Immunoinformatic Analysis of T- and B-Cell Epitopes for SARS-CoV-2 Vaccine Design


Abstract
:1. Introduction
2. Materials and Methods
2.1. B-Cell Epitope Prediction
2.2. T-Cell Epitope Prediction
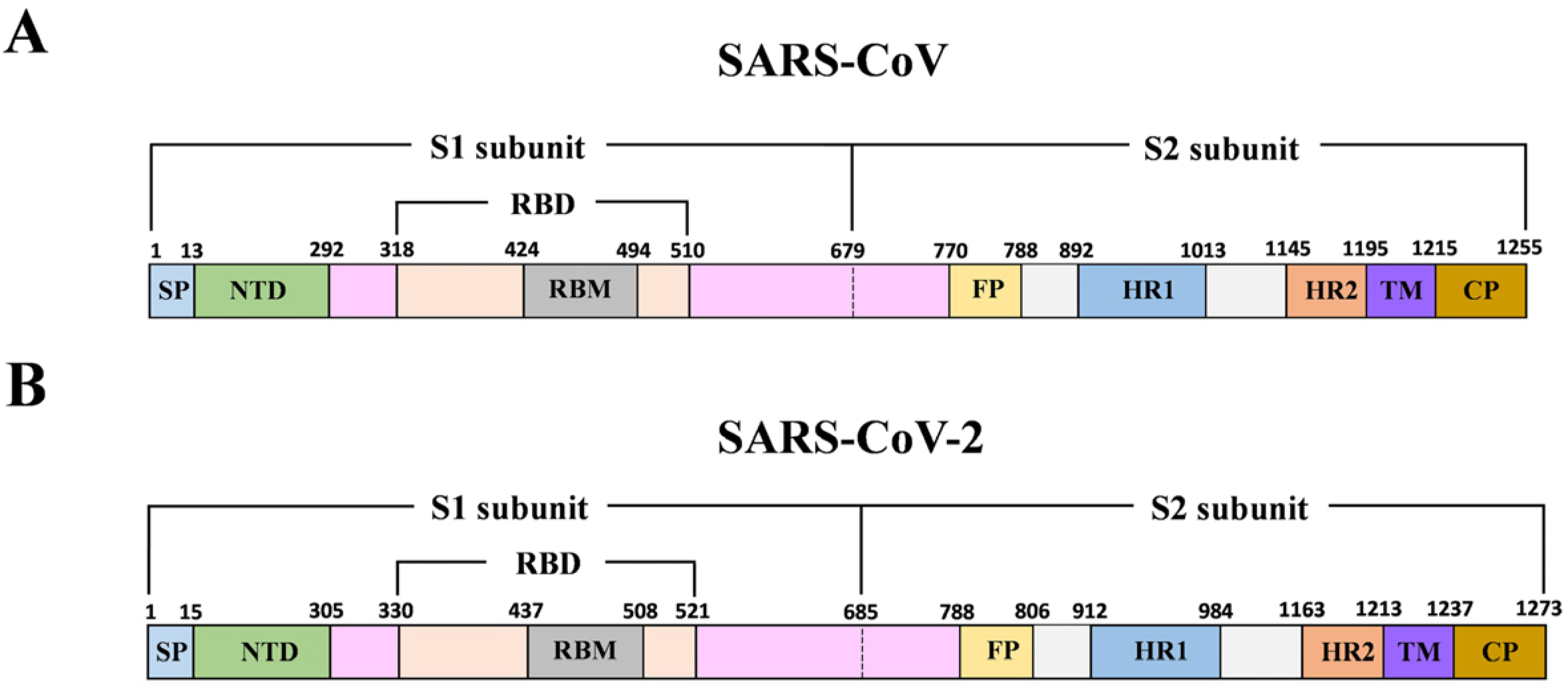
2.3. SARS-CoV S Protein Epitope Acquisition
2.4. Peptide Modeling and Molecular Docking
3. Results
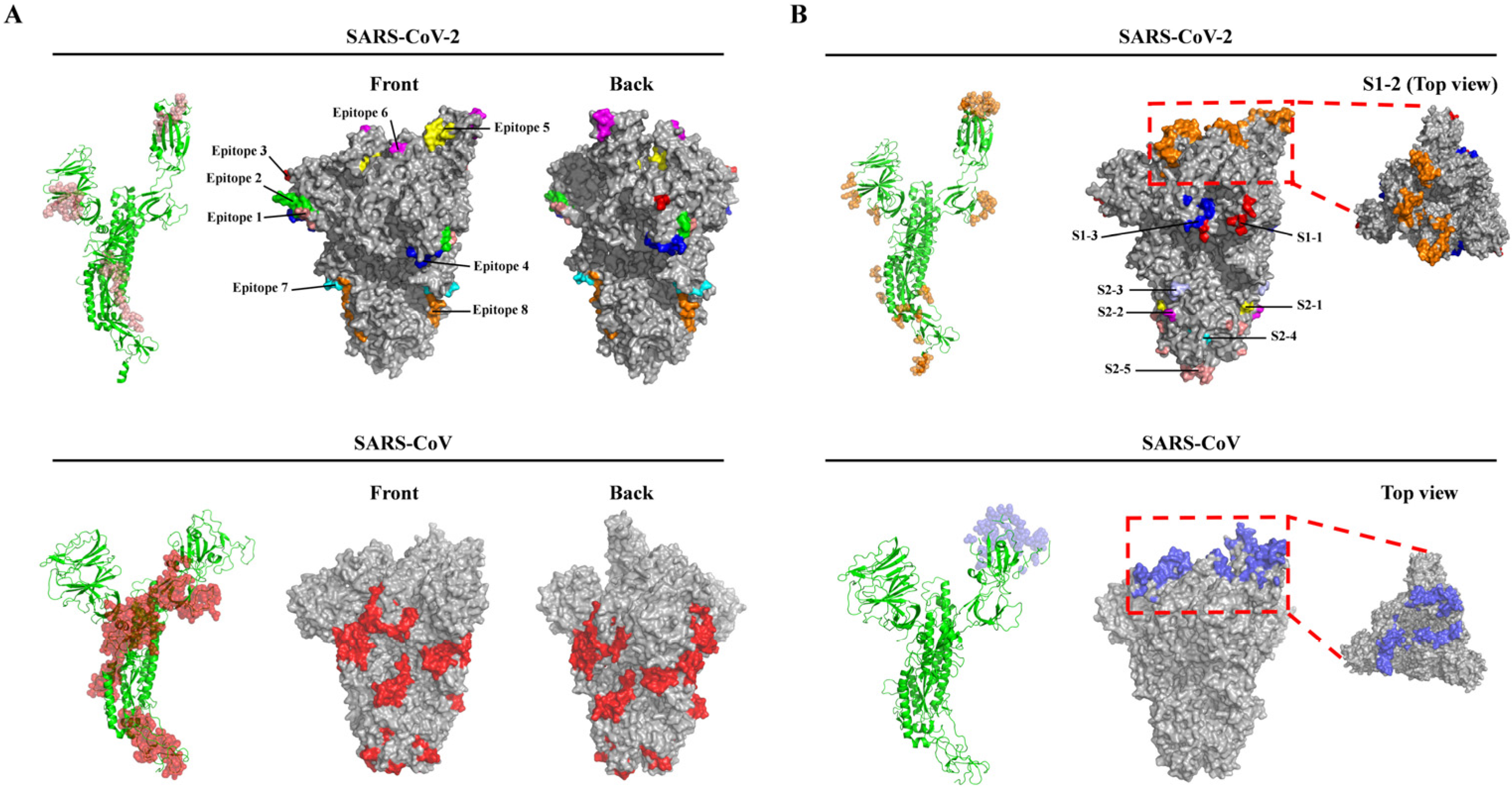
3.1. B-Cell Epitope Prediction and Analysis of Spike Glycoprotein
3.2. T-Cell Epitope Prediction for the SARS-CoV-2 S Protein
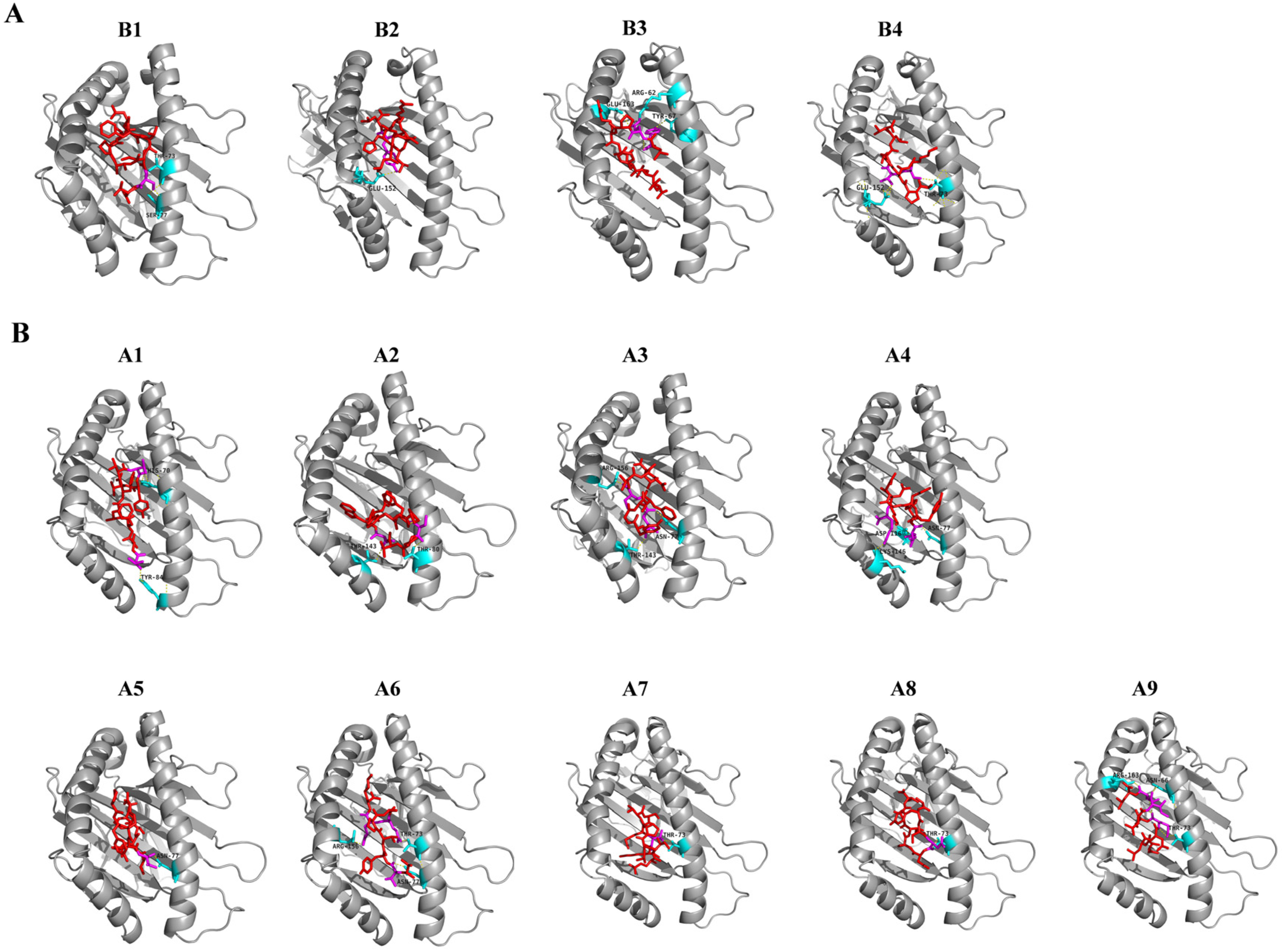
3.3. Molecular Docking of Predicted CD8 T-Cell Epitopes with HLA Alleles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Guo, Y.; Pan, Y.; Zhao, Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 2020, 291, 135–145. [Google Scholar] [CrossRef]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.W.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Fouchier, R.A.M. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Tang, J.; Ma, Y.; Liang, X.; Yang, Y.; Peng, G.; Qi, Q.; Jiang, S.; Li, J.; Du, L.; et al. Receptor usage and cell entry of porcine epidemic diarrhea coronavirus. J. Virol. 2015, 89, 6121–6125. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.K.; Li, W.; Moore, M.J.; Choe, H.; Farzan, M. A 193-Amino Acid Fragment of the SARS Coronavirus S Protein Efficiently Binds Angiotensin-converting Enzyme 2. J. Biol. Chem. 2004, 279, 3197–3201. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Berardi, M.; Li, W.; Farzan, M.; Dormitzer, P.R.; Harrison, S.C. Conformational States of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein Ectodomain. J. Virol. 2006, 80, 6794–6800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Gao, X.-M. Immunological Responses against SARS-Coronavirus Infection in Humans. Cell. Mol. Immunol. 2004, 1, 119–122. [Google Scholar] [PubMed]
- Zhong, X.; Yang, H.; Guo, Z.-F.; Sin, W.-Y.F.; Chen, W.; Xu, J.; Fu, L.; Wu, J.; Mak, C.-K.G.; Cheng, C.-S.S.; et al. B-Cell Responses in Patients Who Have Recovered from Severe Acute Respiratory Syndrome Target a Dominant Site in the S2 Domain of the Surface Spike Glycoprotein. J. Virol. 2005, 79, 3401–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Saleem, S.; Ashfaq, U.A.; Bari, A.; Anwar, F.; Alqahtani, S. Epitope-based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: An immune-informatics study. J. Transl. Med. 2019, 17, 362. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskopf, D.; Angelo, M.A.; Azeredo, E.L.D.; Sidney, J.; Sette, A. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8(+) T cells. Proc. Natl. Acad. Sci. USA 2013, 110, E2046–E2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Arlehamn, C.S.L.; Scriba, T.J.; Dillon, M.B.C.; Oseroff, C.; Hinz, D.; McKinney, D.M.; Pro, S.C.; Sidney, J.; Peters, B.; et al. Development and validation of a broad scheme for prediction of HLA class II restricted T cell epitopes. J. Immunol. Methods 2015, 422, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexis, L.; Pierre, T.; Julien, R.; Marek, V.; Philippe, D.; Pierre, T.J.N.A.R. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast Interaction Refinement in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Efrat, M.; Dina, S.D.; Nelly, A.; Ruth, N.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar]
- Sui, J.; Li, W.; Murakami, A.; Tamin, A.; Matthews, L.J.; Wong, S.K.; Moore, M.J.; Tallarico, A.S.C.; Olurinde, M.; Choe, H.; et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. USA 2004, 101, 2536–2541. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Chakraborti, S.; He, Y.; Roberts, A.; Sheahan, T.; Xiao, X.; Hensley, L.E.; Prabakaran, P.; Rockx, B.; Sidorov, I.A.; et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. USA 2007, 104, 12123–12128. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Ruan, Z.; Wang, L.; Ni, B.; Wu, Y. Identification of a novel conserved HLA-A*0201-restricted epitope from the spike protein of SARS-CoV. BMC Immunol. 2009, 10, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, Y.P.; Lin, J.Y.; Jan, J.T.; Leng, C.H.; Chen, S.L. HLA-A*0201 T-cell epitopes in severe acute respiratory syndrome (SARS) coronavirus nucleocapsid and spike proteins. Biochem. Biophys. Res. Commun. 2006, 344, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, H.; Jiang, X.; Zhang, M.; Cao, X. Identification of an HLA-A*0201-restricted CD8+ T-cell epitope Ssp-1 of SARS-CoV spike protein. Blood 2004, 104, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Eddie, J.; Michelle, R.; Laurie, H.; Gebe, J.A.; Kwok, W.W. Searching immunodominant epitopes prior to epidemic: HLA class II-restricted SARS-CoV spike protein epitopes in unexposed individuals. Int. Immunol. 2008, 21, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Bisht, H.; Roberts, A.; Vogel, L.; Bukreyev, A.; Moss, B. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc. Natl. Acad. Sci. USA 2004, 101, 6641–6646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czub, M.; Weingartl, H.; Czub, S.; He, R.; Cao, J. Evaluation of modified vaccinia virus Ankara based recombinant SARS vaccine in ferrets. Vaccine 2005, 23, 2273–2279. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Y.H.; Luo, B.; Chen, J.; Li, W.; Jiang, S. Identification of immunodominant sites on the spike protein of severe acute respiratory syndrome (SARS) coronavirus: Implication for developing SARS diagnostics and vaccines. J. Immunol. 2004, 173, 4050–4057. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, W.; Yuan, Z.; Jia, R.; Zhao, Z.; Xu, X.; Lv, P.; Zhang, Y.; Jiang, C.; Gao, X.M. A study on antigenicity and receptor-binding ability of fragment 450–650 of the spike protein of SARS coronavirus. Virology 2007, 359, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, G.; Li, J.; Nie, Y.; Shi, X.; Lian, G.; Wang, W.; Yin, X.; Zhao, Y.; Qu, X. Identification of an Antigenic Determinant on the S2 Domain of the Severe Acute Respiratory Syndrome Coronavirus Spike Glycoprotein Capable of Inducing Neutralizing Antibodies. J. Virol. 2004, 78, 6938–6945. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Li, L.; Kao, R.Y.; Kou, B.; Wang, Z.; Zhang, L.; Zhang, H.; Hao, Z.; Tsui, W.H.; Ni, A.; et al. Screening and Identification of Linear B-Cell Epitopes and Entry-Blocking Peptide of Severe Acute Respiratory Syndrome (SARS)-Associated Coronavirus Using Synthetic Overlapping Peptide Library. J. Comb. Chem. 2005, 7, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Xiong, X.; Park, Y.J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A.; et al. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 2019, 176, 1026–1039 e1015. [Google Scholar] [CrossRef] [Green Version]
- Poh, C.M.; Carissimo, G.; Wang, B.; Amrun, S.N.; Lee, C.Y.; Chee, R.S.; Fong, S.W.; Yeo, N.K.; Lee, W.H.; Torres-Ruesta, A.; et al. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat. Commun. 2020, 11, 2806. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.C.; Lin, Y.; Santelli, E.; Sui, J.; Jaroszewski, L.; Stec, B.; Farzan, M.; Marasco, W.A.; Liddington, R.C. Structural Basis of Neutralization by a Human Anti-severe Acute Respiratory Syndrome Spike Protein Antibody, 80R. J. Biol. Chem. 2006, 281, 34610–34616. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, P.; Gan, J.; Feng, Y.; Zhu, Z.; Choudhry, V.; Xiao, X.; Ji, X.; Dimitrov, D.S. Structure of Severe Acute Respiratory Syndrome Coronavirus Receptor-binding Domain Complexed with Neutralizing Antibody. J. Biol. Chem. 2006, 281, 15829–15836. [Google Scholar] [CrossRef] [Green Version]
- De Groot, A.S.; Moise, L.; Terry, F.; Gutierrez, A.H.; Hindocha, P.; Richard, G.; Hoft, D.F.; Ross, T.M.; Noe, A.R.; Takahashi, Y.; et al. Better Epitope Discovery, Precision Immune Engineering, and Accelerated Vaccine Design Using Immunoinformatics Tools. Front. Immunol. 2020, 11, 442. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.H.; Loving, C.; Moise, L.; Terry, F.E.; Brockmeier, S.L.; Hughes, H.R.; Martin, W.D.; De Groot, A.S. In Vivo Validation of Predicted and Conserved T Cell Epitopes in a Swine Influenza Model. PLoS ONE 2016, 11, e0159237. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, J.S.; Karuppannan, A.K.; Tan, S.; Gauger, P.; Halbur, P.G.; Gerber, P.F.; De Groot, A.S.; Moise, L.; Opriessnig, T. A prime-boost concept using a T-cell epitope-driven DNA vaccine followed by a whole virus vaccine effectively protected pigs in the pandemic H1N1 pig challenge model. Vaccine 2019, 37, 4302–4309. [Google Scholar] [CrossRef]
- Song, P.; Li, W.; Xie, J.; Hou, Y.; You, C. Cytokine Storm Induced by SARS-CoV-2. Clin. Chim. Acta Int. J. Clin. Chem. 2020. [Google Scholar] [CrossRef]
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013, 11, 836–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, C.Y.; Ong, H.K.; Yeap, S.K.; Ho, K.L.; Tan, W.S. Recent Advances in the Vaccine Development Against Middle East Respiratory Syndrome-Coronavirus. Front. Microbiol. 2019, 10, 1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Epitope | Position | Sequence | Length | VaxiJen Score | Identity | Domain or Motif |
|---|---|---|---|---|---|---|
| 1 | 15–31 | CVNLTTRTQLPPAYTNS | 17 | 1.2219 | 100% | NTD |
| 2 | 62–75 | VTWFHAIHVSGTNG | 14 | 0.5786 | 100% | NTD |
| 3 | 141–152 | LGVYYHKNNKSW | 13 | 0.8156 | 100% | NTD |
| 4 | 208–220 | TPINLVRDLPQGF | 13 | 0.4768 | 100% | NTD |
| 5 | 405–418 | DEVRQIAPGQTGKI | 14 | 0.9312 | 100% | RBD |
| 6 | 441–448 | LDSKVGGN | 8 | 0.8773 | 100% | RBM |
| 7 | 657–664 | NNSYECDI | 8 | 0.6539 | 100% | S1 (C-terminal) |
| 8 | 696–709 | TMSLGAENSVAYSN | 14 | 0.6780 | 100% | S2 (N-terminal) |
| 9 | 1154–1169 | KYFKNHTSPDVDLGDI | 16 | 0.7333 | 100% | S2 (C-terminal) |
| Region | Sequences | Domain/Motif |
|---|---|---|
| S1–1 | K97, S98, K187, P209, N211, E281, N282 | NTD |
| S1–2 | T415, K417, D420, Y421, N439, N440, S443, K444, V445, G446, G447, N448, * Y449, * N450, L452, R454, K458, S459, N460, K462, S477, P491, * L492, Q493, S494, * G496, F497, Q498, P499, * T500, N501, * G502, V503, G504, * Y505 | RBD |
| S1–3 | N556, K558, L560, P561, F562, Q563, L582 | C-terminal |
| S2–1 | N703, S704, V705 | N-terminal |
| S2–2 | P793, I794 | FP |
| S2–3 | P809, S810, K811 | Between FP and HR1 |
| S2–4 | Y917, E918 | HR1 |
| S2–5 | Q1071, N1074, T1100, L1141, Q1142, P1143, E1144, L1145, D1146, S1147 | Between HR1 and HR2 |
| Start | End | Length | Peptide | Median Consensus Percentile | VaxiJen Score |
|---|---|---|---|---|---|
| 5 | 19 | 15 | LVLLPLVSSQCVNLT | 20 | 1.2086 |
| 52 | 66 | 15 | QDLFLPFFSNVTWFH | 16 | 0.4159 |
| 57 | 71 | 15 | PFFSNVTWFHAIHVS | 16 | 0.7656 |
| 113 | 127 | 15 | KTQSLLIVNNATNVV | 13 | 0.6303 |
| 116 | 130 | 15 | SLLIVNNATNVVIKV | 14 | 0.4707 |
| 139 | 153 | 15 | PFLGVYYHKNNKSWM | 20 | 0.6641 |
| 199 | 213 | 15 | GYFKIYSKHTPINLV | 18 | 0.9278 |
| 209 | 223 | 15 | PINLVRDLPQGFSAL | 15 | 0.6086 |
| 230 | 244 | 15 | PIGINITRFQTLLAL | 13 | 0.8877 |
| 239 | 253 | 15 | QTLLALHRSYLTPGD | 16 | 0.6708 |
| 263 | 277 | 15 | AAYYVGYLQPRTFLL | 16 | 0.6073 |
| 309 | 323 | 15 | EKGIYQTSNFRVQPT | 18 | 0.9243 |
| 312 | 326 | 15 | IYQTSNFRVQPTESI | 11 | 0.7459 |
| 345 | 359 | 15 | TRFASVYAWNRKRIS | 16 | 0.4963 |
| 390 | 404 | 15 | LCFTNVYADSFVIRG | 17 | 0.5950 |
| 506 | 520 | 15 | QPYRVVVLSFELLHA | 14 | 0.9109 |
| 821 | 835 | 15 | LLFNKVTLADAGFIK | 19 | 0.6327 |
| 896 | 910 | 15 | IPFAMQMAYRFNGIG | 6.5 | 1.2828 |
| 1013 | 1027 | 15 | IRAAEIRASANLAAT | 14 | 0.6785 |
| 1016 | 1030 | 15 | AEIRASANLAATKMS | 13 | 0.8255 |
| 1060 | 1074 | 15 | VVFLHVTYVPAQEKN | 20 | 1.1720 |
| 1212 | 1226 | 15 | WPWYIWLGFIAGLIA | 19 | 0.7293 |
| Number | Start | End | Peptide | Global Energy (kcal/mol) | vdW Energy (kcal/mol) | H-Bonding Energy (kcal/mol) | Interacting Residues | VaxiJen Score | Alleles |
|---|---|---|---|---|---|---|---|---|---|
| B1 | 714 | 722 | IPTNFTISV | −26.14 | −22.96 | −0.83 | Thr73, Ser77 | 0.882 | HLA-B7 |
| B2 | 241 | 249 | LLALHRSYL | −25.81 | −25.38 | −2.01 | Glu152 | 0.5241 | |
| B3 | 269 | 277 | YLQPRTFLL | −17.28 | −21.98 | −0.41 | Arg62, Tyr67, Glu163 | 0.4532 | |
| B4 | 526 | 534 | GPKKSTNLV | −11.55 | −18.15 | −1.99 | Thr73, Glu152 | 0.6828 | |
| A1 | 1060 | 1068 | VVFLHVTYV | −46.66 | −21.71 | −1.89 | His70, Tyr84 | 1.5122 | HLA-A*01:01 |
| A2 | 57 | 65 | PFFSNVTWF | −37.65 | −25.71 | −1.94 | Thr80, Thr143 | 0.6638 | |
| A3 | 755 | 763 | QYGSFCTQL | −26.61 | −20.71 | −0.82 | Asn77, Thr143, Arg156 | 1.2906 | |
| A4 | 142 | 150 | GVYYHKNNK | −26.01 | −22.48 | −0.62 | Asn77, Asp116, Lys146 | 0.8264 | |
| A5 | 507 | 515 | PYRVVVLSF | −17.96 | −16.51 | −1.87 | Asn77 | 1.0281 | |
| A6 | 1065 | 1073 | VTYVPAQEK | −14.92 | −24.24 | −3.52 | Thr73, Asn77, Arg156 | 0.8132 | |
| A7 | 725 | 733 | EILPVSMTK | −13.80 | −12.10 | −1.87 | Thr73 | 1.6842 | |
| A8 | 706 | 714 | AYSNNSIAI | −13.29 | −13.02 | −1.81 | Thr73 | 0.8274 | |
| A9 | 1192 | 1200 | NLNESLIDL | −12.90 | −19.85 | −4.32 | Asn66, Thr73, Arg163 | 0.6827 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Mai, J.; Zhou, W.; Yu, W.; Zhan, Y.; Wang, N.; Epstein, N.D.; Yang, Y. Immunoinformatic Analysis of T- and B-Cell Epitopes for SARS-CoV-2 Vaccine Design. Vaccines 2020, 8, 355. https://doi.org/10.3390/vaccines8030355
Wang D, Mai J, Zhou W, Yu W, Zhan Y, Wang N, Epstein ND, Yang Y. Immunoinformatic Analysis of T- and B-Cell Epitopes for SARS-CoV-2 Vaccine Design. Vaccines. 2020; 8(3):355. https://doi.org/10.3390/vaccines8030355
Chicago/Turabian StyleWang, Dongliang, Jinhui Mai, Wenfeng Zhou, Wanting Yu, Yang Zhan, Naidong Wang, Neal D. Epstein, and Yi Yang. 2020. "Immunoinformatic Analysis of T- and B-Cell Epitopes for SARS-CoV-2 Vaccine Design" Vaccines 8, no. 3: 355. https://doi.org/10.3390/vaccines8030355
APA StyleWang, D., Mai, J., Zhou, W., Yu, W., Zhan, Y., Wang, N., Epstein, N. D., & Yang, Y. (2020). Immunoinformatic Analysis of T- and B-Cell Epitopes for SARS-CoV-2 Vaccine Design. Vaccines, 8(3), 355. https://doi.org/10.3390/vaccines8030355