Identification of Vaccinia Virus Inhibitors and Cellular Functions Necessary for Efficient Viral Replication by Screening Bioactives and FDA-Approved Drugs

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Virus Infection
2.3. Gaussia Luciferase (Gluc) Reporter Screening Assay
2.4. Cell Viability Assays
2.5. Determination of the Effects of Compounds on Gluc Enzyme Reaction
2.6. High-Throughput Screening Data Processing and Statistical Analysis
3. Results
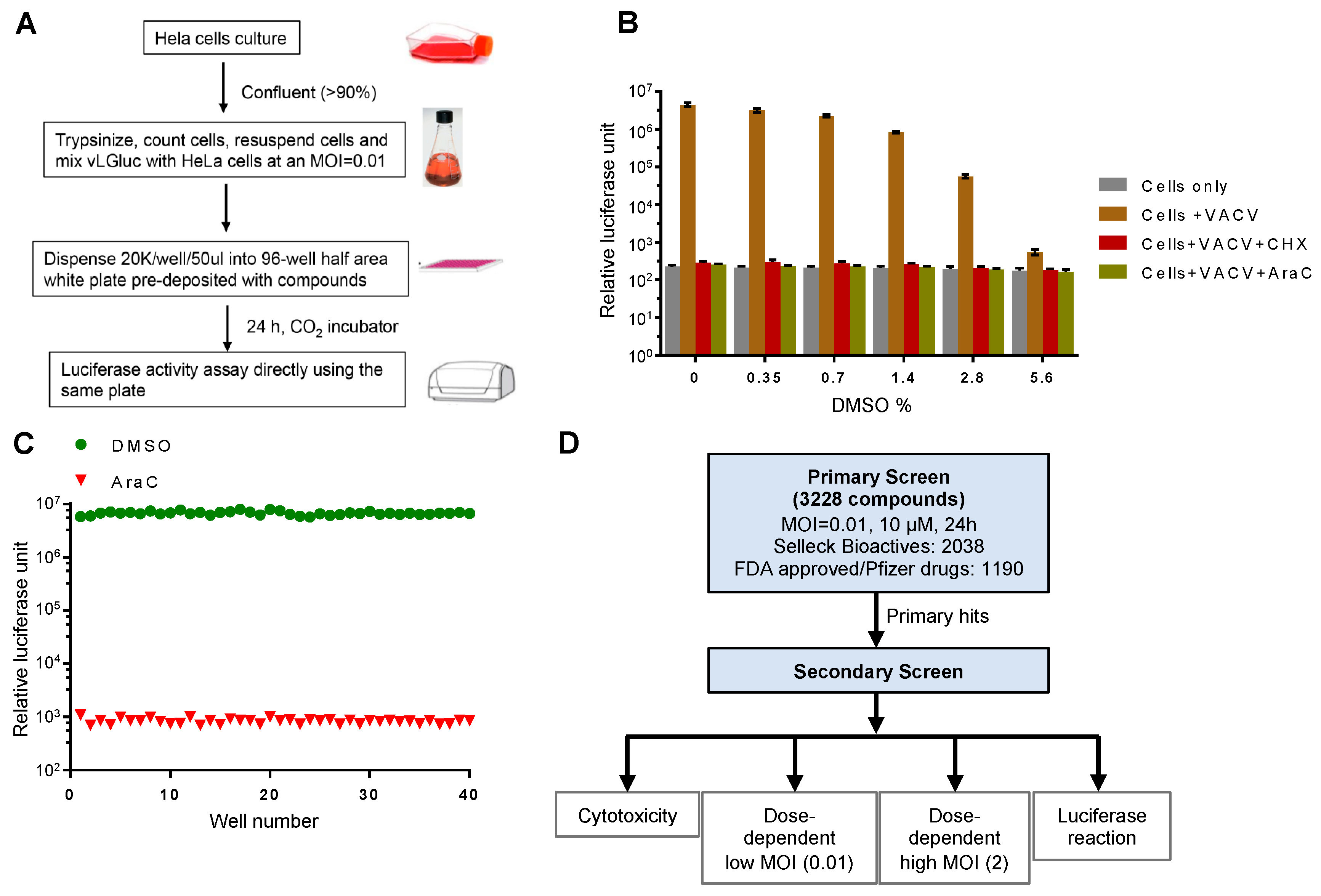
3.1. Development and Optimization of a Secreted Gluc-Based Assay Procedure to Screen VACV Inhibitors
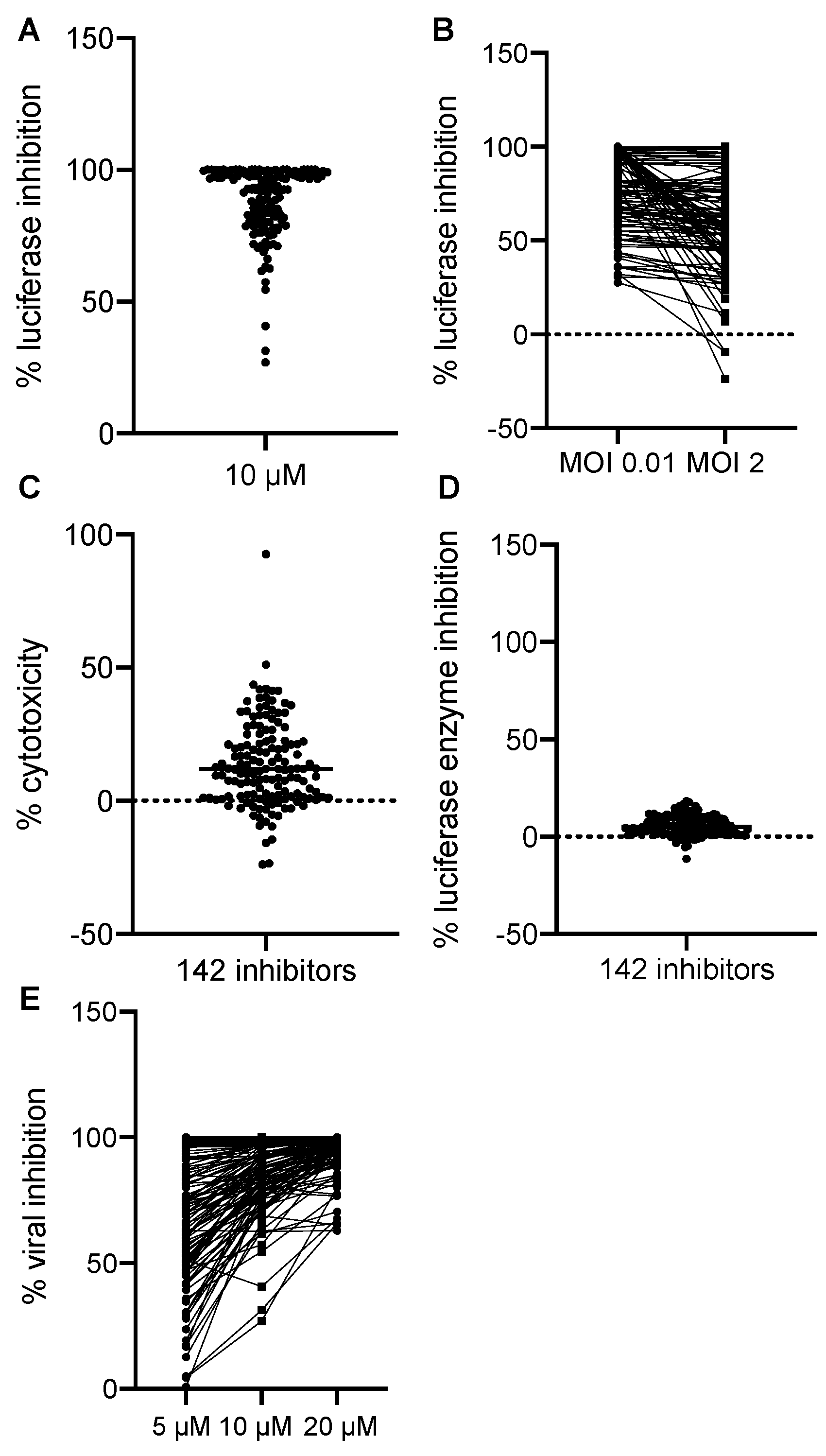
3.2. Identification of VACV Inhibitors Targeting a Broad Range of Cellular Targets and Processes
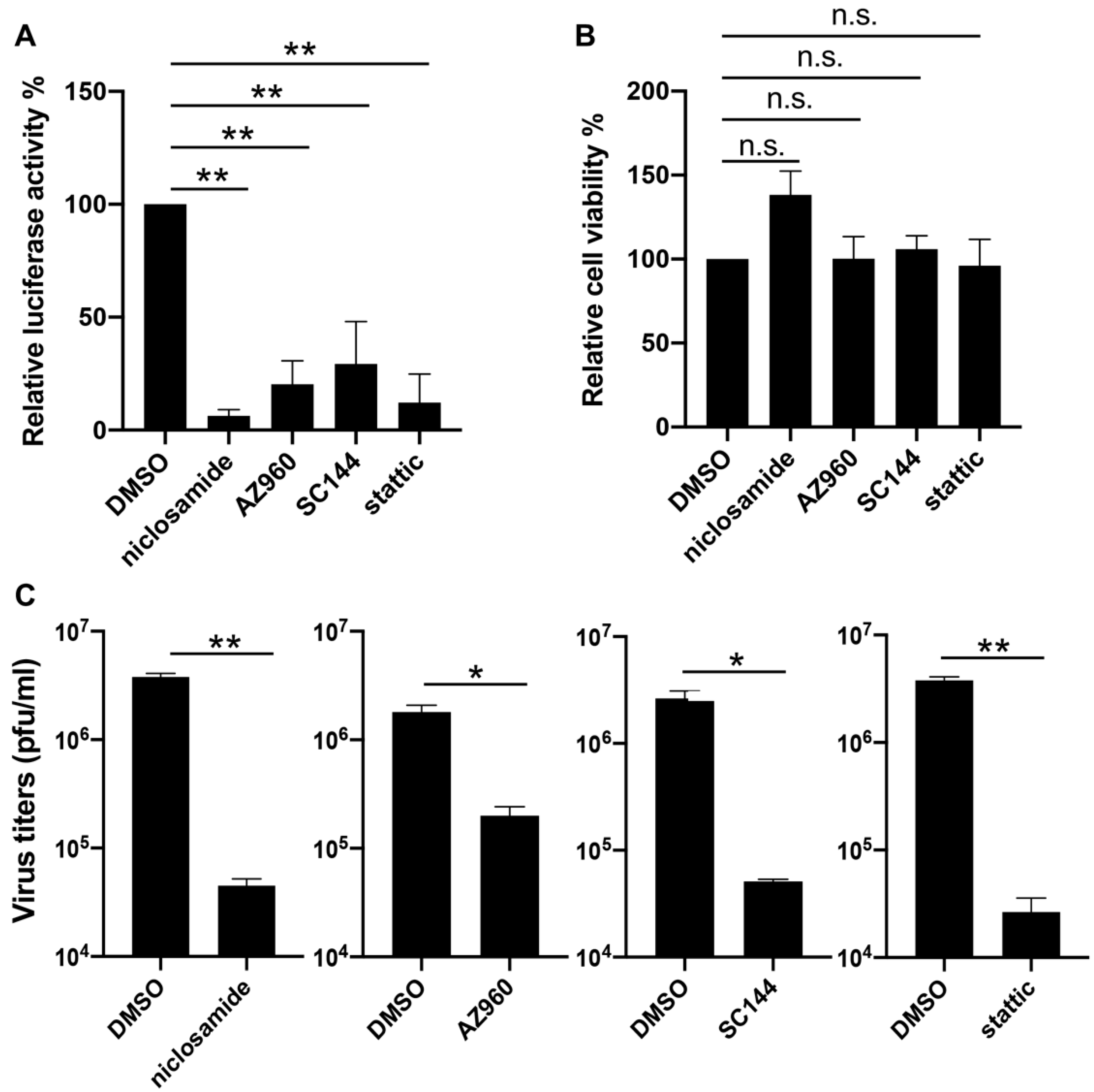
3.3. Verification of the Suppressing Effects of JAK-STAT3 Pathway Inhibitors on VACV Replication
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Chapter 1: Smallpox: Eradicating an ancient scourge. In Bugs, Drugs and Smoke; WHO Press: Geneva, Switzerland, 2011; pp. 3–21. [Google Scholar]
- Moss, B. Poxviridae, Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2129–2159. [Google Scholar]
- Noyce, R.S.; Lederman, S.; Evans, D.H. Construction of an infectious horsepox virus vaccine from chemically synthesized DNA fragments. PLoS ONE 2018, 13, e0188453. [Google Scholar] [CrossRef] [PubMed]
- Beer, E.M.; Rao, V.B. A systematic review of the epidemiology of human monkeypox outbreaks and implications for outbreak strategy. PLoS Negl. Trop. Dis. 2019, 13, e0007791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, E.; Kantele, A.; Koopmans, M.; Asogun, D.; Yinka-Ogunleye, A.; Ihekweazu, C.; Zumla, A. Human Monkeypox: Epidemiologic and Clinical Characteristics, Diagnosis, and Prevention. Infect. Dis. Clin. N. Am. 2019, 33, 1027–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention (CDC). Update: Multistate outbreak of monkeypox—Illinois, Indiana, Kansas, Missouri, Ohio, and Wisconsin, 2003. MMWR Morb. Mortal Wkly Rep. 2003, 52, 642–646. [Google Scholar]
- Vaughan, A.; Aarons, E.; Astbury, J.; Balasegaram, S.; Beadsworth, M.; Beck, C.R.; Chand, M.; O’Connor, C.; Dunning, J.; Ghebrehewet, S.; et al. Two cases of monkeypox imported to the United Kingdom, September 2018. Eurosurveillance 2018, 23, 1800509. [Google Scholar] [CrossRef] [PubMed]
- Ng, O.T.; Lee, V.; Marimuthu, K.; Vasoo, S.; Chan, G.; Lin, R.T.P.; Leo, S.Y. A case of imported Monkeypox in Singapore. Lancet Infect. Dis. 2019, 19, 1166. [Google Scholar] [CrossRef]
- Erez, N.; Achdout, H.; Milrot, E.; Schwartz, Y.; Wiener-Well, Y.; Paran, N.; Politi, B.; Tamir, H.; Israely, T.; Weiss, S.; et al. Diagnosis of Imported Monkeypox, Israel, 2018. Emerg. Infect. Dis. 2019, 25, 980–983. [Google Scholar] [CrossRef] [Green Version]
- Meza-Romero, R.; Navarrete-Dechent, C.; Downey, C. Molluscum contagiosum: An update and review of new perspectives in etiology, diagnosis, and treatment. Clin. Cosmet. Investig. Dermatol. 2019, 12, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Lewis-Jones, S. Zoonotic poxvirus infections in humans. Curr. Opin. Infect. Dis. 2004, 17, 81–89. [Google Scholar] [CrossRef]
- Oliveira, G.P.; Rodrigues, R.A.L.; Lima, M.T.; Drumond, B.P.; Abrahão, J.S. Poxvirus Host Range Genes and Virus-Host Spectrum: A Critical Review. Viruses 2017, 9, 331. [Google Scholar] [CrossRef] [Green Version]
- Essbauer, S.; Pfeffer, M.; Meyer, H. Zoonotic poxviruses. Vet. Microbiol. 2010, 140, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Merchlinsky, M.; Albright, A.; Olson, V.; Schiltz, H.; Merkeley, T.; Hughes, C.; Petersen, B.; Challberg, M. The development and approval of tecoviromat (TPOXX®), the first antiviral against smallpox. Antivir. Res. 2019, 168, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Chan-Tack, K.M.; Harrington, P.R.; Choi, S.Y.; Myers, L.; O’Rear, J.; Seo, S.; McMillan, D.; Ghantous, H.; Birnkrant, D.; Sherwat, A.I. Assessing a drug for an eradicated human disease: US Food and Drug Administration review of tecovirimat for the treatment of smallpox. Lancet Infect. Dis. 2019, 19, e221–e224. [Google Scholar] [CrossRef]
- Pires, M.A.; Rodrigues, N.F.S.; de Oliveira, D.B.; de Assis, F.L.; Costa, G.B.; Kroon, E.G.; Mota, B.E.F. In vitro susceptibility to ST-246 and Cidofovir corroborates the phylogenetic separation of Brazilian Vaccinia virus into two clades. Antivir. Res. 2018, 152, 36–44. [Google Scholar] [CrossRef]
- Delaune, D.; Iseni, F. Drug Development against Smallpox: Present and Future. Antimicrob. Agents Chemother. 2020, 64, e01683–e01719. [Google Scholar] [CrossRef]
- Duraffour, S.; Andrei, G.; Topalis, D.; Krečmerová, M.; Crance, J.M.; Garin, D.; Snoeck, R. Mutations conferring resistance to viral DNA polymerase inhibitors in camelpox virus give different drug-susceptibility profiles in vaccinia virus. J. Virol. 2012, 86, 7310–7325. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef]
- Pant, A.; Cao, S.; Yang, Z. Asparagine Is a Critical Limiting Metabolite for Vaccinia Virus Protein Synthesis during Glutamine Deprivation. J. Virol. 2019, 93, e01834–e01918. [Google Scholar] [CrossRef] [Green Version]
- Renis, H.E.; Johnson, H.G. Inhibition of plaque formation of vaccinia virus by cytosine arabinoside hydrochloride. Bacteriol. Proc. 1962, 45, 45. [Google Scholar]
- Hung, J.J.; Chung, C.S.; Chang, W. Molecular chaperone Hsp90 is important for vaccinia virus growth in cells. J. Virol. 2002, 76, 1379–1390. [Google Scholar] [CrossRef] [Green Version]
- Beerli, C.; Yakimovich, A.; Kilcher, S.; Reynoso, G.V.; Fläschner, G.; Müller, D.J.; Hickman, H.D.; Mercer, J. Vaccinia virus hijacks EGFR signalling to enhance virus spread through rapid and directed infected cell motility. Nat. Microbiol. 2019, 4, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.; Way, M. The role of signalling and the cytoskeleton during Vaccinia Virus egress. Virus Res. 2015, 209, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Meade, N.; King, M.; Munger, J.; Walsh, D. mTOR Dysregulation by Vaccinia Virus F17 Controls Multiple Processes with Varying Roles in Infection. J. Virol. 2019, 93, e00784–e00819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meade, N.; Furey, C.; Li, H.; Verma, R.; Chai, Q.; Rollins, M.G.; DiGiuseppe, S.; Naghavi, M.H.; Walsh, D. Poxviruses Evade Cytosolic Sensing through Disruption of an mTORC1-mTORC2 Regulatory Circuit. Cell 2018, 174, 1143–1157. [Google Scholar] [CrossRef] [Green Version]
- Slezak, K.; Michalik, M.; Kowalczyk, A.; Rokita, H. YY1 is recruited to the cytoplasm of vaccinia virus-infected human macrophages by the Crm1 system. Virus Res. 2004, 102, 177–784. [Google Scholar] [CrossRef]
- Satheshkumar, P.S.; Anton, L.C.; Sanz, P.; Moss, B. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 2009, 83, 2469–2479. [Google Scholar] [CrossRef] [Green Version]
- Sivan, G.; Martin, S.E.; Myers, T.G.; Buehler, E.; Szymczyk, K.H.; Ormanoglu, P.; Moss, B. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3519–3524. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Snijder, B.; Sacher, R.; Burkard, C.; Bleck, C.K.E.; Stahlberg, H.; Pelkmans, L.; Helenius, A. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Rep. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- Maluquer de Motes, C.; Smith, G.L. Vaccinia virus protein A49 activates Wnt signalling by targetting the E3 ligase β-TrCP. J. Gen. Virol. 2017, 98, 3086–3092. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.R.; Vega, F.M.; Blanco, S.; Barcia, R.; Lazo, P.A. The vaccinia virus B1R kinase induces p53 downregulation by an Mdm2-dependent mechanism. Virology 2004, 328, 254–265. [Google Scholar] [CrossRef]
- Remichkova, M.; Petrov, N.; Galabov, A.S. Synergistic Combination Effect of Cidofovir and Idoxuridine on Vaccinia Virus Replication. Antivir. Chem. Chemother. 2016, 17, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, B. Poxvirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smee, D.F. Orthopoxvirus inhibitors that are active in animal models: An update from 2008 to 2012. Future Virol. 2013, 8, 891–901. [Google Scholar] [CrossRef] [Green Version]
- Gammon, D.B.; Gowrishankar, B.; Duraffour, S.; Andrei, G.; Upton, C.; Evans, D.H. Vaccinia virus-encoded ribonucleotide reductase subunits are differentially required for replication and pathogenesis. PLoS Pathog. 2010, 6, e1000984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Child, S.J.; Franke, C.A.; Hruby, D.E. Inhibition of vaccinia virus replication by nicotinamide: Evidence for ADP-ribosylation of viral proteins. Virus Res. 1988, 9, 119–132. [Google Scholar] [CrossRef]
- Ryerson, M.R.; Richards, M.M.; Kvansakul, M.; Hawkins, C.J.; Shisler, J.L. Vaccinia Virus Encodes a Novel Inhibitor of Apoptosis That Associates with the Apoptosome. J. Virol. 2017, 91, e01385–e01417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baixeras, E.; Cebrián, A.; Albar, J.P.; Salas, J.; Martínez, A.C.; Viñuela, E.; Revilla, Y. Vaccinia virus-induced apoptosis in immature B lymphocytes: Role of cellular Bcl-2. Virus Res. 1998, 58, 107–113. [Google Scholar] [CrossRef]
- Piróg, K.A.; Kowalczyk, A.K.; Rokita, H.B. Changes in Bcl-2 expression in vaccinia virus-infected human peripheral blood monocytes. Viral Immunol. 2005, 18, 224–231. [Google Scholar] [CrossRef]
- Liu, Y.; Wolff, K.C.; Jacobs, B.L.; Samuel, C.E. Vaccinia virus E3L interferon resistance protein inhibits the interferon-induced adenosine deaminase A-to-I editing activity. Virology 2001, 289, 378–387. [Google Scholar] [CrossRef] [Green Version]
- Connor, J.D.; Sweetman, L.; Carey, S.; Stuckey, M.A.; Buchanan, R. Effect of adenosine deaminase upon the antiviral activity in vitro of adenine arabinoside for vaccinia virus. Antimicrob. Agents Chemother. 1974, 6, 630–636. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.A.J.; Ryzhakov, G.; Cooray, S.; Randow, F.; Smith, G.L. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 2008, 4, e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Stuart, J.H.; Talbot-Cooper, C.; Agrawal-Singh, S.; Huntly, B.; Smid, A.I.; Snowden, J.S.; Dupont, L.; Smith, G.L. Histone deacetylase 4 promotes type I interferon signaling, restricts DNA viruses, and is degraded via vaccinia virus protein C6. Proc. Natl. Acad. Sci. USA 2019, 116, 11997–12006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greseth, M.D.; Traktman, P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog. 2014, 10, e1004021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reading, P.C.; Moore, J.B.; Smith, G.L. Steroid hormone synthesis by vaccinia virus suppresses the inflammatory response to infection. J. Exp. Med. 2003, 197, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, G.C.; Rodger, G.; Murphy, B.J.; Law, M.; Krauss, O.; Hollinshead, M.; Smith, G.L. Vaccinia virus cores are transported on microtubules. J. Gen. Virol. 2003, 84, 2443–2458. [Google Scholar] [CrossRef]
- Ward, B.M.; Moss, B. Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J. Virol. 2001, 75, 11651–11663. [Google Scholar] [CrossRef] [Green Version]
- Rietdorf, J.; Ploubidou, A.; Reckmann, I.; Holmström, A.; Frischknecht, F.; Zettl, M.; Zimmermann, T.; Way, M. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat. Cell Biol. 2001, 3, 992–1000. [Google Scholar] [CrossRef]
- Hollinshead, M.; Rodger, G.; Van Eijl, H.; Law, M.; Hollinshead, R.; Vaux, D.J.; Smith, G.L. Vaccinia virus utilizes microtubules for movement to the cell surface. J. Cell Biol. 2001, 154, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Bonjardim, C.A. Viral exploitation of the MEK/ERK pathway—A tale of vaccinia virus and other viruses. Virology 2017, 507, 267–275. [Google Scholar] [CrossRef]
- Torres, A.A.; Albarnaz, J.D.; Bonjardim, C.A.; Smith, G.L. Multiple Bcl-2 family immunomodulators from vaccinia virus regulate MAPK/AP-1 activation. J. Gen. Virol. 2016, 97, 2346–2351. [Google Scholar] [CrossRef]
- Andrade, A.A.; Silva, P.N.G.; Pereira, A.C.T.C.; De Sousa, L.P.; Ferreira, P.C.P.; Gazzinelli, R.T.; Kroon, E.G.; Ropert, C.; Bonjardim, C.A. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 2004, 381, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizopoulos, Z.; Balistreri, G.; Kilcher, S.; Martin, C.K.; Syedbasha, M.; Helenius, A.; Mercer, J. Vaccinia Virus Infection Requires Maturation of Macropinosomes. Traffic 2015, 16, 814–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbar, R.; Rogers, E.; Murooka, T.; Kislinger, T.; Fish, E.N. Glomulin: A permissivity factor for vaccinia virus infection. J. Interferon Cytokine Res. 2012, 32, 127–137. [Google Scholar] [CrossRef]
- Singh, P.; Yao, Y.; Weliver, A.; Broxmeyer, H.E.; Hong, S.C.; Chang, C.H. Vaccinia virus infection modulates the hematopoietic cell compartments in the bone marrow. Stem Cells 2008, 26, 1009–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, F.G.G.; Torres, A.A.; De Oliveira, L.C.; Da Cruz, A.F.P.; Soares-Martins, J.A.P.; Pereira, A.C.T.C.; Trindade, G.S.; Abrahao, J.S.; Kroon, E.G.; Ferreira, P.C.P.; et al. c-Jun integrates signals from both MEK/ERK and MKK/JNK pathways upon vaccinia virus infection. Arch. Virol. 2017, 162, 2971–2981. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Hofstetter, W.; Guo, W.; Li, H.; Pataer, A.; Peng, H.H.; Huo, Z.S.; Bartlett, D.L.; Lin, A.; Swisher, S.G.; et al. JNK-deficiency enhanced oncolytic vaccinia virus replication and blocked activation of double-stranded RNA-dependent protein kinase. Cancer Gene Ther. 2008, 15, 616–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izmailyan, R.; Hsao, J.C.; Chung, C.S.; Chen, C.H.; Hsu, P.W.C.; Liao, C.L.; Chang, W. Integrin β1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 2012, 86, 6677–6687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNulty, S.; Bornmann, W.; Schriewer, J.; Werner, C.; Smith, S.K.; Olson, V.A.; Damon, I.K.; Buller, R.M.; Heuser, J.; Kalman, D. Multiple phosphatidylinositol 3-kinases regulate vaccinia virus morphogenesis. PLoS ONE 2010, 5, e10884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, J.A.P.; Leite, F.G.G.; Andrade, L.G.; Torres, A.A.; De Sousa, L.P.; Barcelos, L.S.; Teixeira, M.M.; Ferreira, P.C.P.; Kroon, E.G.; Souto-Padron, T.; et al. Activation of the PI3K/Akt pathway early during vaccinia and cowpox virus infections is required for both host survival and viral replication. J. Virol. 2009, 83, 6883–6899. [Google Scholar] [CrossRef] [Green Version]
- Zaborowska, I.; Walsh, D. PI3K signaling regulates rapamycin-insensitive translation initiation complex formation in vaccinia virus-infected cells. J. Virol. 2009, 83, 3988–3992. [Google Scholar] [CrossRef] [Green Version]
- Newsome, T.P.; Weisswange, I.; Frischknecht, F.; Way, M. Abl collaborates with Src family kinases to stimulate actin-based motility of vaccinia virus. Cell Microbiol. 2006, 8, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Newsome, T.P.; Scaplehorn, N.; Way, M. SRC mediates a switch from microtubule- to actin-based motility of vaccinia virus. Science 2004, 306, 124–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, F.I.; Bleck, C.K.E.; Reh, L.; Novy, K.; Wollscheid, B.; Helenius, A.; Stahlberg, H.; Mercer, J. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep. 2013, 4, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.C.J.; Li, J.; Irwin, C.R.; Jenkins, H.; DeLange, L.; Evans, D.H. Vaccinia virus DNA ligase recruits cellular topoisomerase II to sites of viral replication and assembly. J. Virol. 2008, 82, 5922–5932. [Google Scholar] [CrossRef] [Green Version]
- Allen-Mersh, T.G.; Earlam, S.; Fordy, C.; Abrams, K.; Houghton, J. Quality of life and survival with continuous hepatic-artery floxuridine infusion for colorectal liver metastases. Lancet 1994, 344, 1255–1260. [Google Scholar] [CrossRef]
- Mazzucconi, M.G.; Baldacci, E.; Latagliata, R.; Breccia, M.; Paoloni, F.; Di Veroli, A.; Cedrone, M.; Anaclerico, B.; Villivà, N.; Porrini, R.; et al. Anagrelide in Essential Thrombocythemia (ET): Results from 150 patients over 25 years by the ’Ph1-negative Myeloproliferative Neoplasms Latium Group’. Eur. J. Haematol. 2020, ejh.13454. [Google Scholar] [CrossRef]
- Kulkarni, N.S.; Parvathaneni, V.; Shukla, S.K.; Barasa, L.; Perron, J.C.; Yoganathan, S.; Muth, A.; Gupta, V. Tyrosine kinase inhibitor conjugated quantum dots for non-small cell lung cancer (NSCLC) treatment. Eur. J. Pharm. Sci. 2019, 133, 145–159. [Google Scholar] [CrossRef]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef]
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT signaling in immunity and disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, A.; Guzik, K.; Slezak, K.; Dziedzic, J.; Rokita, H. Heat shock protein and heat shock factor 1 expression and localization in vaccinia virus infected human monocyte derived macrophages. J. Inflamm. (Lond.) 2005, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A.J. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses 2016, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Fisher, R.; Chowdhury, S.; Sultana, I.; Pereira, C.P.; Bray, M. Vaccinia virus induces rapid necrosis in keratinocytes by a STAT3-dependent mechanism. PLoS ONE 2014, 9, e113690. [Google Scholar] [CrossRef] [Green Version]
- Osborne, J.D.; Da Silva, M.; Frace, A.M.; Sammons, S.A.; Olsen-Rasmussen, M.; Upton, C.; Buller, R.M.L.; Chen, N.; Feng, Z.; Roper, R.L.; et al. Genomic differences of Vaccinia virus clones from Dryvax smallpox vaccine: The Dryvax-like ACAM2000 and the mouse neurovirulent Clone-3. Vaccine 2007, 25, 8807–8832. [Google Scholar] [CrossRef]
- Melamed, S.; Wyatt, L.S.; Kastenmayer, R.J.; Moss, B. Attenuation and immunogenicity of host-range extended modified vaccinia virus Ankara recombinants. Vaccine 2013, 31, 4569–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antivir. Res. 2009, 84, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Prazsák, I.; Tombácz, D.; Szűcs, A.; Dénes, B.; Snyder, M.; Boldogkői, Z. Full Genome Sequence of the Western Reserve Strain of Vaccinia Virus Determined by Third-Generation Sequencing. Genome Announc. 2018, 6, e01570–e01617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Mendez-Rios, J.D.; Peng, C.; Xiao, W.; Weisberg, A.S.; Wyatt, L.S.; Moss, B. SPI-1 is a missing host-range factor required for replication of the attenuated modified vaccinia Ankara (MVA) vaccine vector in human cells. PLoS Pathog. 2019, 15, e1007710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Moss, B. Repair of a previously uncharacterized second host-range gene contributes to full replication of modified vaccinia virus Ankara (MVA) in human cells. Proc. Natl. Acad. Sci. USA 2020, 117, 3759–3767. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.S.; Jones, R.G.; Thompson, C.B.; Coyne, C.B.; Cherry, S. A kinome RNAi screen identified AMPK as promoting poxvirus entry through the control of actin dynamics. PLoS Pathog. 2010, 6, e1000954. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Compound Name | Approved by the FDA (Y/N) | Primary Screen (% of Inhibition) | Secondary Screen (% of Inhibition) | Cytotoxicity Effect (% of Reduction) | Direct Gluc Effect (% of Reduction) | Cellular Target | Cellular Target Known to Interact with VACV? | ||
|---|---|---|---|---|---|---|---|---|---|
| MOI 0.01 | MOI 0.01 | MOI 2 | 24 h | 48 h | 2 h | ||||
| StemRegenin 1 | N | 67.1 | 68.1 | 55.6 | 3.3 | 2.0 | −8.4 | AhR: aryl hydrocarbon receptor | No |
| Niclosamide (Niclocide) | Y | 94.9 | 43.7 | 32.7 | 20.4 | 42.0 | −6.4 | Signal transducer and activator of transcription-3 (STAT3) | No |
| ICG-001 | N | 76.2 | 80.9 | 78.4 | 29.4 | 35.9 | 5.3 | Wnt/β-catenin/TCF-mediated transcription a | [31] |
| Nifuroxazide | N | 78.5 | 48.8 | 42.0 | 20.7 | 18.5 | −5.9 | STAT1/3/5 | No |
| NSC 319726 | N | 99.9 | 99.8 | 99.9 | 25.3 | 28.0 | 2.0 | p53 | [32] |
| Tenovin-1 | N | 96.9 | 32.0 | 26.7 | −1.2 | 5.0 | −6.9 | ||
| RITA (NSC 652287) | N | 83.6 | 50.2 | 43.9 | 16.8 | 23.3 | −1.2 | ||
| Ciclopirox ethanolamine | N | 100.0 | 99.8 | 99.9 | 9.0 | 0.4 | −15.9 | Iron chelator | No |
| Econazole nitrate (Spectazole) | Y | 85.8 | 55.1 | 56.7 | 2.8 | 10.9 | −2.8 | Antifungal | No |
| Miconazole nitrate | Y | 76.1 | 58.3 | 59.9 | 12.0 | 13.0 | −2.3 | ||
| Monensin sodium salt (Coban) | N | 92.2 | 96.7 | 96.6 | 24.3 | 34.4 | 7.2 | Antibiotic | No |
| Idoxuridine | Y | 82.3 | 85.8 | 85.3 | −5.9 | −3.9 | 0.4 | Antiviral | [33] |
| 3-Deazaneplanocin A (DZNeP) | N | 97.4 | 76.8 | 75.7 | 1.0 | 42.5 | −5.1 | Purine analog | No |
| Fludarabine (Fludara) | Y | 99.6 | 99.1 | 99.1 | −9.4 | 10.8 | −1.0 | ||
| Clofarabine | Y | 100.0 | 99.8 | 100.0 | −5.9 | 1.3 | 4.3 | ||
| Azaguanine-8 | N | 99.6 | 12.7 | 6.7 | 2.9 | 21.9 | 0.7 | ||
| Cytarabine | Y | 99.5 | 99.8 | 100.0 | 2.1 | 13.3 | 0.0 | Antimetabolic agent and DNA synthesis inhibitor | [34] |
| Gemcitabine | Y | 100.0 | 99.9 | 100.0 | 7.7 | 9.0 | 6.8 | Nucleoside analog | [35] |
| Cyclocytidine HCl | N | 100.0 | 99.8 | 100.0 | −0.6 | 17.2 | 7.2 | ||
| Trifluridine (Viroptic) | Y | 100.0 | 99.9 | 100.0 | −5.7 | 9.3 | −3.1 | ||
| Azacitidine (Vidaza) | Y | 98.8 | 99.9 | 100.0 | 9.2 | 7.1 | 3.8 | DNA methylation | No |
| Floxuridine | Y | 96.7 | 62.6 | 48.4 | 14.4 | 73.2 | 0.0 | MDCK/PEPT1. | No |
| Abitrexate (Methotrexate) | Y | 93.4 | 62.2 | 67.5 | −12.6 | 56.4 | 1.3 | antifolate antimetabolite | No |
| Triapine | N | 100.0 | 99.9 | 100.0 | 9.9 | 14.2 | −22.3 | ribonucleotide reductase; | [36] |
| Vidarabine (Vira-A) | Y | 88.1 | 99.7 | 100.0 | −4.4 | −4.3 | 5.6 | adenosine with the D-ribose | [37] |
| Apigenin | N | 89.8 | 54.8 | 53.7 | 17.4 | 32.1 | 5.6 | Cytochrome P450 | No |
| Avasimibe | N | 63.3 | 50.9 | 47.8 | 7.1 | 19.6 | 3.6 | ||
| Cyclosporin A | N | 73.2 | 67.2 | 46.8 | 27.4 | 24.5 | −5.1 | ||
| ML130 | N | 70.6 | 35.9 | 23.4 | 6.9 | 13.6 | −5.3 | NOD1 | No |
| PAC-1 | N | 95.1 | 95.5 | 95.0 | 14.3 | 16.5 | −7.7 | procaspase-3 activator | [38] |
| ABT-263 (Navitoclax) | N | 64.1 | 33.1 | 73.9 | −2.2 | 5.5 | −7.6 | BCL protein | [39,40] |
| ABT-737 | N | 72.1 | 26.6 | 55.7 | 4.7 | 16.3 | −4.7 | ||
| PF-2545920 | N | 99.6 | 33.7 | 41.1 | 5.5 | 11.2 | −1.0 | PDE10A | No |
| Cladribine | Y | 99.8 | 99.8 | 100.0 | −8.8 | −4.1 | 1.4 | adenosine deaminase | [41,42] |
| Adefovir Dipivoxil (Preveon, Hepsera) | Y | 100.0 | 99.8 | 100.0 | 9.7 | 8.1 | −2.1 | reverse transcriptase | No |
| TPCA-1 | N | 78.8 | 78.5 | 77.1 | 18.5 | 30.7 | −3.7 | IKK-2 | [43] |
| Mycophenolic (Mycophenolate) | Y | 98.3 | 96.7 | 97.1 | 27.7 | 35.3 | −2.3 | inosine monophosphate dehydrogenase | No |
| PTC-209 | N | 89.3 | 90.7 | 91.5 | 31.6 | 38.5 | −2.3 | BMI-1, polycomb complex protein | No |
| EX 527 (Selisistat) | N | 71.7 | 30.5 | 29.6 | 17.2 | 41.4 | 0.0 | Sirtuin 1 (SIRT1) | No |
| Salinomycin (Procoxacin) | N | 95.9 | 92.5 | 96.8 | 19.5 | 35.1 | −1.7 | Aromatase | No |
| Dapivirine | N | 70.1 | 41.7 | 34.2 | 37.3 | 70.3 | 8.3 | Nonnucleoside reverse transcriptase | No |
| CCG 50014 | N | 85.0 | 35.8 | 30.2 | 5.7 | 2.4 | 5.8 | RGS4, G-protein signaling 4 | No |
| Mocetinostat (MGCD0103) | 75.9 | 46.9 | 27.0 | 33.0 | 74.1 | −0.3 | HDAC | [44] | |
| Atorvastatin calcium (Lipitor) | Y | 65.7 | 25.4 | 30.6 | 10.2 | 42.3 | −3.2 | HMG-CoA reductase | No |
| UK 383367 | N | 73.7 | 17.0 | 33.6 | 17.0 | 29.4 | −3.0 | BMP-1 (C-proteinase) peptidase | No |
| Lonafarnib (SCH66336) | N | 66.1 | 36.2 | 71.2 | 10.7 | 13.5 | −5.9 | FPTase, prenyltransferases | No |
| BX-912 | N | 68.0 | 42.3 | 45.6 | 34.3 | 55.5 | 0.3 | PDK1: pyruvate dehydrogenase kinase 1 | No |
| URB597 | N | 60.7 | 21.7 | −9.2 | −5.0 | −3.7 | −5.6 | FAAH: fatty acid amide hydrolase | [45] |
| Anagrelide HCl | Y | 97.4 | 58.4 | 52.9 | 34.1 | 89.3 | −5.5 | phosphodiesterase | No |
| Drospirenone | Y | 98.0 | 57.4 | 56.3 | 31.7 | 87.3 | 8.9 | Hormone | [46] |
| Ethinyl Estradiol | Y | 98.0 | 53.3 | 44.3 | 42.9 | 89.0 | 0.8 | ||
| Norethindrone (Norethisterone) | N | 98.8 | 48.4 | 58.4 | 42.2 | 90.8 | −4.6 | ||
| Ganetespib (STA-9090) | N | 88.8 | 99.7 | 100.0 | 32.9 | 28.6 | −96.8 | Heat shock protein 90 (HSP90) | [22] |
| PF-04929113 (SNX-5422) | N | 95.4 | 99.4 | 99.7 | 33.0 | 26.2 | 1.0 | ||
| NMS-E973 | N | 96.0 | 99.1 | 99.3 | 33.8 | 21.5 | −3.5 | ||
| XL888 | N | 95.5 | 99.6 | 99.7 | 38.7 | 38.9 | −3.4 | ||
| 17-AAG | N | 97.3 | 99.4 | 99.5 | 39.9 | 50.9 | 10.0 | ||
| VER-49009 | N | 95.7 | 98.9 | 99.0 | 30.7 | 26.5 | −8.9 | ||
| SNX2112 | N | 95.3 | 99.4 | 99.6 | 29.3 | 26.1 | 1.4 | ||
| BIIB021 | N | 87.0 | 99.8 | 99.9 | 34.5 | 28.6 | −5.6 | ||
| AT13387 | N | 87.3 | 99.8 | 99.9 | 37.9 | 31.7 | −68.5 | ||
| NVP-BEP800 | N | 87.6 | 98.8 | 98.6 | 28.7 | 13.2 | −7.4 | ||
| AUY922 (NVP-AUY922) | N | 89.5 | 99.8 | 100.0 | 34.6 | 40.8 | −72.5 | ||
| KW-2478 | N | 81.9 | 92.7 | 92.7 | 33.7 | 25.7 | −2.7 | ||
| Vincristine | Y | 95.3 | 96.6 | 96.4 | 45.8 | 76.9 | −7.2 | Microtubules | [47,48,49,50] |
| ABT-751 (E7010) | N | 74.3 | 81.5 | 82.7 | 41.4 | 74.5 | −117.1 | ||
| Epothilone A | N | 68.0 | 27.3 | 71.6 | 32.5 | 84.1 | −3.4 | ||
| Amygdalin | N | 73.3 | 58.3 | 53.9 | 4.7 | 11.7 | −0.3 | Natural product, Vit B17 | No |
| Kaempferol | N | 84.8 | 64.7 | 60.2 | 7.0 | 14.7 | 1.4 | Natural product, flavonoid antioxidant | No |
| ENMD-2076 | N | 66.5 | 72.3 | 73.4 | 47.7 | 81.7 | −7.3 | Aurora A and B | No |
| AMG-900 | N | 69.7 | 50.4 | 45.4 | 3.5 | 15.4 | −7.5 | ||
| MLN8054 | N | 80.1 | 36.2 | 30.7 | −2.8 | 5.2 | 3.3 | ||
| PCI-32765 (Ibrutinib) | Y | 99.7 | 74.9 | 82.8 | 6.9 | −7.7 | −2.4 | Bruton’s tyrosine kinase (BTK), | No |
| CNX-774 | N | 90.1 | 70.0 | 62.8 | 39.6 | 60.3 | 3.4 | ||
| AR-A014418 | N | 97.0 | 47.9 | 46.5 | 0.4 | 17.8 | 0.6 | GSK3β | No |
| CGP 57380 | N | 70.1 | 58.1 | 53.7 | 17.6 | 21.3 | −2.4 | MNK1, Ser-Thr PK | No |
| Genistein | N | 88.6 | 40.6 | 27.0 | −6.7 | 18.2 | −4.9 | protein tyrosine kinase (PTK) | No |
| HMN-214 | N | 77.0 | 58.9 | 64.6 | 20.0 | 84.6 | 1.7 | Polo-like kinase (Plk)1 | No |
| TG003 | N | 79.3 | 46.7 | 46.1 | 8.1 | 25.3 | −1.9 | Cdc2-like kinase (Clk) | No |
| Skepinone-L | N | 60.7 | 47.1 | 39.9 | −1.5 | −3.0 | 5.9 | MAPK | [51,52,53] |
| TAK-632 | N | 98.9 | 65.2 | 52.8 | 1.2 | 34.0 | 1.6 | ||
| YM201636 | N | 81.6 | 56.3 | 44.8 | 45.9 | 62.1 | 6.3 | PIKfyve | [54] |
| MK-2461 | N | 67.0 | 57.0 | 56.5 | 30.1 | 41.8 | −2.1 | c-Met | [55] |
| OSI-930 | N | 90.6 | 43.5 | 26.5 | 8.3 | 15.1 | 10.1 | Kit and KDR | [56] |
| SP600125 | N | 95.7 | 50.5 | 53.3 | 30.7 | 59.4 | 2.3 | JNK | [57,58] |
| SKI II | N | 72.2 | 65.1 | 63.4 | 13.2 | 29.0 | −3.4 | PI3K/mTOR (mammalian target of rapamycin) | [25,59,60,61,62] |
| PKI-402 | N | 74.1 | 44.9 | 50.4 | 43.1 | 51.8 | −0.2 | ||
| BEZ235 (NVP-BEZ235) | N | 72.9 | 37.4 | 32.7 | 33.7 | 17.7 | −1.5 | ||
| WYE-125132 | N | 87.6 | 67.6 | 54.9 | 45.8 | 58.2 | −13.0 | ||
| OSI-027 | N | 74.1 | 68.3 | 62.2 | 36.1 | 27.3 | −1.0 | ||
| Torin 1 | N | 64.6 | 57.3 | 65.9 | 36.1 | 37.8 | −5.0 | ||
| Ku-0063794 | N | 69.3 | 55.3 | 57.9 | 28.3 | 25.5 | 3.0 | ||
| AZD2014 | N | 74.8 | 70.3 | 73.3 | 30.6 | 36.3 | −3.9 | ||
| Nocodazole | N | 92.4 | 76.2 | 76.5 | 48.3 | 77.3 | −3.0 | Abl and src | [63,64] |
| Dasatinib (BMS-354825) | Y | 64.7 | 74.2 | 84.5 | 20.3 | 45.9 | −0.8 | ||
| KX2-391 | N | 93.5 | 65.1 | 84.4 | 34.7 | 78.3 | −4.2 | ||
| Bosutinib (SKI-606) | Y | 72.8 | 81.8 | 77.1 | 20.3 | 28.5 | 5.3 | ||
| PP1 | N | 84.3 | 35.8 | 37.8 | 37.6 | 56.3 | −0.2 | ||
| AZ 960 | N | 84.1 | 82.1 | 82.4 | 45.5 | 77.6 | 1.4 | Janus kinase-2 (JAK2) | No |
| LY2784544 | N | 71.9 | 65.7 | 67.0 | 25.5 | 74.6 | −0.2 | ||
| WHI-P154 | N | 83.6 | 66.3 | 62.0 | 10.3 | 16.5 | −2.7 | JAK3 | No |
| Cyt387 | N | 62.4 | 45.2 | 41.9 | 26.3 | 35.6 | 4.3 | JAK1 and JAK2 | No |
| KPT-276 | N | 88.8 | 27.6 | 11.3 | 25.2 | 43.0 | 9.2 | Chromosomal maintenance 1 (CRM1) | [27] |
| KPT-185 | N | 79.3 | 26.2 | 18.9 | 33.4 | 68.0 | 0.5 | ||
| Phloretin | N | 68.3 | −27.0 | −23.8 | 8.6 | 24.4 | −3.9 | Sodium/glucose cotransporter 1 and 2 | No |
| Ivacaftor (VX-770) | Y | 82.6 | 75.5 | 77.1 | 37.5 | 46.3 | −3.3 | CFTR, chloride channel | No |
| Tolbutamide | Y | 99.8 | 99.9 | 100.0 | 62.0 | 81.2 | −2.9 | Potassium channel blocker | No |
| Fexofenadine HCl | Y | 63.8 | 10.5 | 27.6 | −3.0 | −1.8 | −6.4 | histamine H1 receptor | No |
| Bergapten | N | 76.4 | 7.6 | 10.4 | 14.4 | 36.1 | −0.4 | DNA crosslinks | No |
| SC144 | N | 99.9 | 98.4 | 98.6 | 15.7 | 17.7 | −4.2 | gp130, cytokine receptors | No |
| Daidzein | N | 78.5 | 32.2 | −9.2 | 7.8 | 22.2 | −3.0 | Peroxisome proliferator-activated receptor | No |
| CEP-18770 (Delanzomib) | N | 99.5 | 99.7 | 99.8 | 30.9 | 94.6 | −4.5 | Proteasome | [28,65] |
| MLN9708 | N | 99.1 | 99.6 | 99.7 | 39.9 | 92.0 | −1.9 | ||
| Oprozomib (ONX 0912) | N | 99.8 | 99.5 | 99.6 | 40.7 | 96.4 | −6.1 | ||
| MG-132 | N | 99.5 | 98.9 | 99.3 | 48.6 | 97.5 | −2.7 | ||
| MLN2238 | N | 99.5 | 99.7 | 99.8 | 39.4 | 93.3 | −3.2 | ||
| ONX-0914 (PR-957) | N | 99.7 | 99.5 | 99.7 | 52.5 | 95.2 | 1.7 | ||
| Amonafide | N | 98.5 | 99.3 | 99.8 | 30.5 | 43.8 | −4.6 | Topoisomerase II | [66] |
| Teniposide (Vumon) | Y | 96.4 | 97.9 | 98.8 | 27.5 | 92.8 | 7.4 | ||
| Etoposide (VP-16) | Y | 84.6 | 60.1 | 63.5 | 0.0 | 32.9 | 2.6 | ||
| Golvatinib (E7050) | N | 70.4 | 79.8 | 72.2 | 28.9 | 32.3 | −9.6 | VEGFR and epidermal growth factor receptor (EGFR) | [23] |
| Sorafenib (Nexavar) | Y | 78.5 | 72.6 | 89.4 | 23.1 | 45.1 | −5.3 | ||
| Tyrphostin AG 1296 (AG 1296) | N | 99.0 | 53.2 | 48.6 | 36.4 | 91.9 | −19.3 | ||
| NVP-TAE226 | N | 93.1 | 91.5 | 90.7 | 41.5 | 68.2 | −2.9 | ||
| Cabozantinib malate | Y | 77.9 | 79.3 | 81.3 | 27.4 | 39.3 | 0.3 | ||
| XL-184 (Cabozantinib) | Y | 50.4 | 64.2 | 64.6 | −29.2 | 45.5 | −1.4 | ||
| Linifanib (ABT-869) | N | 79.5 | 60.5 | 55.6 | 20.7 | 35.3 | −13.9 | ||
| Dovitinib (TKI-258) Dilactic Acid | N | 97.3 | 67.6 | 57.9 | 11.8 | 32.8 | 8.2 | ||
| AZD4547 | N | 75.7 | 81.5 | 80.0 | 42.3 | 66.4 | 8.7 | ||
| AG-1024 | N | 91.6 | 71.8 | 66.4 | 5.4 | 9.7 | 17.0 | ||
| BMS-754807 | N | 90.0 | 61.8 | 53.2 | 43.6 | 61.5 | 9.3 | ||
| Afatinib (BIBW2992) | Y | 89.2 | 95.2 | 94.2 | 34.8 | 69.9 | −0.3 | ||
| Butein | N | 92.3 | 62.6 | 60.1 | 42.7 | 64.3 | −4.5 | ||
| CO-1686 (AVL-301) | N | 96.7 | 74.3 | 76.9 | 45.5 | 67.9 | −6.1 | ||
| PD168393 | N | 80.9 | 67.2 | 57.1 | 17.0 | 37.0 | −5.8 | ||
| PD153035 HCl | N | 72.2 | 74.7 | 65.4 | 13.9 | 14.2 | −4.1 | ||
| Gefitinib (Iressa) | N | 77.5 | 91.2 | 89.3 | 17.8 | −10.2 | −3.1 | ||
| Desmethyl Erlotinib (CP-473420) | Y | 89.1 | 75.5 | 70.1 | 16.3 | 28.1 | 6.0 | ||
| OSI-420 (Desmethyl Erlotinib) | N | 86.2 | 46.7 | 42.4 | 16.2 | 26.8 | 5.1 | ||
| Neratinib (HKI-272) | Y | 84.7 | 56.7 | 55.6 | 10.4 | 25.8 | 0.3 | ||
| Erlotinib HCl | N | 76.9 | 66.8 | 62.8 | 8.7 | 14.5 | −0.6 | ||
| WZ8040 | N | 99.2 | 96.4 | 96.9 | 78.7 | 92.0 | 1.4 | ||
| Compound | Target |
|---|---|
| Niclosamide | STAT3 |
| Nifuroxazide | STAT1/3/5 |
| AZ 960 | JAK2 |
| LY2784544 | JAK2 |
| Cyt387 | JAK1/2 |
| Fludarabine | STAT1 |
| WHI-P154 | JAK3 |
| SC144 | IL-6 receptor |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, C.; Zhou, Y.; Cao, S.; Pant, A.; Campos Guerrero, M.L.; McDonald, P.; Roy, A.; Yang, Z. Identification of Vaccinia Virus Inhibitors and Cellular Functions Necessary for Efficient Viral Replication by Screening Bioactives and FDA-Approved Drugs. Vaccines 2020, 8, 401. https://doi.org/10.3390/vaccines8030401
Peng C, Zhou Y, Cao S, Pant A, Campos Guerrero ML, McDonald P, Roy A, Yang Z. Identification of Vaccinia Virus Inhibitors and Cellular Functions Necessary for Efficient Viral Replication by Screening Bioactives and FDA-Approved Drugs. Vaccines. 2020; 8(3):401. https://doi.org/10.3390/vaccines8030401
Chicago/Turabian StylePeng, Chen, Yanan Zhou, Shuai Cao, Anil Pant, Marlene L. Campos Guerrero, Peter McDonald, Anuradha Roy, and Zhilong Yang. 2020. "Identification of Vaccinia Virus Inhibitors and Cellular Functions Necessary for Efficient Viral Replication by Screening Bioactives and FDA-Approved Drugs" Vaccines 8, no. 3: 401. https://doi.org/10.3390/vaccines8030401
APA StylePeng, C., Zhou, Y., Cao, S., Pant, A., Campos Guerrero, M. L., McDonald, P., Roy, A., & Yang, Z. (2020). Identification of Vaccinia Virus Inhibitors and Cellular Functions Necessary for Efficient Viral Replication by Screening Bioactives and FDA-Approved Drugs. Vaccines, 8(3), 401. https://doi.org/10.3390/vaccines8030401