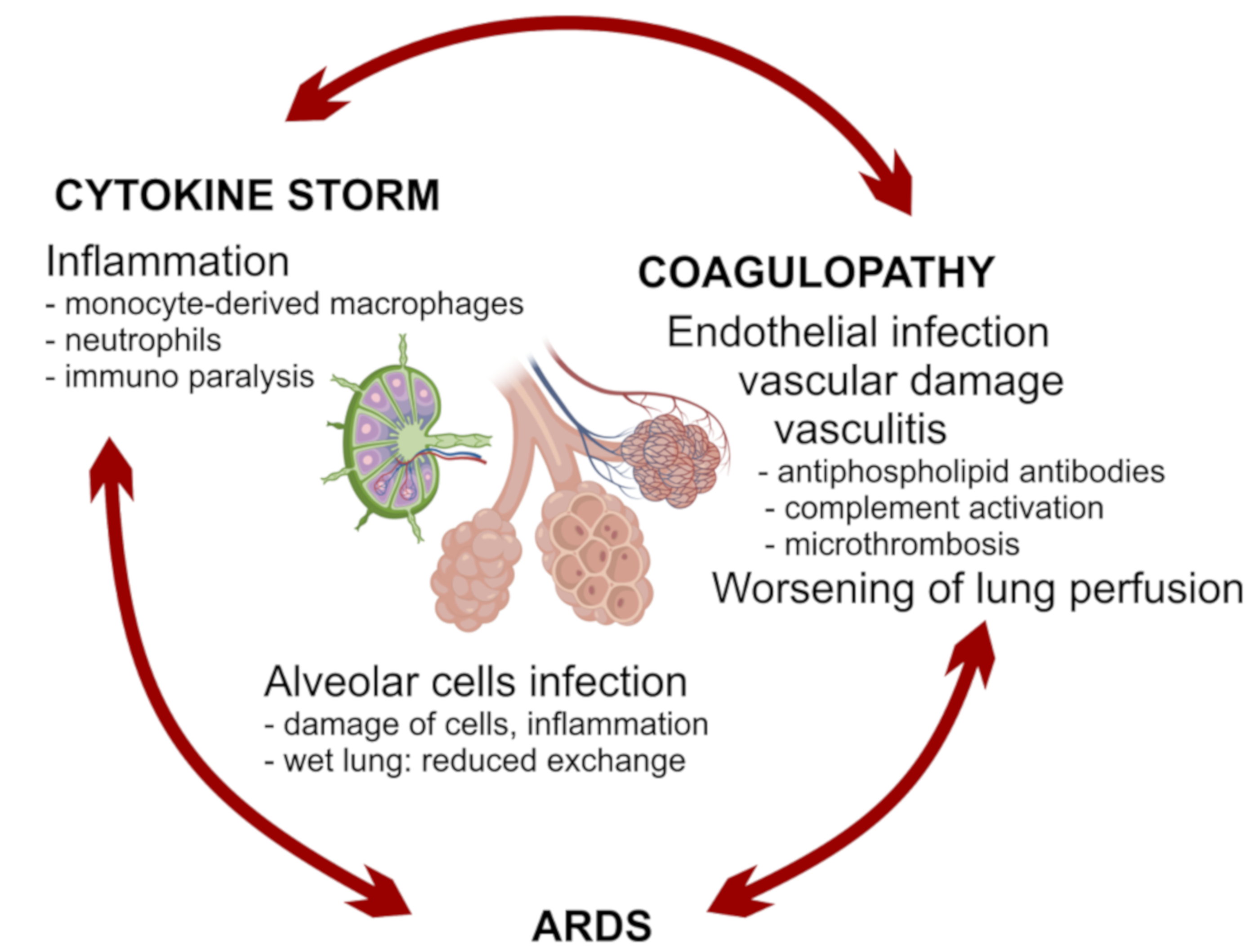
2.2. A Possible Scenario of Immune Response in Severe Covid-19
2.2.1. Insufficient Recruitment of Virus-Specific T Cells (Figure 1b VI)
2.2.2. Virus and Inflammation Induced Tissue Damage (Figure 1b VII)
3. Immunity to SARS-CoV-2 and Factors Determining Disease Progression
3.1. The Age Gradient
3.1.1. ACE2 Density and Distribution Changes with Age
3.1.2. T Cell Repertoire and Aging
3.1.3. Age-Related Changes in Antibody Production
3.1.4. Innate Immune System and Ageing
3.2. Why Do Some Young People Become Critically Ill?
3.2.1. Viral Load
3.2.2. Variants in Innate Immunity Genes
3.2.3. Immunodeficiencies Affecting T Cell Repertoire
3.2.4. Immunodeficiencies Affecting Cytotoxic Functions
3.2.5. HLA Haplotypes Correlations
4. Why Bats Don’t Get Sick and What They Can Tell Us
5. Critical Mechanisms of Potential Relevance to Determine the Course of Infection and Possible Targets of Therapies
6. Therapies under Evaluation
6.1. Antivirals
6.2. Chloroquine and Hydroxychloroquine
6.3. SARS-CoV2-Specific Monoclonal Antibodies
6.4. Immunomodulatory Agents
6.5. Convalescent Plasma, Hyperimmune Globulins, and Intravenous Immunoglobulins (IVIG)
6.6. Interferons and JAK-Inhibitors
6.7. Corticosteroids
6.8. Other Therapeutic Strategies
7. Acquired Protection and Development of Vaccines
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Coronavirus Disease (COVID-19) Dashboard. Available online: https://www.covid19.who.int/2020 (accessed on 5 May 2020).
- Zhang, S.; Diao, M.; Yu, W.; Pei, L.; Lin, Z.; Chen, D. Estimation of the reproductive number of novel coronavirus (COVID-19) and the probable outbreak size on the Diamond Princess cruise ship: A data-driven analysis. Int. J. Infect. Dis. 2020, 93, 201–204. [Google Scholar] [CrossRef]
- Sanche, S.; Lin, Y.T.; Xu, C.; Romero-Severson, E.; Hengartner, N.; Ke, R. High Contagiousness and Rapid Spread of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26. [Google Scholar] [CrossRef]
- Verity, R.; Okell, L.C.; Dorigatti, I.; Winskill, P.; Whittaker, C.; Imai, N.; Cuomo-Dannenburg, G.; Thompson, H.; Walker, P.G.T.; Fu, H.; et al. Estimates of the severity of coronavirus disease 2019: A model-based analysis. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- McCombs, A.; Kadelka, C. A model-based evaluation of the efficacy of COVID-19 social distancing, testing and hospital triage policies. medRxiv 2020. [Google Scholar] [CrossRef]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 11, 11. [Google Scholar] [CrossRef]
- Wu, F.; Wang, A.; Liu, M.; Wang, Q.; Chen, J.; Xia, S.; Ling, Y.; Zhang, Y.; Xun, J.; Lu, L.; et al. Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications. medRxiv 2020. [Google Scholar] [CrossRef]
- Garg, S.; Kim, L.; Whitaker, M.; O’Halloran, A.; Cummings, C.; Holstein, R.; Prill, M.; Chai, S.J.; Kirley, P.D.; Alden, N.B.; et al. Hospitalization Rates and Characteristics of Patients Hospitalized with Laboratory-Confirmed Coronavirus Disease 2019—COVID-NET, 14 States, March 1–30, 2020. MMWR Morb. Mortal Wkly. Rep. 2020, 69, 458–464. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020. [Google Scholar] [CrossRef]
- Cristiani, L.; Mancino, E.; Matera, L.; Nenna, R.; Pierangeli, A.; Scagnolari, C.; Midulla, F. Will children reveal their secret? The coronavirus dilemma. Eur. Respir. J. 2020. [Google Scholar] [CrossRef] [Green Version]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudbjartsson, D.F.; Helgason, A.; Jonsson, H.; Magnusson, O.T.; Melsted, P.; Norddahl, G.L.; Saemundsdottir, J.; Sigurdsson, A.; Sulem, P.; Agustsdottir, A.B.; et al. Spread of SARS-CoV-2 in the Icelandic Population. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Rose-John, S. The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clin. Pharm. 2017, 102, 591–598. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov. bioRxiv 2020. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020. [Google Scholar] [CrossRef]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; Paul van Schayck, J.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Jiang, N.; Zhou, Q.; Ma, W.L. Persistence of intestinal SARS-CoV-2 infection in patients with COVID-19 leads to re-admission after pneumonia resolved. Int. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.D.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-up. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Chiumello, D. Covid-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Cheung, C.Y.; Poon, L.L.; Ng, I.H.; Luk, W.; Sia, S.F.; Wu, M.H.; Chan, K.H.; Yuen, K.Y.; Gordon, S.; Guan, Y.; et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: Possible relevance to pathogenesis. J. Virol. 2005, 79, 7819–7826. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J. SARS-CoV-2: An Emerging Coronavirus that Causes a Global Threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, W.; Hu, G.; Xia, S.; Sun, Z.; Liu, Z.; Xie, Y.; Zhang, R.; Jiang, S.; Lu, L. SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion. Cell. Mol. Immunol. 2020, 1–3. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Decroly, E.; Imbert, I.; Coutard, B.; Bouvet, M.; Selisko, B.; Alvarez, K.; Gorbalenya, A.E.; Snijder, E.J.; Canard, B. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2’O)-methyltransferase activity. J. Virol. 2008, 82, 8071–8084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zust, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Xing, Y.; Chen, X.; Zheng, Y.; Yang, Y.; Nichols, D.B.; Clementz, M.A.; Banach, B.S.; Li, K.; Baker, S.C.; et al. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS ONE 2012, 7, e30802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menachery, V.D.; Eisfeld, A.J.; Schafer, A.; Josset, L.; Sims, A.C.; Proll, S.; Fan, S.; Li, C.; Neumann, G.; Tilton, S.C.; et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 2014, 5, e01174-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieman, M.; Yount, B.; Heise, M.; Kopecky-Bromberg, S.A.; Palese, P.; Baric, R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 2007, 81, 9812–9824. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-Y.; Liu, H.M.; Chang, M.-F.; Chang, S.C. Middle East respiratory syndrome coronavirus nucleocapsid protein suppresses type I and type III interferon induction by targeting RIG-I signaling. J. Virol. 2020. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B., Jr.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 130, 3625–3639. [Google Scholar] [CrossRef]
- Jing, H.; Su, H.C. New immunodeficiency syndromes that help us understand the IFN-mediated antiviral immune response. Curr. Opin. Pediatr. 2019, 31, 815–820. [Google Scholar] [CrossRef]
- Doms, A.; Sanabria, T.; Hansen, J.N.; Altan-Bonnet, N.; Holm, G.H. 25-Hydroxycholesterol Production by the Cholesterol-25-Hydroxylase Interferon-Stimulated Gene Restricts Mammalian Reovirus Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrensch, F.; Winkler, M.; Pohlmann, S. IFITM proteins inhibit entry driven by the MERS-coronavirus spike protein: Evidence for cholesterol-independent mechanisms. Viruses 2014, 6, 3683–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everitt, A.R.; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; Adams, D.J.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trouillet-Assant, S.; Viel, S.; Gaymard, A.; Pons, S.; Richard, J.C.; Perret, M.; Villard, M.; Brengel-Pesce, K.; Lina, B.; Mezidi, M.; et al. Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, S.N.; Reighard, S.D.; Gyurova, I.E.; Cranert, S.A.; Mahl, S.E.; Karmele, E.P.; McNally, J.P.; Moran, M.T.; Brooks, T.R.; Yaqoob, F.; et al. Roles of natural killer cells in antiviral immunity. Curr. Opin. Virol. 2016, 16, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The involvement of natural killer cells in the pathogenesis of severe acute respiratory syndrome. Am. J. Clin. Pathol. 2004, 121, 507–511. [CrossRef]
- Wang, F.; Nie, J.; Wang, H.; Zhao, Q.; Xiong, Y.; Deng, L.; Song, S.; Ma, Z.; Mo, P.; Zhang, Y. Characteristics of peripheral lymphocyte subset alteration in COVID-19 pneumonia. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Law, H.K.; Cheung, C.Y.; Ng, H.Y.; Sia, S.F.; Chan, Y.O.; Luk, W.; Nicholls, J.M.; Peiris, J.S.; Lau, Y.L. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood 2005, 106, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Cervantes-Barragan, L.; Zust, R.; Weber, F.; Spiegel, M.; Lang, K.S.; Akira, S.; Thiel, V.; Ludewig, B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 2007, 109, 1131–1137. [Google Scholar] [CrossRef] [Green Version]
- Legge, K.L.; Braciale, T.J. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity 2003, 18, 265–277. [Google Scholar] [CrossRef] [Green Version]
- Lau, Y.L.; Peiris, J.S.; Law, H.K. Role of dendritic cells in SARS coronavirus infection. Hong Kong Med. J. 2012, 18 (Suppl. 3), 28–30. [Google Scholar] [PubMed]
- Park, M.D. Macrophages: A Trojan horse in COVID-19? Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Chen, L.; Li, J.; Wang, X.; Wang, F.; et al. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Dienz, O.; Rud, J.G.; Eaton, S.M.; Lanthier, P.A.; Burg, E.; Drew, A.; Bunn, J.; Suratt, B.T.; Haynes, L.; Rincon, M. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. 2012, 5, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.S.; Kim, Y.; Kim, G.; Lee, J.Y.; Jeong, I.; Joh, J.S.; Kim, H.; Chang, E.; Sim, S.Y.; Park, J.S.; et al. Immune Responses to Middle East Respiratory Syndrome Coronavirus During the Acute and Convalescent Phases of Human Infection. Clin. Infect. Dis. 2019, 68, 984–992. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, M.K.; Moon, J.J. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J. Immunol. 2012, 188, 4135–4140. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhao, J.; Mangalam, A.K.; Channappanavar, R.; Fett, C.; Meyerholz, D.K.; Agnihothram, S.; Baric, R.S.; David, C.S.; Perlman, S. Airway Memory CD4(+) T Cells Mediate Protective Immunity against Emerging Respiratory Coronaviruses. Immunity 2016, 44, 1379–1391. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.Y.; Sohn, Y.; Lee, S.H.; Cho, Y.; Hyun, J.H.; Baek, Y.J.; Jeong, S.J.; Kim, J.H.; Ku, N.S.; Yeom, J.S.; et al. Use of Convalescent Plasma Therapy in Two COVID-19 Patients with Acute Respiratory Distress Syndrome in Korea. J. Korean Med. Sci. 2020, 35, e149. [Google Scholar] [CrossRef] [Green Version]
- Soresina, A.; Moratto, D.; Chiarini, M.; Paolillo, C.; Baresi, G.; Foca, E.; Bezzi, M.; Baronio, B.; Giacomelli, M.; Badolato, R. Two X-linked agammaglobulinemia patients develop pneumonia as COVID-19 manifestation but recover. Pediatr. Allergy Immunol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Quinti, I.; Lougaris, V.; Milito, C.; Cinetto, F.; Pecoraro, A.; Mezzaroma, I.; Mastroianni, C.M.; Turriziani, O.; Bondioni, M.P.; Filippini, M.; et al. A possible role for B cells in COVID-19?: Lesson from patients with Agammaglobulinemia. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Huisman, W.; Martina, B.E.; Rimmelzwaan, G.F.; Gruters, R.A.; Osterhaus, A.D. Vaccine-induced enhancement of viral infections. Vaccine 2009, 27, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, L.; Kuwahara, K.; Li, L.; Liu, Z.; Li, T.; Zhu, H.; Liu, J.; Xu, Y.; Xie, J.; et al. Immunodominant SARS Coronavirus Epitopes in Humans Elicited both Enhancing and Neutralizing Effects on Infection in Non-human Primates. ACS Infect. Dis. 2016, 2, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Lin, C.Y.; Chang, K.; Urbina, A.N.; Assavalapsakul, W.; Thitithanyanont, A.; Lu, P.L.; Chen, Y.H.; Wang, S.F. A clinical and epidemiological survey of the largest dengue outbreak in Southern Taiwan in 2015. Int. J. Infect. Dis. 2019, 88, 88–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, R.A.; de Oliveira-Filho, E.F.; Fernandes, A.I.; Brito, C.A.; Marques, E.T.; Tenorio, M.C.; Gil, L.H. Previous dengue or Zika virus exposure can drive to infection enhancement or neutralisation of other flaviviruses. Mem. Inst. Oswaldo Cruz 2019, 114, e190098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetro, J.A. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect. 2020, 22, 72–73. [Google Scholar] [CrossRef]
- Liu, L.; Wei, Q.; Lin, Q.; Fang, J.; Wang, H.; Kwok, H.; Tang, H.; Nishiura, K.; Peng, J.; Tan, Z.; et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Harzallah, I.; Debliquis, A.; Drenou, B. Lupus anticoagulant is frequent in patients with Covid-19. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Cheng, Y.; Cheng, G.; Chui, C.H.; Lau, F.Y.; Chan, P.K.; Ng, M.H.; Sung, J.J.; Wong, R.S. ABO blood group and susceptibility to severe acute respiratory syndrome. JAMA 2005, 293, 1450–1451. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, Y.; Huang, H.-P.; Li, D.; Gu, D.-F.; Lu, X.-F.; Zhang, Z.; Liu, L.; Liu, T.; Liu, Y.-K.; et al. Relationship between the ABO Blood Group and the COVID-19 Susceptibility. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Guillon, P.; Clement, M.; Sebille, V.; Rivain, J.G.; Chou, C.F.; Ruvoen-Clouet, N.; Le Pendu, J. Inhibition of the interaction between the SARS-CoV spike protein and its cellular receptor by anti-histo-blood group antibodies. Glycobiology 2008, 18, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouse, B.T.; Sehrawat, S. Immunity and immunopathology to viruses: What decides the outcome? Nat. Rev. Immunol. 2010, 10, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Park, W.B.; Kwon, N.J.; Choi, S.J.; Kang, C.K.; Choe, P.G.; Kim, J.Y.; Yun, J.; Lee, G.W.; Seong, M.W.; Kim, N.J.; et al. Virus Isolation from the First Patient with SARS-CoV-2 in Korea. J. Korean Med. Sci. 2020, 35. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensiv. Care Med. 2020, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Wynants, L.; Van Calster, B.; Bonten, M.M.J.; Collins, G.S.; Debray, T.P.A.; De Vos, M.; Haller, M.C.; Heinze, G.; Moons, K.G.M.; Riley, R.D.; et al. Prediction models for diagnosis and prognosis of covid-19 infection: Systematic review and critical appraisal. BMJ 2020, 369. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.S.; Wu, A.; To, K.F.; Lee, N.; Lam, C.W.; Wong, C.K.; Chan, P.K.; Ng, M.H.; Yu, L.M.; Hui, D.S.; et al. Haematological manifestations in patients with severe acute respiratory syndrome: Retrospective analysis. BMJ 2003, 326, 1358–1362. [Google Scholar] [CrossRef] [Green Version]
- Summerfield, A.; Alves, M.; Ruggli, N.; de Bruin, M.G.; McCullough, K.C. High IFN-alpha responses associated with depletion of lymphocytes and natural IFN-producing cells during classical swine fever. J. Interferon Cytokine Res. 2006, 26, 248–255. [Google Scholar] [CrossRef]
- Kamphuis, E.; Junt, T.; Waibler, Z.; Forster, R.; Kalinke, U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood 2006, 108, 3253–3261. [Google Scholar] [CrossRef]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, X.; Qiu, Y.; Feng, F.; Feng, J.; Jia, Y.; Zhu, H.; Hu, K.; Liu, J.; Liu, Z.; et al. Clinical characteristics of 82 death cases with COVID-19. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Chu, K.H.; Tsang, W.K.; Tang, C.S.; Lam, M.F.; Lai, F.M.; To, K.F.; Fung, K.S.; Tang, H.L.; Yan, W.W.; Chan, H.W.; et al. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int. 2005, 67, 698–705. [Google Scholar] [CrossRef] [Green Version]
- Kollias, A.; Kyriakoulis, K.G.; Dimakakos, E.; Poulakou, G.; Stergiou, G.S.; Syrigos, K. Thromboembolic risk and anticoagulant therapy in COVID-19 patients: Emerging evidence and call for action. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020. [Google Scholar] [CrossRef]
- Castagnoli, R.; Votto, M.; Licari, A.; Brambilla, I.; Bruno, R.; Perlini, S.; Rovida, F.; Baldanti, F.; Marseglia, G.L. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection in Children and Adolescents: A Systematic Review. JAMA Pediatr. 2020. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Jiang, Q.; Xia, X.; Liu, K.; Yu, Z.; Tao, W.; Gong, W.; Han, J.J. Individual Variation of the SARS-CoV2 Receptor ACE2 Gene Expression and Regulation. Preprints 2020, 2020030191. [Google Scholar]
- Cai, G.; Bosse, Y.; Xiao, F.; Kheradmand, F.; Amos, C.I. Tobacco Smoking Increases the Lung Gene Expression of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mehra, M.R.; Desai, S.S.; Kuy, S.; Henry, T.D.; Patel, A.N. Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; Rea, F.; Ludergnani, M.; Apolone, G.; Corrao, G. Renin-Angiotensin-Aldosterone System Blockers and the Risk of Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, H.R.; Adhikari, S.; Pulgarin, C.; Troxel, A.B.; Iturrate, E.; Johnson, S.B.; Hausvater, A.; Newman, J.D.; Berger, J.S.; Bangalore, S.; et al. Renin-Angiotensin-Aldosterone System Inhibitors and Risk of Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Chinn, I.K.; Blackburn, C.C.; Manley, N.R.; Sempowski, G.D. Changes in primary lymphoid organs with aging. Semin. Immunol. 2012, 24, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Yager, E.J.; Ahmed, M.; Lanzer, K.; Randall, T.D.; Woodland, D.L.; Blackman, M.A. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J. Exp. Med. 2008, 205, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Bermejo-Martin, J.F.; Martin-Loeches, I.; Rello, J.; Anton, A.; Almansa, R.; Xu, L.; Lopez-Campos, G.; Pumarola, T.; Ran, L.; Ramirez, P.; et al. Host adaptive immunity deficiency in severe pandemic influenza. Crit. Care 2010, 14, R167. [Google Scholar] [CrossRef] [Green Version]
- Thompson, H.L.; Smithey, M.J.; Surh, C.D.; Nikolich-Zugich, J. Functional and Homeostatic Impact of Age-Related Changes in Lymph Node Stroma. Front. Immunol. 2017, 8, 706. [Google Scholar] [CrossRef] [Green Version]
- Shakerian, L.; Pourpak, Z.; Shamlou, S.; Domsgen, E.; Kazemnejad, A.; Dalili, H.; Nourizadeh, M. Determining Laboratory Reference Values of TREC and KREC in Different Age Groups of Iranian Healthy Individuals. Iran. J. Allergy Asthma Immunol. 2019, 18, 143–152. [Google Scholar] [CrossRef]
- Carlock, M.A.; Ingram, J.G.; Clutter, E.F.; Cecil, N.C.; Ramgopal, M.; Zimmerman, R.K.; Warren, W.; Kleanthous, H.; Ross, T.M. Impact of age and pre-existing immunity on the induction of human antibody responses against influenza B viruses. Hum. Vaccines Immunother. 2019, 15, 2030–2043. [Google Scholar] [CrossRef] [Green Version]
- Gorse, G.J.; Donovan, M.M.; Patel, G.B. Antibodies to coronaviruses are higher in older compared with younger adults and binding antibodies are more sensitive than neutralizing antibodies in identifying coronavirus-associated illnesses. J. Med. Virol. 2020, 92, 512–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Shang, J.; Sun, S.; Tai, W.; Chen, J.; Geng, Q.; He, L.; Chen, Y.; Wu, J.; Shi, Z.; et al. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J. Virol. 2020, 94, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kam, Y.W.; Kien, F.; Roberts, A.; Cheung, Y.C.; Lamirande, E.W.; Vogel, L.; Chu, S.L.; Tse, J.; Guarner, J.; Zaki, S.R.; et al. Antibodies against trimeric S glycoprotein protect hamsters against SARS-CoV challenge despite their capacity to mediate FcgammaRII-dependent entry into B cells in vitro. Vaccine 2007, 25, 729–740. [Google Scholar] [CrossRef]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Wang, X.; Zhang, L.; Lin, A.; Zhao, H.; Fikrig, E.; Montgomery, R.R. Impaired interferon signaling in dendritic cells from older donors infected in vitro with West Nile virus. J. Infect. Dis. 2011, 203, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Legge, K.L.; Braciale, T.J. Lymph node dendritic cells control CD8+ T cell responses through regulated FasL expression. Immunity 2005, 23, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; McCaffery, J.M.; Eichelberger, M.C. Dose-dependent changes in influenza virus-infected dendritic cells result in increased allogeneic T-cell proliferation at low, but not high, doses of virus. J. Virol. 2000, 74, 5460–5469. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.; Jardeleza, M.S.; Kim, I.; Durand, M.L.; Kim, L.; Lobo, A.M. Varicella Zoster Virus Necrotizing Retinitis in Two Patients with Idiopathic CD4 Lymphocytopenia. Ocul. Immunol. Inflamm. 2016, 24, 544–548. [Google Scholar] [CrossRef]
- Steele, R.W.; Britton, H.A.; Anderson, C.T.; Kniker, W.T. Severe combined immunodeficiency with cartilage-hair hypoplasa: In vitro response to thymosin and attempted reconstitution. Pediatr. Res. 1976, 10, 1003–1005. [Google Scholar] [CrossRef] [Green Version]
- Blanco, J.L.; Ambrosioni, J.; Garcia, F.; Martinez, E.; Soriano, A.; Mallolas, J.; Miro, J.M. COVID-19 in patients with HIV: Clinical case series. Lancet HIV 2020. [Google Scholar] [CrossRef]
- Schulert, G.S.; Zhang, M.; Fall, N.; Husami, A.; Kissell, D.; Hanosh, A.; Zhang, K.; Davis, K.; Jentzen, J.M.; Napolitano, L.; et al. Whole-Exome Sequencing Reveals Mutations in Genes Linked to Hemophagocytic Lymphohistiocytosis and Macrophage Activation Syndrome in Fatal Cases of H1N1 Influenza. J. Infect. Dis. 2016, 213, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Bracaglia, C.; Prencipe, G.; De Benedetti, F. Macrophage Activation Syndrome: Different mechanisms leading to a one clinical syndrome. Pediatr. Rheumatol. Online J. 2017, 15, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human leukocyte antigen susceptibility map for SARS-CoV-2. J. Virol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Tseng, H.K.; Trejaut, J.A.; Lee, H.L.; Loo, J.H.; Chu, C.C.; Chen, P.J.; Su, Y.W.; Lim, K.H.; Tsai, Z.U.; et al. Association of HLA class I with severe acute respiratory syndrome coronavirus infection. BMC Med. Genet. 2003, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Delicat, A.; Paweska, J.T.; Gonzalez, J.P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef]
- Clayton, B.A.; Wang, L.F.; Marsh, G.A. Henipaviruses: An updated review focusing on the pteropid reservoir and features of transmission. Zoonoses Public Health 2013, 60, 69–83. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Mohd, H.A.; Al-Tawfiq, J.A.; Memish, Z.A. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) origin and animal reservoir. Virol. J. 2016, 13, 87. [Google Scholar] [CrossRef] [Green Version]
- Pavlovich, S.S.; Lovett, S.P.; Koroleva, G.; Guito, J.C.; Arnold, C.E.; Nagle, E.R.; Kulcsar, K.; Lee, A.; Thibaud-Nissen, F.; Hume, A.J.; et al. The Egyptian Rousette Genome Reveals Unexpected Features of Bat Antiviral Immunity. Cell 2018, 173, 1098–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Cruz-Rivera, P.C.; Kanchwala, M.; Liang, H.; Kumar, A.; Wang, L.F.; Xing, C.; Schoggins, J.W. The IFN Response in Bats Displays Distinctive IFN-Stimulated Gene Expression Kinetics with Atypical RNASEL Induction. J. Immunol. 2018, 200, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Rapin, N.; Bollinger, T.; Misra, V. Lack of inflammatory gene expression in bats: A unique role for a transcription repressor. Sci. Rep. 2017, 7, 2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, M.; Anderson, D.E.; Zhang, Q.; Tan, C.W.; Lim, B.L.; Luko, K.; Wen, M.; Chia, W.N.; Mani, S.; Wang, L.C.; et al. Dampened NLRP3-mediated inflammation in bats and implications for a special viral reservoir host. Nat. Microbiol. 2019, 4, 789–799. [Google Scholar] [CrossRef]
- Ren, R.; Wu, S.; Cai, J.; Yang, Y.; Ren, X.; Feng, Y.; Chen, L.; Qin, B.; Xu, C.; Yang, H.; et al. The H7N9 influenza A virus infection results in lethal inflammation in the mammalian host via the NLRP3-caspase-1 inflammasome. Sci. Rep. 2017, 7, 7625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Li, G.; De, W.; Luo, Z.; Pan, P.; Tian, M.; Wang, Y.; Xiao, F.; Li, A.; Wu, K.; et al. Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin-1beta secretion. Nat. Commun. 2018, 9, 106. [Google Scholar] [CrossRef]
- Coates, B.M.; Staricha, K.L.; Ravindran, N.; Koch, C.M.; Cheng, Y.; Davis, J.M.; Shumaker, D.K.; Ridge, K.M. Inhibition of the NOD-Like Receptor Protein 3 Inflammasome Is Protective in Juvenile Influenza A Virus Infection. Front. Immunol. 2017, 8, 782. [Google Scholar] [CrossRef] [Green Version]
- Tate, M.D.; Ong, J.D.H.; Dowling, J.K.; McAuley, J.L.; Robertson, A.B.; Latz, E.; Drummond, G.R.; Cooper, M.A.; Hertzog, P.J.; Mansell, A. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci. Rep. 2016, 6, 27912. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef]
- Peiris, J.S.; Chu, C.M.; Cheng, V.C.; Chan, K.S.; Hung, I.F.; Poon, L.L.; Law, K.I.; Tang, B.S.; Hon, T.Y.; Chan, C.S.; et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: A prospective study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.M.; Cheng, V.C.; Hung, I.F.; Wong, M.M.; Chan, K.H.; Chan, K.S.; Kao, R.Y.; Poon, L.L.; Wong, C.L.; Guan, Y.; et al. Role of lopinavir/ritonavir in the treatment of SARS: Initial virological and clinical findings. Thorax 2004, 59, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020. [Google Scholar] [CrossRef]
- Cai, Q.; Yang, M.; Liu, D.; Chen, J.; Shu, D.; Xia, J.; Liao, X.; Gu, Y.; Cai, Q.; Yang, Y.; et al. Experimental Treatment with Favipiravir for COVID-19: An Open-Label Control Study. Engineering 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, J.; Cheng, Z.; Wu, J.; Chen, S.; Zhang, Y.; Chen, B.; Lu, M.; Luo, Y.; Zhang, J.; et al. Favipiravir versus Arbidol for COVID-19: A Randomized Clinical Trial. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Khalili, J.S.; Zhu, H.; Mak, A.; Yan, Y.; Zhu, Y. Novel coronavirus treatment with ribavirin: Groundwork for evaluation concerning COVID-19. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockman, L.J.; Bellamy, R.; Garner, P. SARS: Systematic review of treatment effects. PLoS Med. 2006, 3, e343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Wu, Z.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6. [Google Scholar] [CrossRef] [Green Version]
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.J.; Sparks, J.A.; Liew, J.W.; Putman, M.S.; Berenbaum, F.; Duarte-Garcia, A.; Graef, E.R.; Korsten, P.; Sattui, S.E.; Sirotich, E.; et al. A Rush to Judgment? Rapid Reporting and Dissemination of Results and Its Consequences Regarding the Use of Hydroxychloroquine for COVID-19. Ann. Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Hu, J.; Zhang, Z.; Shan Jiang, S.; Han, S.; Yan, D.; Zhuang, R.; Hu, B.; Zhang, Z. Efficacy of hydroxychloroquine in patients with COVID-19: Results of a randomized clinical trial. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Magagnoli, J.; Narendran, S.; Pereira, F.; Cummings, T.; Hardin, J.W.; Sutton, S.S.; Ambati, J. Outcomes of hydroxychloroquine usage in United States veterans hospitalized with Covid-19. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Mahevas, M.; Tran, V.-T.; Roumier, M.; Chabrol, A.; Paule, R.; Guillaud, C.; Gallien, S.; Lepeule, R.; Szwebel, T.-A.; Lescure, X.; et al. No evidence of clinical efficacy of hydroxychloroquine in patients hospitalized for COVID-19 infection with oxygen requirement: Results of a study using routinely collected data to emulate a target trial. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.M.A.; van Haperen, R.; Osterhaus, A.D.M.E.; van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.-J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020, 11, 2251. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell 4Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Luo, P.; Liu, Y.; Qiu, L.; Liu, X.; Liu, D.; Li, J. Tocilizumab treatment in COVID-19: A single center experience. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Di Giambenedetto, S.; Ciccullo, A.; Borghetti, A.; Gambassi, G.; Landi, F.; Visconti, E.; Zileri Dal Verme, L.; Bernabei, R.; Tamburrini, E.; Cauda, R.; et al. Off-label Use of Tocilizumab in Patients with SARS-CoV-2 Infection. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Toniati, P.; Piva, S.; Cattalini, M.; Garrafa, E.; Regola, F.; Castelli, F.; Franceschini, F.; Focà, E.; Andreoli, L.; Latronico, N. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gritti, G.; Raimondi, F.; Ripamonti, D.; Riva, I.; Landi, F.; Alborghetti, L.; Frigeni, M.; Damiani, M.; Micò, C.; Fagiuoli, S.; et al. Use of siltuximab in patients with COVID-19 pneumonia requiring ventilatory support. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G.; De Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Tassan Din, C.; Boffini, N.; et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2020. [Google Scholar] [CrossRef]
- Aouba, A.; Baldolli, A.; Geffray, L.; Verdon, R.; Bergot, E.; Martin-Silva, N.; Justet, A. Targeting the inflammatory cascade with anakinra in moderate to severe COVID-19 pneumonia: Case series. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pontali, E.; Volpi, S.; Antonucci, G.; Castellaneta, M.; Buzzi, D.; Tricerri, F.; Angelelli, A.; Caorsi, R.; Feasi, M.; Calautti, F.; et al. Safety and efficacy of early high-dose IV anakinra in severe COVID-19 lung disease. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the alert for cytokine storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, M.; Maini, R.N.; Woody, J.N.; Holgate, S.T.; Winter, G.; Rowland, M.; Richards, D.; Hussell, T. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 2020, 395, 1407–1409. [Google Scholar] [CrossRef]
- Charles, P.; Elliott, M.J.; Davis, D.; Potter, A.; Kalden, J.R.; Antoni, C.; Breedveld, F.C.; Smolen, J.S.; Eberl, G.; deWoody, K.; et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J. Immunol. 1999, 163, 1521–1528. [Google Scholar]
- Hussell, T.; Pennycook, A.; Openshaw, P.J. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur. J. Immunol. 2001, 31, 2566–2573. [Google Scholar] [CrossRef]
- McDermott, J.E.; Mitchell, H.D.; Gralinski, L.E.; Eisfeld, A.J.; Josset, L.; Bankhead, A.; Neumann, G.; Tilton, S.C.; Schafer, A.; Li, C.; et al. The effect of inhibition of PP1 and TNFalpha signaling on pathogenesis of SARS coronavirus. BMC Syst. Biol. 2016, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Hung, I.F.; To, K.K.; Lee, C.K.; Lee, K.L.; Chan, K.; Yan, W.W.; Liu, R.; Watt, C.L.; Chan, W.M.; Lai, K.Y.; et al. Convalescent plasma treatment reduced mortality in patients with severe pandemic influenza A (H1N1) 2009 virus infection. Clin. Infect. Dis. 2011, 52, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Mair-Jenkins, J.; Saavedra-Campos, M.; Baillie, J.K.; Cleary, P.; Khaw, F.M.; Lim, W.S.; Makki, S.; Rooney, K.D.; Nguyen-Van-Tam, J.S.; Beck, C.R.; et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: A systematic review and exploratory meta-analysis. J. Infect. Dis. 2015, 211, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Xiong, J.; Bao, L.; Shi, Y. Convalescent plasma as a potential therapy for COVID-19. Lancet Infect. Dis. 2020, 20, 398–400. [Google Scholar] [CrossRef]
- Shen, C.; Wang, Z.; Zhao, F.; Yang, Y.; Li, J.; Yuan, J.; Wang, F.; Li, D.; Yang, M.; Xing, L.; et al. Treatment of 5 Critically Ill Patients With COVID-19 With Convalescent Plasma. JAMA 2020. [Google Scholar] [CrossRef]
- Duan, K.; Liu, B.; Li, C.; Zhang, H.; Yu, T.; Qu, J.; Zhou, M.; Chen, L.; Meng, S.; Hu, Y.; et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, J.; Zhao, X.; Liu, C.; Wang, W.; Wang, D.; Xu, W.; Zhang, C.; Yu, J.; Jiang, B.; et al. Clinical Characteristics of Imported Cases of COVID-19 in Jiangsu Province: A Multicenter Descriptive Study. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Minoia, F.; Davi, S.; Horne, A.; Bovis, F.; Demirkaya, E.; Akikusa, J.; Ayaz, N.A.; Al-Mayouf, S.M.; Barone, P.; Bica, B.; et al. Dissecting the heterogeneity of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J. Rheumatol. 2015, 42, 994–1001. [Google Scholar] [CrossRef]
- Sedaghat, Z.; Karimi, N. Guillain Barre syndrome associated with COVID-19 infection: A case report. J. Clin. Neurosci. 2020. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Shalhoub, S.; Mandourah, Y.; Al-Hameed, F.; Al-Omari, A.; Al Qasim, E.; Jose, J.; Alraddadi, B.; Almotairi, A.; Al Khatib, K.; et al. Ribavirin and Interferon Therapy for Critically Ill Patients with Middle East Respiratory Syndrome: A Multicenter Observational Study. Clin. Infect. Dis. 2020, 70, 1837–1844. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef] [Green Version]
- Cantini, F.; Niccoli, L.; Matarrese, D.; Nicastri, E.; Stobbione, P.; Goletti, D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Arabi, Y.M.; Mandourah, Y.; Al-Hameed, F.; Sindi, A.A.; Almekhlafi, G.A.; Hussein, M.A.; Jose, J.; Pinto, R.; Al-Omari, A.; Kharaba, A.; et al. Corticosteroid Therapy for Critically Ill Patients with Middle East Respiratory Syndrome. Am. J. Respir. Crit. Care Med. 2018, 197, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.C.; Ooi, G.C.; Mok, T.Y.; Chan, J.W.; Hung, I.; Lam, B.; Wong, P.C.; Li, P.C.; Ho, P.L.; Lam, W.K.; et al. High-dose pulse versus nonpulse corticosteroid regimens in severe acute respiratory syndrome. Am. J. Respir. Crit. Care Med. 2003, 168, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- So, L.K.; Lau, A.C.; Yam, L.Y.; Cheung, T.M.; Poon, E.; Yung, R.W.; Yuen, K.Y. Development of a standard treatment protocol for severe acute respiratory syndrome. Lancet 2003, 361, 1615–1617. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.J.; Wu, A.; Joynt, G.M.; Yuen, K.Y.; Lee, N.; Chan, P.K.; Cockram, C.S.; Ahuja, A.T.; Yu, L.M.; Wong, V.W.; et al. Severe acute respiratory syndrome: Report of treatment and outcome after a major outbreak. Thorax 2004, 59, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, C.; Leonardi-Bee, J.; Nguyen-Van-Tam, J.; Lim, W.S. Corticosteroids as adjunctive therapy in the treatment of influenza. Cochrane Database Syst. Rev. 2016, 3. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- NIH Covid-19 Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov (accessed on 5 May 2020).
- Abbadi, A.; Loftis, J.; Wang, A.; Yu, M.; Wang, Y.; Shakya, S.; Li, X.; Maytin, E.; Hascall, V. Heparin inhibits proinflammatory and promotes anti-inflammatory macrophage polarization under hyperglycemic stress. J. Biol. Chem. 2020, 295, 4849–4857. [Google Scholar] [CrossRef]
- Chen, D.; Xu, W.; Lei, Z.; Huang, Z.; Liu, J.; Gao, Z.; Peng, L. Recurrence of positive SARS-CoV-2 RNA in COVID-19: A case report. Int. J. Infect. Dis. 2020, 93, 297–299. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, C.; Tang, L.; Hong, Z.; Zhou, J.; Dong, X.; Yin, H.; Xiao, Q.; Tang, Y.; Qu, X.; et al. Prolonged presence of SARS-CoV-2 viral RNA in faecal samples. Lancet Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Mo, H.; Zeng, G.; Ren, X.; Li, H.; Ke, C.; Tan, Y.; Cai, C.; Lai, K.; Chen, R.; Chan-Yeung, M.; et al. Longitudinal profile of antibodies against SARS-coronavirus in SARS patients and their clinical significance. Respirology 2006, 11, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Fontanet, A.; Zhang, P.H.; Zhan, L.; Xin, Z.T.; Baril, L.; Tang, F.; Lv, H.; Cao, W.C. Two-year prospective study of the humoral immune response of patients with severe acute respiratory syndrome. J. Infect. Dis. 2006, 193, 792–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, D.C.; Iblan, I.; Rha, B.; Alqasrawi, S.; Haddadin, A.; Al Nsour, M.; Alsanouri, T.; Ali, S.S.; Harcourt, J.; Miao, C.; et al. Persistence of Antibodies against Middle East Respiratory Syndrome Coronavirus. Emerg. Infect. Dis. 2016, 22, 1824–1826. [Google Scholar] [CrossRef]
- Channappanavar, R.; Zhao, J.; Perlman, S. T cell-mediated immune response to respiratory coronaviruses. Immunol. Res. 2014, 59, 118–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Quan, Y.; Xin, Z.T.; Wrammert, J.; Ma, M.J.; Lv, H.; Wang, T.B.; Yang, H.; Richardus, J.H.; Liu, W.; et al. Lack of peripheral memory B cell responses in recovered patients with severe acute respiratory syndrome: A six-year follow-up study. J. Immunol. 2011, 186, 7264–7268. [Google Scholar] [CrossRef] [Green Version]
- Xiang, F.; Wang, X.; He, X.; Peng, Z.; Yang, B.; Zhang, J.; Zhou, Q.; Ye, H.; Ma, Y.; Li, H.; et al. Antibody Detection and Dynamic Characteristics in Patients with COVID-19. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Liu, W.J.; Zhao, M.; Liu, K.; Xu, K.; Wong, G.; Tan, W.; Gao, G.F. T-cell immunity of SARS-CoV: Implications for vaccine development against MERS-CoV. Antivir. Res. 2017, 137, 82–92. [Google Scholar] [CrossRef]
- Chen, H.; Hou, J.; Jiang, X.; Ma, S.; Meng, M.; Wang, B.; Zhang, M.; Zhang, M.; Tang, X.; Zhang, F.; et al. Response of memory CD8+ T cells to severe acute respiratory syndrome (SARS) coronavirus in recovered SARS patients and healthy individuals. J. Immunol. 2005, 175, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.Y.; Huang, Z.T.; Li, L.; Wu, M.H.; Yu, T.; Koup, R.A.; Bailer, R.T.; Wu, C.Y. Characterization of SARS-CoV-specific memory T cells from recovered individuals 4 years after infection. Arch. Virol. 2009, 154, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Yang, L.T.; Wang, L.Y.; Li, J.; Huang, J.; Lu, Z.Q.; Koup, R.A.; Bailer, R.T.; Wu, C.Y. Long-lived memory T lymphocyte responses against SARS coronavirus nucleocapsid protein in SARS-recovered patients. Virology 2006, 351, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.T.; Peng, H.; Zhu, Z.L.; Li, G.; Huang, Z.T.; Zhao, Z.X.; Koup, R.A.; Bailer, R.T.; Wu, C.Y. Long-lived effector/central memory T-cell responses to severe acute respiratory syndrome coronavirus (SARS-CoV) S antigen in recovered SARS patients. Clin. Immunol. 2006, 120, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Peng, H.; Zhu, Z.; Li, G.; Huang, Z.; Zhao, Z.; Koup, R.A.; Bailer, R.T.; Wu, C. Persistent memory CD4+ and CD8+ T-cell responses in recovered severe acute respiratory syndrome (SARS) patients to SARS coronavirus M antigen. J. Gen. Virol. 2007, 88, 2740–2748. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Vezys, V.; Masopust, D. Sensing and alarm function of resident memory CD8(+) T cells. Nat. Immunol. 2013, 14, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasson, S.C.; Gordon, C.L.; Christo, S.N.; Klenerman, P.; Mackay, L.K. Local heroes or villains: Tissue-resident memory T cells in human health and disease. Cell Mol. Immunol. 2020, 17, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, J.; Perlman, S. T cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus-infected mice. J. Virol. 2010, 84, 9318–9325. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Fett, C.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J. Virol. 2014, 88, 11034–11044. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Immunologic Mechanism Involved in Antiviral Response | Hypothesized Defects Associated with Severe Infection | Effect on Antiviral Response | Proposal of Possible Therapeutic Actions | |
|---|---|---|---|---|
| Innate immunity | Early production of IFNs | Defects in IFN cascade. | Susceptibility to severe course of herpes, VZV and flu | Early Type-I IFN administration. Administration of effective antiviral medications |
| Activity of natural killer cells | Defective cytotoxic functions | Primary hemophagocytic lymphohistiocytosis. Susceptibility to herpetic viruses. | Administration of third-party NK. Blockade of lymphohistiocytosis associated cytokine storm | |
| Late activation of neutrophils | Increased inflammasome signaling | Possible enhancement of inflammatory response to viruses | Anti-cytokine biologic agents | |
| Adaptive immunity | Recruitment and expansion of virus-specific CD4 T cells | Combined immunodeficiencies with defects in the generation of T-cell and B-cell repertoire | Susceptibility to severe infections from various kind of viruses | Administration of third-party specific lymphocytes. Modulation of thymic function |
| Killing of infected cells by virus-specific CD8 T cells | Defective cytotoxic functions and defects in generation of T-cell receptor | Defective response to most viruses | ||
| Production of antibodies | Agammaglobulinemia | Not strictly required to overcome the infection. Reduced memory response to new challenges from the same virus | Hyper-immune plasma from recovered subjects | |
| Production of ADE-capable antibodies | Increased spreading of infection, even to cells not expressing ACE2 | Administration of high dose donor immunoglobulins | ||
| Production of autoantibodies | Vasculitis | Vascular protection; anticoagulation; immunosuppression | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lega, S.; Naviglio, S.; Volpi, S.; Tommasini, A. Recent Insight into SARS-CoV2 Immunopathology and Rationale for Potential Treatment and Preventive Strategies in COVID-19. Vaccines 2020, 8, 224. https://doi.org/10.3390/vaccines8020224
Lega S, Naviglio S, Volpi S, Tommasini A. Recent Insight into SARS-CoV2 Immunopathology and Rationale for Potential Treatment and Preventive Strategies in COVID-19. Vaccines. 2020; 8(2):224. https://doi.org/10.3390/vaccines8020224
Chicago/Turabian StyleLega, Sara, Samuele Naviglio, Stefano Volpi, and Alberto Tommasini. 2020. "Recent Insight into SARS-CoV2 Immunopathology and Rationale for Potential Treatment and Preventive Strategies in COVID-19" Vaccines 8, no. 2: 224. https://doi.org/10.3390/vaccines8020224
APA StyleLega, S., Naviglio, S., Volpi, S., & Tommasini, A. (2020). Recent Insight into SARS-CoV2 Immunopathology and Rationale for Potential Treatment and Preventive Strategies in COVID-19. Vaccines, 8(2), 224. https://doi.org/10.3390/vaccines8020224