Revisiting CD8 T-cell ‘Memory Inflation’: New Insights with Implications for Cytomegaloviruses as Vaccine Vectors

 , ,
, ,  and
and 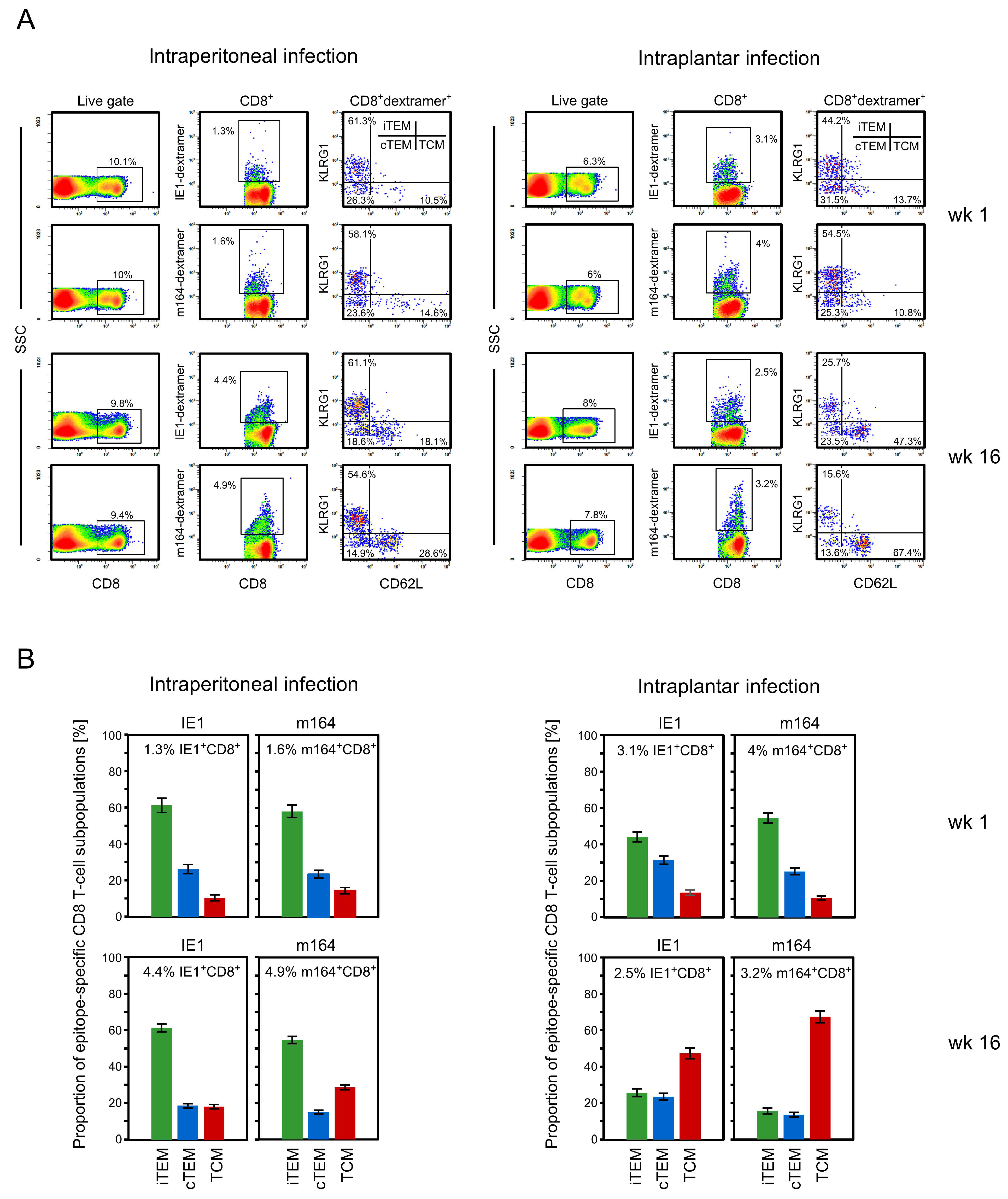{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice, Viruses and Infection Procedures
2.2. Experimental Hematopoietic Cell Transplantation
2.3. Quantitation of Infectious Virus
2.4. Quantitation of Viral Genomes
2.5. Peptides
2.6. Preparation of Single-Cell Suspensions from Lungs and Spleen
2.7. ELISpot Assay
2.8. Cytofluorometric Analyses
2.9. Statistics
3. Results
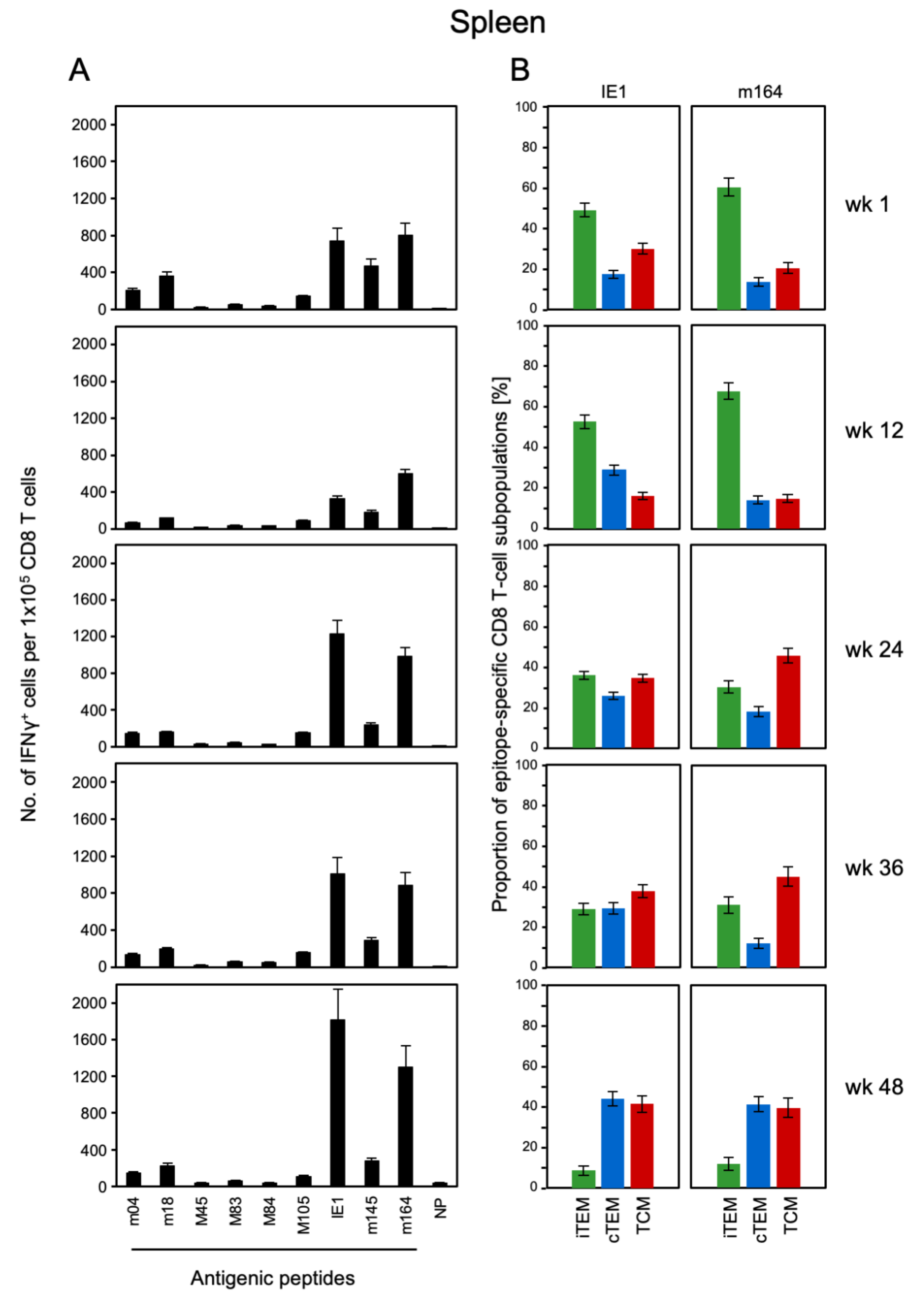
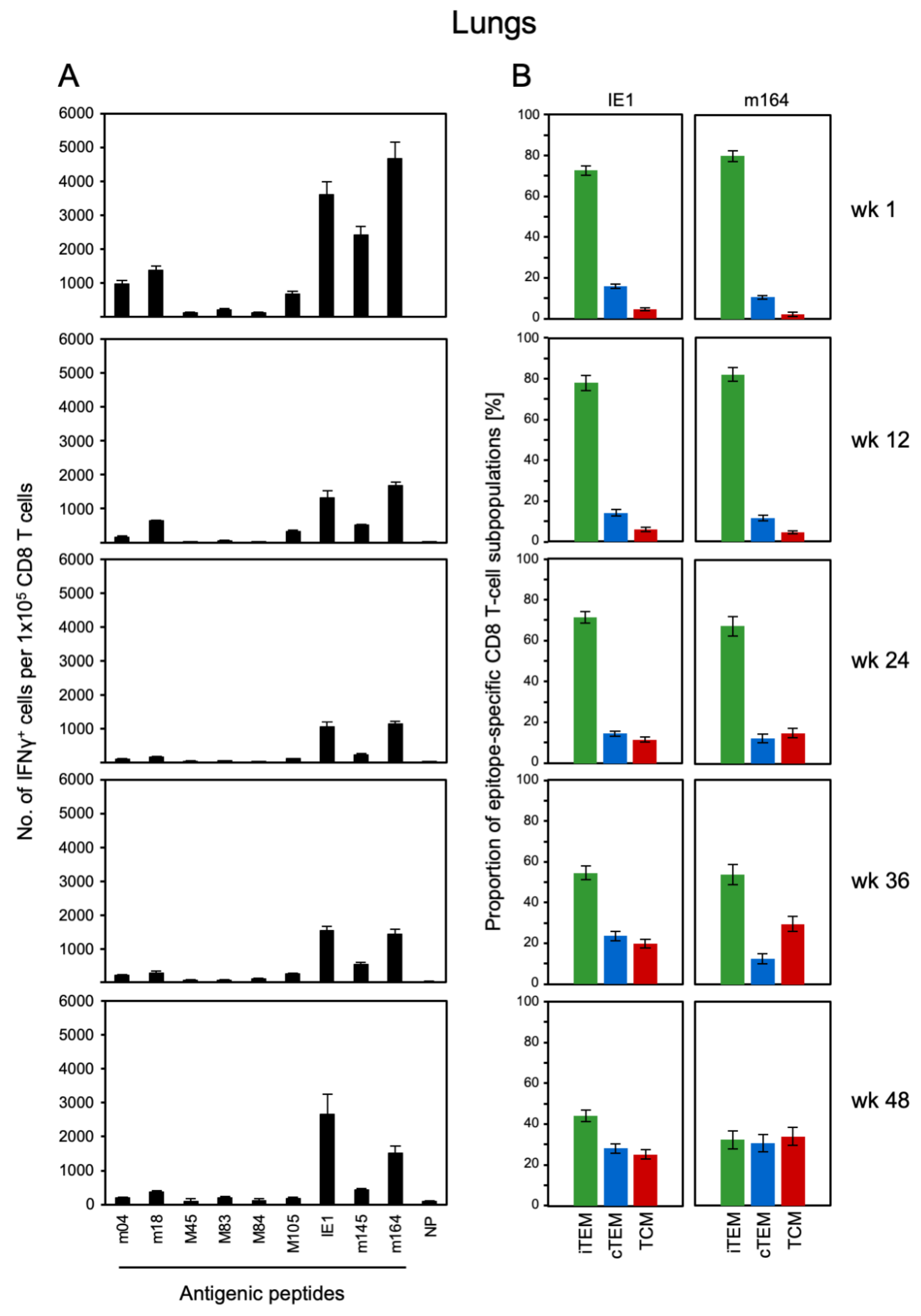
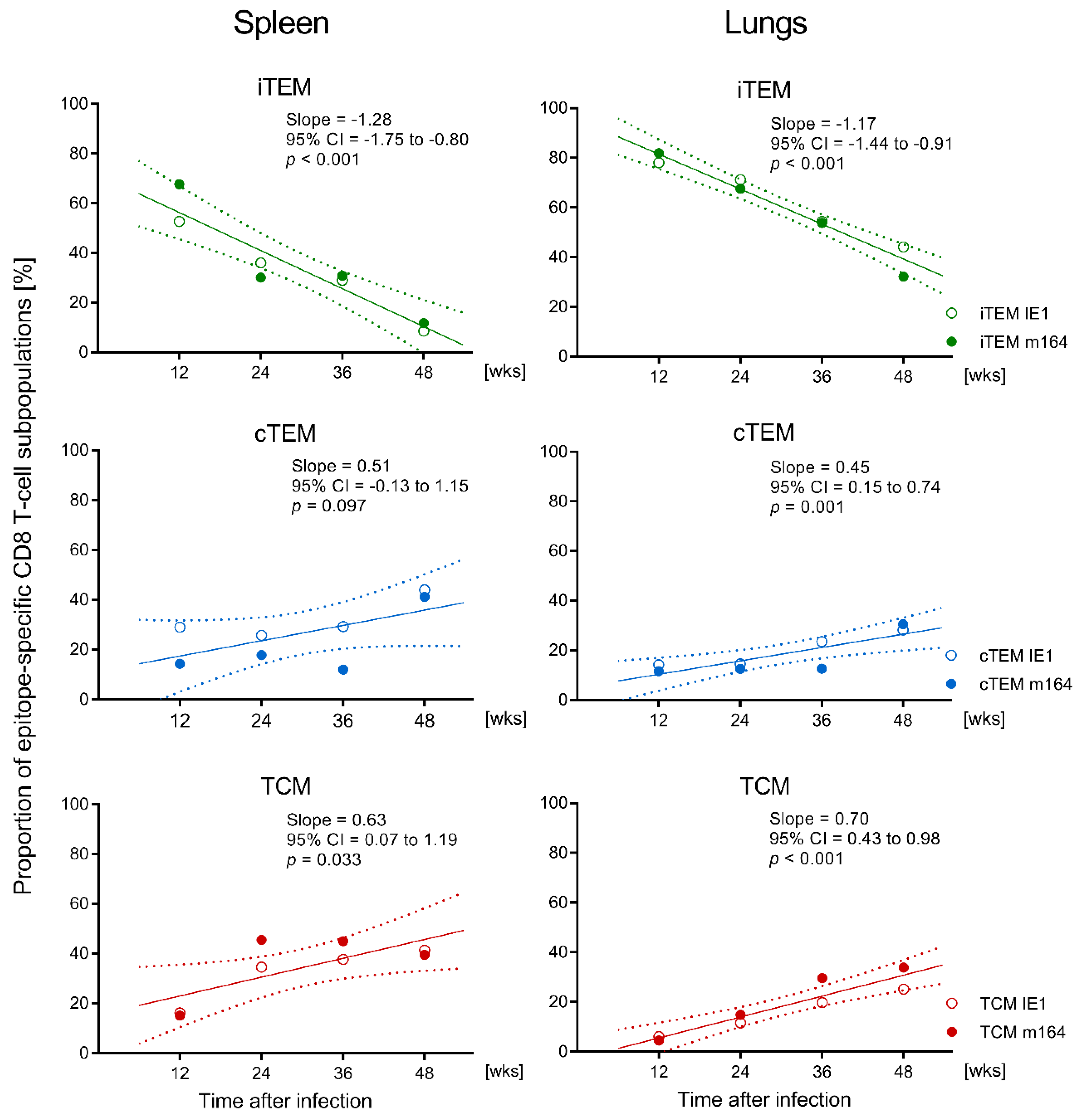
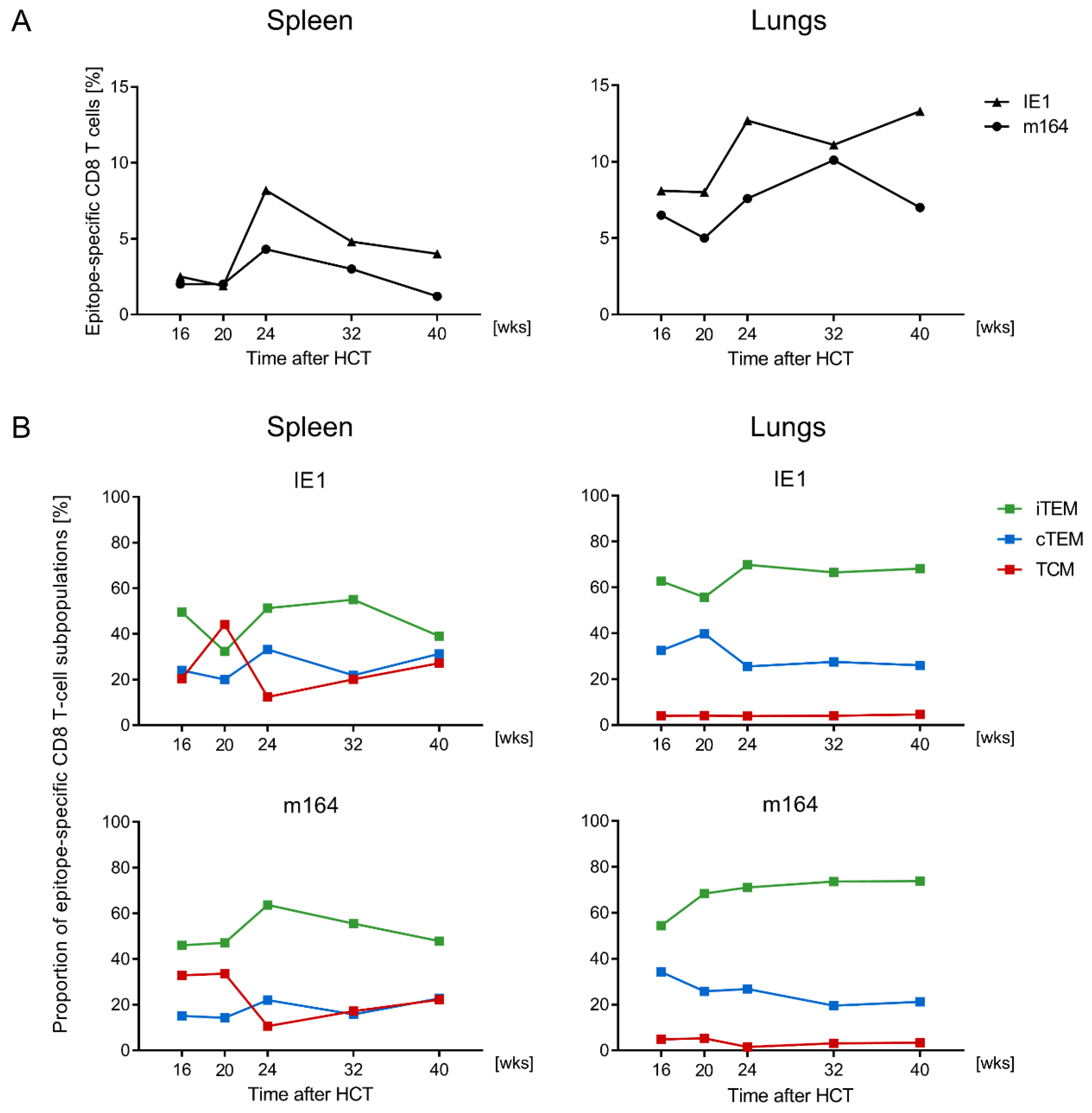
3.1. Memory CD8 T-cell Population Dynamics Reveals Continuous Loss of iTEM and Increase in cTEM and TCM after Local Infection of the Immunocompetent Host
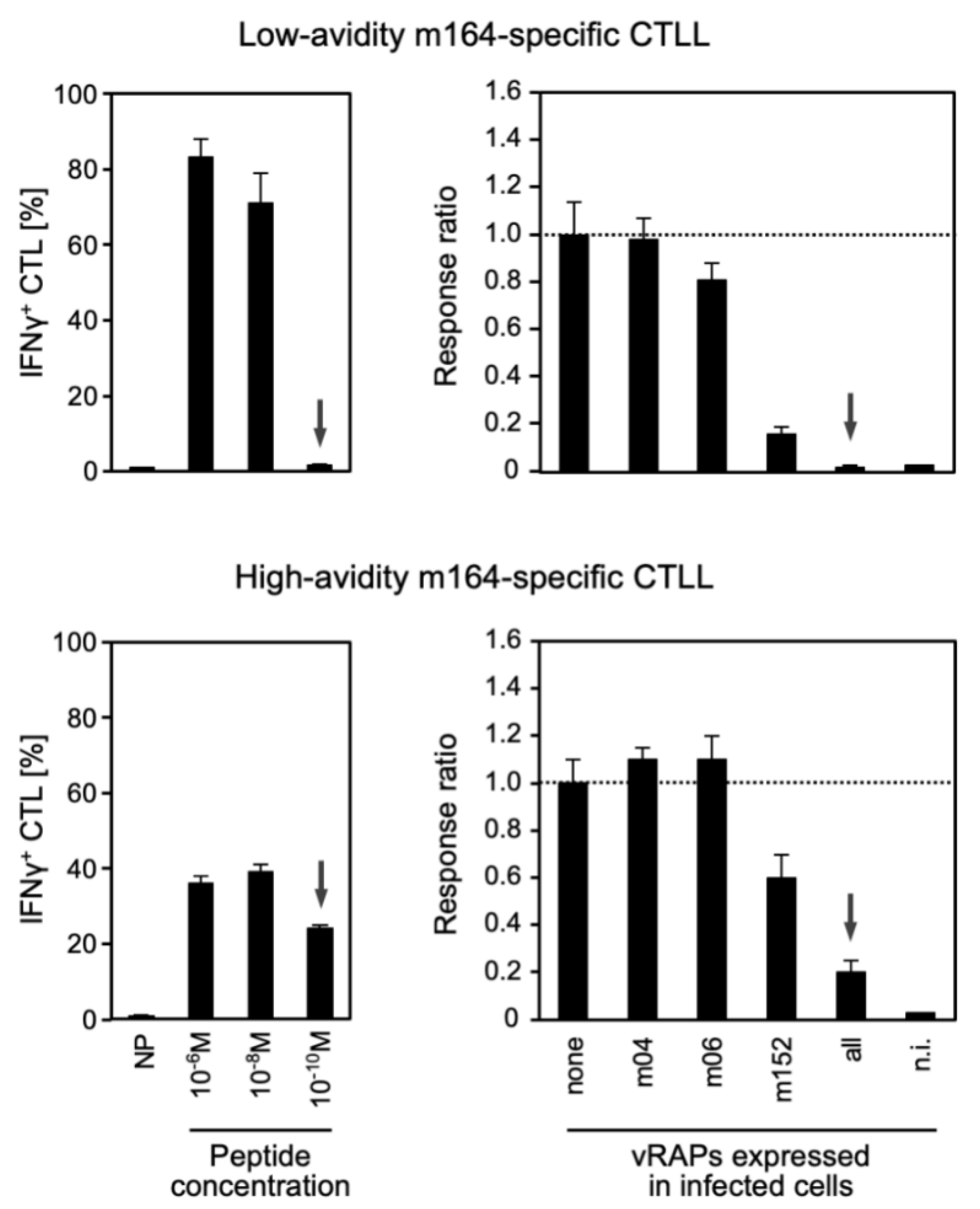
3.2. Functional Avidity of CD8 T cells Is Decisive for Overcoming Viral Immune Evasion
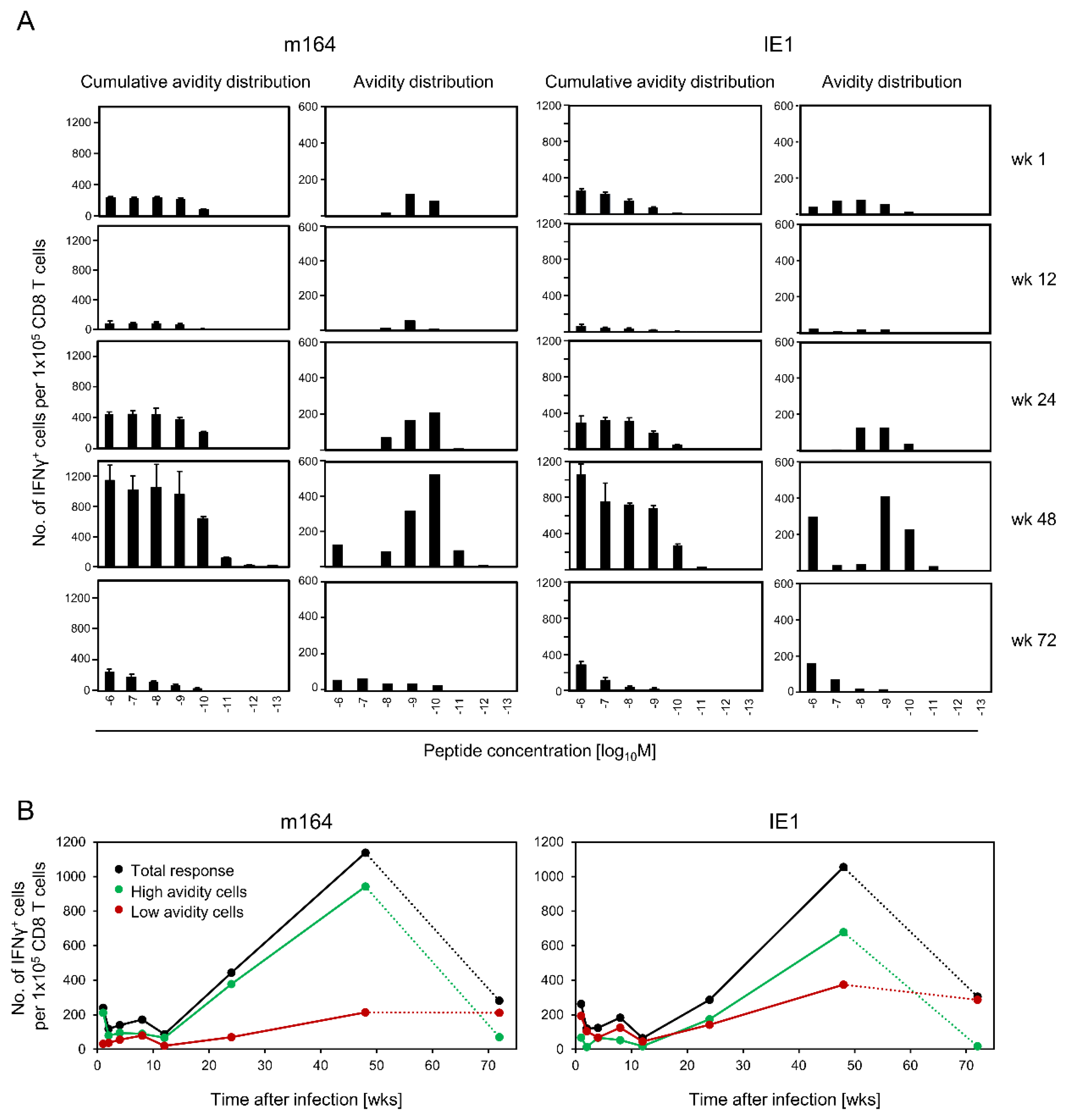
3.3. Avidity Dynamics of Functional Viral Epitope-Specific CD8 T cells
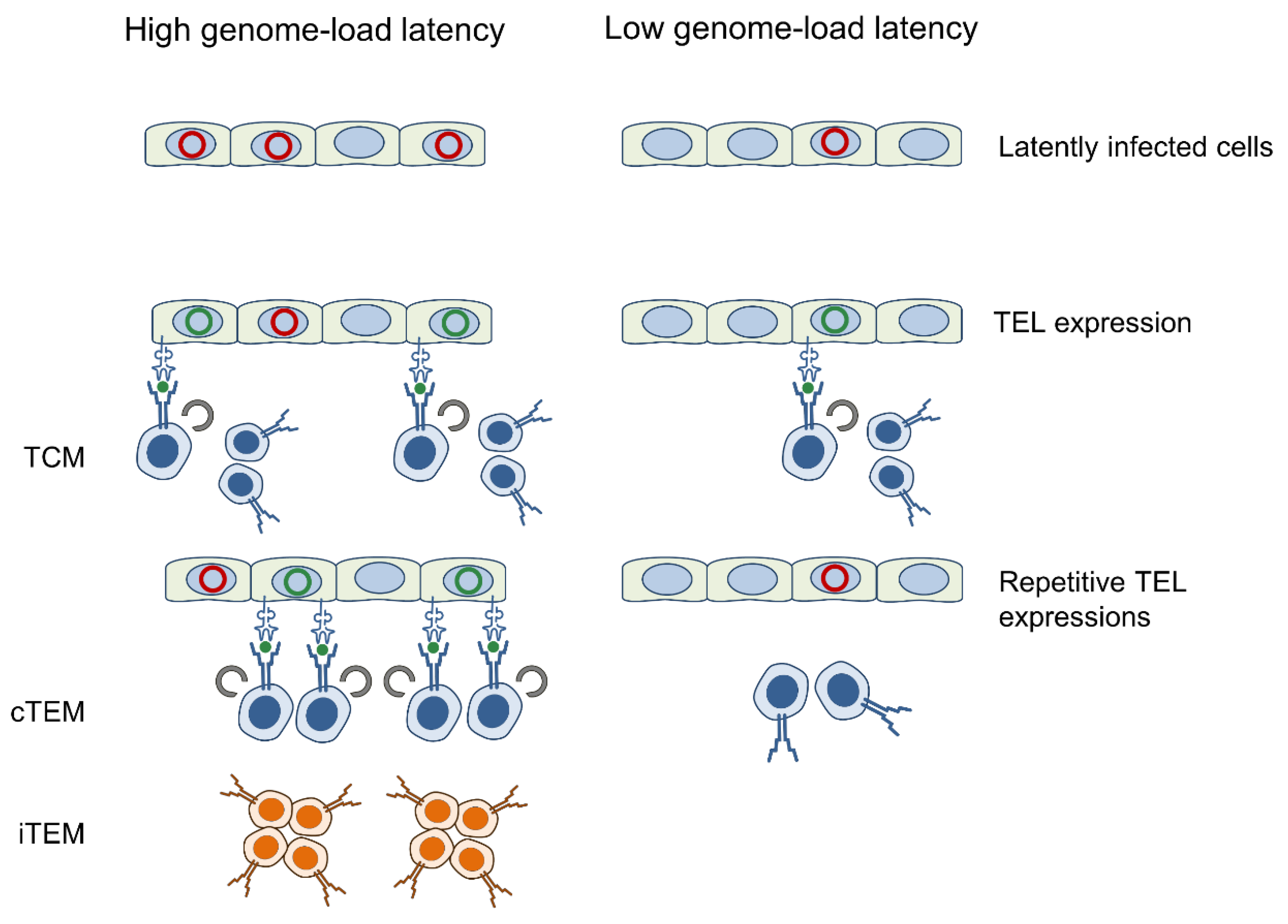
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Reddehase, M.J. Antigens and immunoevasins: Opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2002, 2, 831–844. [Google Scholar] [CrossRef]
- Powers, C.; DeFilippis, V.; Malouli, D.; Früh, K. Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 2008, 325, 333–359. [Google Scholar] [PubMed]
- Lisnić, B.; Lisnić, V.J.; Jonjić, S. NK cell interplay with cytomegaloviruses. Curr. Opin. Virol. 2015, 15, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.; Watson, G.M.; Jonjic, S.; Degli-Esposti, M.A.; Rossjohn, J. Modulation of innate and adaptive immunity by cytomegaloviruses. Nat. Rev. Immunol. 2019, 20, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Stempel, M.; Chan, B.; Brinkmann, M.M. Coevolution pays off: Herpesviruses have the license to escape the DNA sensing pathway. Med. Microbiol. Immunol. 2019, 208, 495–512. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Lemmermann, N.A.W. Cellular reservoirs of latent cytomegaloviruses. Med. Microbiol. Immunol. 2019, 208, 391–403. [Google Scholar] [CrossRef]
- Poole, E.; Sinclair, J. Sleepless latency of human cytomegalovirus. Med. Microbiol. Immunol. 2015, 204, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Goodrum, F. Human cytomegalovirus latency: Approaching the gordian knot. Annu. Rev. Virol. 2016, 3, 333–357. [Google Scholar] [CrossRef] [Green Version]
- Elder, E.; Sinclair, J. HCMV latency: What regulates the regulators? Med. Microbiol. Immunol. 2019, 208, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M. The history of cytomegalovirus and its diseases. Med. Microbiol. Immunol. 2008, 197, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boppana, S.B.; Britt, W.J. Synopsis of clinical aspects of human cytomegalovirus disease. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 1–25. [Google Scholar]
- Söderberg-Nauclér, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Reeves, M.; Sinclair, J. Epigenetic regulation of human cytomegalovirus gene expression: Impact an latency and reactivation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume I, pp. 330–346. [Google Scholar]
- Collins-McMillen, D.; Buehler, J.; Peppenelli, M.; Goodrum, F. Molecular determinants and the regulation of human cytomegalovirus latency and reactivation. Viruses 2018, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.; Boeckh, M. Clinical cytomegalovirus research: Hematopoietic cell transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 337–353. [Google Scholar]
- Emery, V.C.; Milne, R.S.B.; Griffiths, P.D. Clinical cytomegalovirus research: Liver and kidney transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 301–311. [Google Scholar]
- Stern, L.; Withers, B.; Avdic, S.; Gottlieb, D.; Abendroth, A.; Blyth, E.; Slobedman, B. Human cytomegalovirus latency and reactivation in allogeneic hematopoietic stem cell transplant recipients. Front. Microbiol. 2019, 10, 1186. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J.; Lemmermann, N.A.W. Mouse model of cytomegalovirus disease and immunotherapy in the immunocompromised host: Predictions for medical translation that survived the "test of time". Viruses 2018, 10, 693. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, G.A.; Welten, S.P.M.; Klenerman, P.; Arens, R. Memory T cell inflation: Understanding cause and effect. Trends Immunol. 2012, 33, 84–90. [Google Scholar] [CrossRef]
- Klenerman, P.; Oxenius, A. T cell responses to cytomegalovirus. Nat. Rev. Immunol. 2016, 16, 367–377. [Google Scholar] [CrossRef]
- Cicin-Sain, L.; Arens, R. Exhaustion and inflation at antipodes of T cell responses to chronic virus infection. Trends Microbiol. 2018, 26, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Welten, S.P.M.; Sandu, I.; Baumann, N.S.; Oxenius, A. Memory CD8 T cell inflation vs tissue-resident memory T cells: Same patrollers, same controllers? Immunol. Rev. 2018, 283, 161–175. [Google Scholar] [CrossRef]
- Cicin-Sain, L. Cytomegalovirus memory inflation and immune protection. Med. Microbiol. Immunol. 2019, 208, 339–347. [Google Scholar] [CrossRef]
- Welten, S.P.M.; Baumann, N.S.; Oxenius, A. Fuel and brake of memory T cell inflation. Med. Microbiol. Immunol. 2019, 208, 329–338. [Google Scholar] [CrossRef]
- Seckert, C.K.; Griessl, M.; Büttner, J.K.; Scheller, S.; Simon, C.O.; Kropp, K.A.; Renzaho, A.; Kühnapfel, B.; Grzimek, N.K.A.; Reddehase, M.J. Viral latency drives ‘memory inflation’: A unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 551–566. [Google Scholar] [CrossRef]
- Ebert, S.; Podlech, J.; Gillert-Marien, D.; Gergely, K.M.; Büttner, J.K.; Fink, A.; Freitag, K.; Thomas, D.; Reddehase, M.J.; Holtappels, R. Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Holtappels, R.; Ebert, S.; Podlech, J.; Fink, A.; Böhm, V.; Lemmermann, N.A.W.; Freitag, K.; Renzaho, A.; Thomas, D.; Reddehase, M.J. Murine model for cytoimmunotherapy of CMV disease after haematopoietic cell transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 352–379. [Google Scholar]
- Nauerth, M.; Weißbrich, B.; Knall, R.; Franz, T.; Dössinger, G.; Bet, J.; Paszkiewicz, P.J.; Pfeifer, L.; Bunse, M.; Uckert, W.; et al. TCR-ligand koff rate correlates with the protective capacity of antigen-specific CD8+ T cells for adoptive transfer. Sci. Transl. Med. 2013, 5, 192ra87. [Google Scholar] [CrossRef] [Green Version]
- Karrer, U.; Wagner, M.; Sierro, S.; Oxenius, A.; Hengel, H.; Dumrese, T.; Freigang, S.; Koszinowski, U.H.; Phillips, R.E.; Klenerman, P. Expansion of protective CD8+ T-cell responses driven by recombinant cytomegaloviruses. J. Virol. 2004, 78, 2255–2264. [Google Scholar] [CrossRef] [Green Version]
- Jarvis, M.A.; Hansen, S.G.; Nelson, J.A.; Picker, L.J.; Früh, K. Vaccine vectors using the unique biology and immunology of cytomegalovirus. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume II, pp. 450–462. [Google Scholar]
- Méndez, A.C.; Rodríguez-Rojas, C.; Del Val, M. Vaccine vectors: The bright side of cytomegalovirus. Med. Microbiol. Immunol. 2019, 208, 349–363. [Google Scholar] [CrossRef]
- Graef, P.; Buchholz, V.R.; Stemberger, C.; Flossdorf, M.; Henkel, L.; Schiemann, M.; Drexler, I.; Höfer, T.; Riddell, S.R.; Busch, D.H. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity 2014, 41, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Buchholz, V.R.; Schumacher, T.N.; Busch, D.H. T cell fate at the single-cell level. Annu. Rev. Immunol. 2016, 34, 65–92. [Google Scholar] [CrossRef] [Green Version]
- Snyder, C.M.; Cho, K.S.; Morrison, E.L.; van Dommelen, S.; Shellam, G.R.; Hill, A.B. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity 2008, 29, 650–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, N.S.; Torti, N.; Welten, S.P.M.; Barnstorf, I.; Borsa, M.; Pallmer, K.; Oduro, J.D.; Cicin-Sain, L.; Ikuta, K.; Ludewig, B.; et al. Tissue maintenance of CMV-specific inflationary memory T cells by IL-15. PLoS Pathog. 2018, 14, e1006993. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Sedikides, G.X.; Okecha, G.; Wills, M.R. Generation, maintenance and tissue distribution of T cell responses to human cytomegalovirus in lytic and latent infection. Med. Microbiol. Immunol. 2019, 208, 375–389. [Google Scholar] [CrossRef] [Green Version]
- Hickman, H.D.; Takeda, K.; Skon, C.N.; Murray, F.R.; Hensley, S.E.; Loomis, J.; Barber, G.N.; Bennink, J.R.; Yewdell, J.W. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat. Immunol. 2008, 9, 155–165. [Google Scholar] [CrossRef]
- Böhm, V.; Simon, C.O.; Podlech, J.; Seckert, C.K.; Gendig, D.; Deegen, P.; Gillert-Marien, D.; Lemmermann, N.A.W.; Holtappels, R.; Reddehase, M.J. The immune evasion paradox: Immunoevasins of murine cytomegalovirus enhance priming of CD8 T cells by preventing negative feedback regulation. J. Virol. 2008, 82, 11637–11650. [Google Scholar] [CrossRef] [Green Version]
- Farrell, H.E.; Davis-Poynter, N.; Bruce, K.; Lawler, C.; Dolken, L.; Mach, M.; Stevenson, P.G. Lymph node macrophages restrict murine cytomegalovirus dissemination. J. Virol. 2015, 89, 7147–7158. [Google Scholar] [CrossRef] [Green Version]
- Farrell, H.E.; Bruce, K.; Lawler, C.; Cardin, R.D.; Davis-Poynter, N.J.; Stevenson, P.G. Type 1 interferons and NK cells limit murine cytomegalovirus escape from the lymph node subcapsular sinus. PLoS Pathog. 2016, 12, e1006069. [Google Scholar] [CrossRef]
- Wagner, M.; Jonjic, S.; Koszinowski, U.H.; Messerle, M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 1999, 73, 7056–7060. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Gutermann, A.; Podlech, J.; Reddehase, M.J.; Koszinowski, U.H. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 2003, 196, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Podlech, J.; Holtappels, R.; Grzimek, N.K.; Reddehase, M.J. Animal models: Murine cytomegalovirus. In Methods in Microbiology; Kaufmann, S.H.E., Kabelitz, D., Eds.; Immunology of infection; Academic Press: London, UK, 2002; Volume 32, pp. 493–525. [Google Scholar]
- Lemmermann, N.A.; Podlech, J.; Seckert, C.K.; Kropp, K.A.; Grzimek, N.K.A.; Reddehase, M.J.; Holtappels, R. CD8 T cell immunotherapy of cytomegalovirus disease in the murine model. In Methods in Microbiology; Kabelitz, D., Kaufmann, S.H.E., Eds.; Immunology of infection; Academic Press: London, UK, 2010; Volume 37, pp. 369–420. [Google Scholar]
- Holtappels, R.; Thomas, D.; Podlech, J.; Reddehase, M.J. Two antigenic peptides from genes m123 and m164 of murine cytomegalovirus quantitatively dominate CD8 T-cell memory in the H-2d haplotype. J. Virol. 2002, 76, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Snyder, C.M.; Cho, K.S.; Bonnett, E.L.; Allan, J.E.; Hill, A.B. Sustained CD8+ T cell memory inflation after infection with a single-cycle cytomegalovirus. PLoS Pathog. 2011, 7, e1002295. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.J.; Venturi, V.; Quigley, M.F.; Turula, H.; Gostick, E.; Ladell, K.; Hill, B.J.; Himelfarb, D.; Quinn, K.M.; Greenaway, H.Y.; et al. Stochastic expansions maintain the clonal stability of CD8+ T cell populations undergoing memory inflation driven by murine cytomegalovirus. J. Immunol. 2020, 204, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Podlech, J.; Holtappels, R.; Pahl-Seibert, M.F.; Steffens, H.P.; Reddehase, M.J. Murine model of interstitial cytomegalovirus pneumonia in syngeneic bone marrow transplantation: Persistence of protective pulmonary CD8-T-cell infiltrates after clearance of acute infection. J. Virol. 2000, 74, 7496–7507. [Google Scholar] [CrossRef] [Green Version]
- Ebert, S.; Lemmermann, N.A.W.; Thomas, D.; Renzaho, A.; Reddehase, M.J.; Holtappels, R. Immune control in the absence of immunodominant epitopes: Implications for immunotherapy of cytomegalovirus infection with with antiviral CD8 T cells. Med. Microbiol. Immunol. 2012, 201, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Holtappels, R.; Lemmermann, N.A.; Podlech, J.; Ebert, S.; Reddehase, M.J. Reconstitution of CD8 T cells protective against cytomegalovirus in a mouse model of hematopoietic cell transplantation: Dynamics and inessentiality of epitope immunodominance. Front. Immunol. 2016, 7, 232. [Google Scholar] [CrossRef] [PubMed]
- Karrer, U.; Sierro, S.; Wagner, M.; Oxenius, A.; Hengel, H.; Koszinowski, U.H.; Phillips, R.E.; Klenerman, P. Memory inflation: Continuous accumulation of antiviral CD8+ T cells over time. J. Immunol. 2003, 170, 2022–2029. [Google Scholar] [CrossRef] [Green Version]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef] [PubMed]
- Lefrançois, L.; Marzo, A.L. The descent of memory T-cell subsets. Nat. Rev. Immunol. 2006, 6, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Holtappels, R.; Gillert-Marien, D.; Thomas, D.; Podlech, J.; Deegen, P.; Herter, S.; Oehrlein-Karpi, S.A.; Strand, D.; Wagner, M.; Reddehase, M.J. Cytomegalovirus encodes a positive regulator of antigen presentation. J. Virol. 2006, 80, 7613–7624. [Google Scholar] [CrossRef] [Green Version]
- Schober, K.; Voit, F.; Grassmann, S.; Müller, T.R.; Eggert, J.; Jarosch, S.; Weißbrich, B.; Hoffmann, P.; Borkner, L.; Nio, E.; et al. Reverse TCR repertoire evolution toward dominant low-affinity clones during chronic CMV infection. Nat. Immunol. 2020, 21, 434–441. [Google Scholar] [CrossRef]
- Pinto, A.K.; Munks, M.W.; Koszinowski, U.H.; Hill, A.B. Coordinated function of murine cytomegalovirus genes completely inhibits CTL lysis. J. Immunol. 2006, 177, 3225–3234. [Google Scholar] [CrossRef]
- Lueder, Y.; Heller, K.; Ritter, C.; Keyser, K.A.; Wagner, K.; Liu, X.; Messerle, M.; Stahl, F.R.; Halle, S.; Förster, R. Control of primary mouse cytomegalovirus infection in lung nodular inflammatory foci by cooperation of interferon-gamma expressing CD4 and CD8 T cells. PLoS Pathog. 2018, 14, e1007252. [Google Scholar] [CrossRef]
- Fink, A.; Lemmermann, N.A.W.; Gillert-Marien, D.; Thomas, D.; Freitag, K.; Böhm, V.; Wilhelmi, V.; Reifenberg, K.; Reddehase, M.J.; Holtappels, R. Antigen presentation under the influence of ’immune evasion’ proteins and its modulation by interferon-gamma: Implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med. Microbiol. Immunol. 2012, 201, 513–525. [Google Scholar] [CrossRef]
- Lucin, P.; Jonjić, S.; Messerle, M.; Polić, B.; Hengel, H.; Koszinowski, U.H. Late phase inhibition of murine cytomegalovirus replication by synergistic action of interferon-gamma and tumour necrosis factor. J. Gen. Virol. 1994, 75, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengel, H.; Lucin, P.; Jonjić, S.; Ruppert, T.; Koszinowski, U.H. Restoration of cytomegalovirus antigen presentation by gamma interferon combats viral escape. J. Virol. 1994, 68, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Baumann, N.S.; Welten, S.P.M.; Torti, N.; Pallmer, K.; Borsa, M.; Barnstorf, I.; Oduro, J.D.; Cicin-Sain, L.; Oxenius, A. Early primed KLRG1- CMV-specific T cells determine the size of the inflationary T cell pool. PLoS Pathog. 2019, 15, e1007785. [Google Scholar] [CrossRef] [Green Version]
- Welten, S.P.M.; Yermanos, A.; Baumann, N.S.; Wagen, F.; Oetiker, N.; Sandu, I.; Pedrioli, A.; Oduro, J.D.; Reddy, S.T.; Cicin-Sain, L.; et al. Tcf1+ cells are required to maintain the inflationary T cell pool upon MCMV infection. Nat. Commun. 2020, 11, 2295. [Google Scholar] [CrossRef] [PubMed]
- Welsh, R.M.; Che, J.W.; Brehm, M.A.; Selin, L.K. Heterologous immunity between viruses. Immunol. Rev. 2010, 235, 244–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, B. Heterologous immunity: Role in natural and vaccine-induced resistance to infections. Front. Immunol. 2019, 10, 2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazza, C.; Malissen, B. What guides MHC-restricted TCR recognition? Semin. Immunol. 2007, 19, 225–235. [Google Scholar] [CrossRef]
- Mazza, C.; Auphan-Anezin, N.; Gregoire, C.; Guimezanes, A.; Kellenberger, C.; Roussel, A.; Kearney, A.; van der Merwe, P.A.; Schmitt-Verhulst, A.-M.; Malissen, B. How much can a T-cell antigen receptor adapt to structurally distinct antigenic peptides? EMBO J. 2007, 26, 1972–1983. [Google Scholar] [CrossRef] [Green Version]
- Schober, K.; Buchholz, V.R.; Busch, D.H. TCR repertoire evolution during maintenance of CMV-specific T-cell populations. Immunol. Rev. 2018, 283, 113–128. [Google Scholar] [CrossRef]
- Pawelec, G.; Akbar, A.; Caruso, C.; Effros, R.; Grubeck-Loebenstein, B.; Wikby, A. Is immunosenescence infectious? Trends Immunol. 2004, 25, 406–410. [Google Scholar] [CrossRef]
- Nikolich-Zugich, J. Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nat. Rev. Immunol. 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Jergović, M.; Contreras, N.A.; Nikolich-Žugich, J. Impact of CMV upon immune aging: Facts and fiction. Med. Microbiol. Immunol. 2019, 208, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Moss, P. ‘From immunosenescence to immune modulation’: A re-appraisal of the role of cytomegalovirus as major regulator of human immune function. Med. Microbiol. Immunol. 2019, 208, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.O.; Holtappels, R.; Tervo, H.; Böhm, V.; Däubner, T.; Oehrlein-Karpi, S.A.; Kühnapfel, B.; Renzaho, A.; Strand, D.; Podlech, J.; et al. CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J. Virol. 2006, 80, 10436–10456. [Google Scholar] [CrossRef] [Green Version]
- Renzaho, A.; Schmiedeke, J.K.; Griessl, M.; Kühnapfel, B.; Seckert, C.K.; Lemmermann, N.A.W. Transcripts expressed in cytomegalovirus latency coding for an antigenic IE/E phase peptide that drives “memory inflation”. Med. Microbiol. Immunol. 2019, 208, 439–446. [Google Scholar] [PubMed]
- Gabel, M.; Baumann, N.S.; Oxenius, A.; Graw, F. Investigating the dynamics of MCMV-specific CD8+ T cell responses in individual hosts. Front. Immunol. 2019, 10, 1358. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Ladell, K.; Wynn, K.K.; Stacey, M.A.; Quigley, M.F.; Gostick, E.; Price, D.A.; Humphreys, I.R. IL-10 restricts memory T cell inflation during cytomegalovirus infection. J. Immunol. 2010, 185, 3583–3592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchholz, V.R.; Flossdorf, M.; Hensel, I.; Kretschmer, L.; Weissbrich, B.; Gräf, P.; Verschoor, A.; Schiemann, M.; Höfer, T.; Busch, D.H. Disparate individual fates compose robust CD8+ T cell immunity. Science 2013, 340, 630–635. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holtappels, R.; Freitag, K.; Renzaho, A.; Becker, S.; Lemmermann, N.A.W.; Reddehase, M.J. Revisiting CD8 T-cell ‘Memory Inflation’: New Insights with Implications for Cytomegaloviruses as Vaccine Vectors. Vaccines 2020, 8, 402. https://doi.org/10.3390/vaccines8030402
Holtappels R, Freitag K, Renzaho A, Becker S, Lemmermann NAW, Reddehase MJ. Revisiting CD8 T-cell ‘Memory Inflation’: New Insights with Implications for Cytomegaloviruses as Vaccine Vectors. Vaccines. 2020; 8(3):402. https://doi.org/10.3390/vaccines8030402
Chicago/Turabian StyleHoltappels, Rafaela, Kirsten Freitag, Angelique Renzaho, Sara Becker, Niels A.W. Lemmermann, and Matthias J. Reddehase. 2020. "Revisiting CD8 T-cell ‘Memory Inflation’: New Insights with Implications for Cytomegaloviruses as Vaccine Vectors" Vaccines 8, no. 3: 402. https://doi.org/10.3390/vaccines8030402
APA StyleHoltappels, R., Freitag, K., Renzaho, A., Becker, S., Lemmermann, N. A. W., & Reddehase, M. J. (2020). Revisiting CD8 T-cell ‘Memory Inflation’: New Insights with Implications for Cytomegaloviruses as Vaccine Vectors. Vaccines, 8(3), 402. https://doi.org/10.3390/vaccines8030402