
Natural Flt3Lg-Based Chimeric Antigen Receptor (Flt3-CAR) T Cells Successfully Target Flt3 on AML Cell Lines

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of CAR and mKate2 Lentiviruses
2.2. Generation of CAR T Cells, CAR Jurkat Cells, and THP-1-mKate2 and U937-mKate2 Cell Lines
2.3. Western Blotting
2.4. Cell Culture
2.5. Flow Cytometry
2.6. CAR T and CAR Jurkat Cell Cytotoxicity Assay
2.7. Cell Proliferation Assay
2.8. Quantitative Data Handling
3. Results
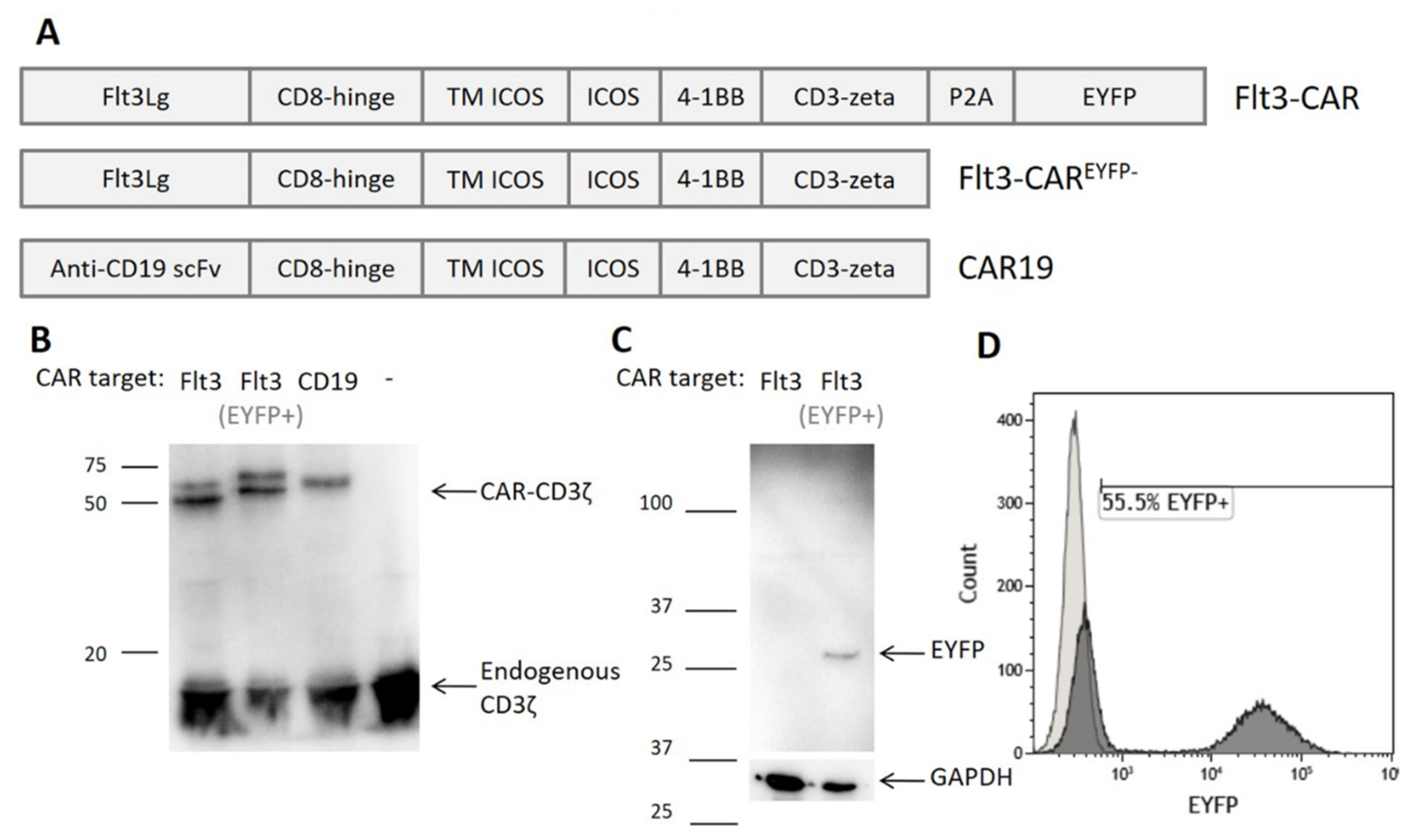
3.1. Generating Flt3-CAR
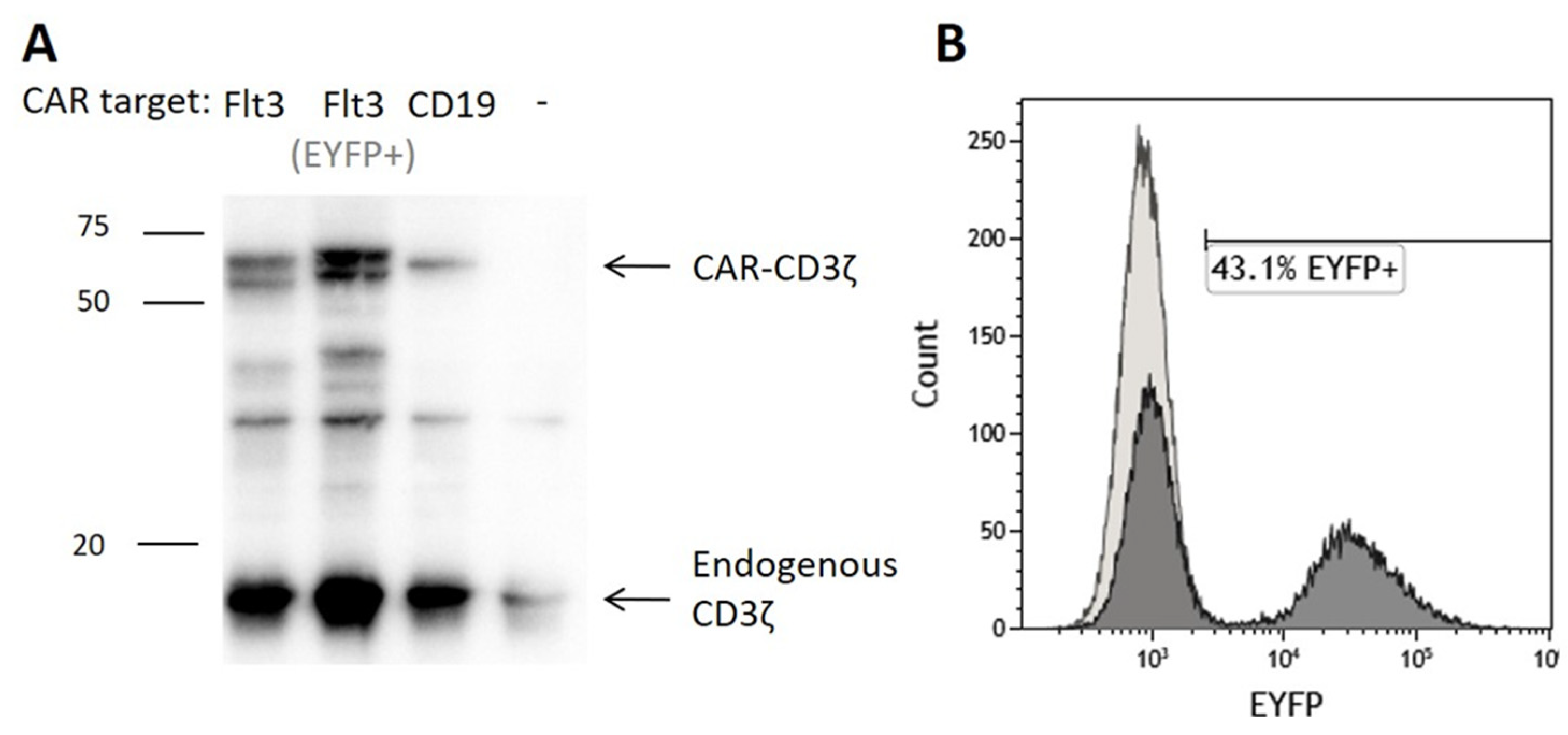
3.2. Flt3-CAR T Cells Specifically Kill Flt3-Positive Cells
3.3. Specific Cytolytic Capacity of T Cells Is Necessary for Flt3-CAR-Mediated Killing
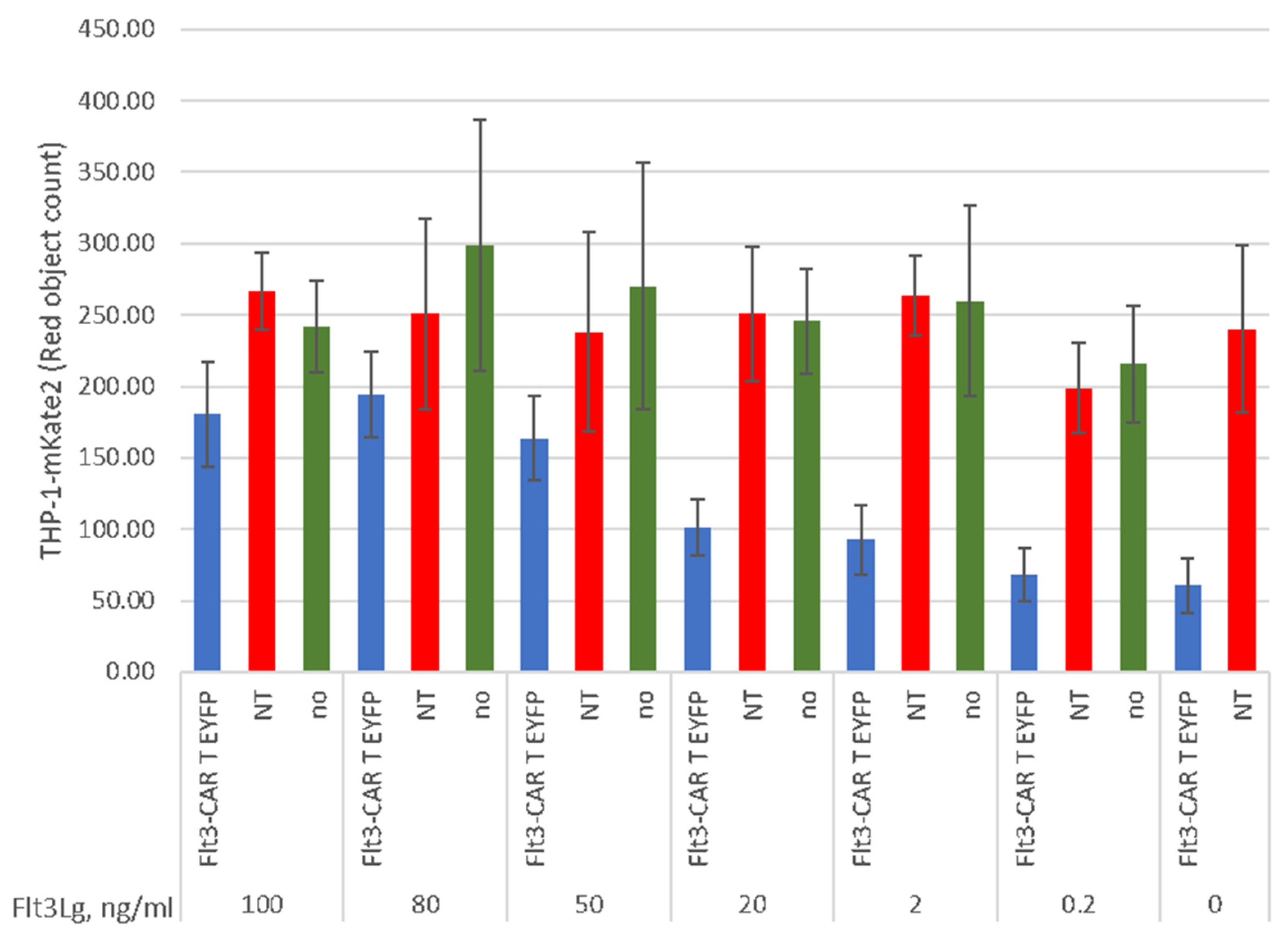
3.4. Soluble Flt3Lg Promotes Proliferation of THP-1 Cells
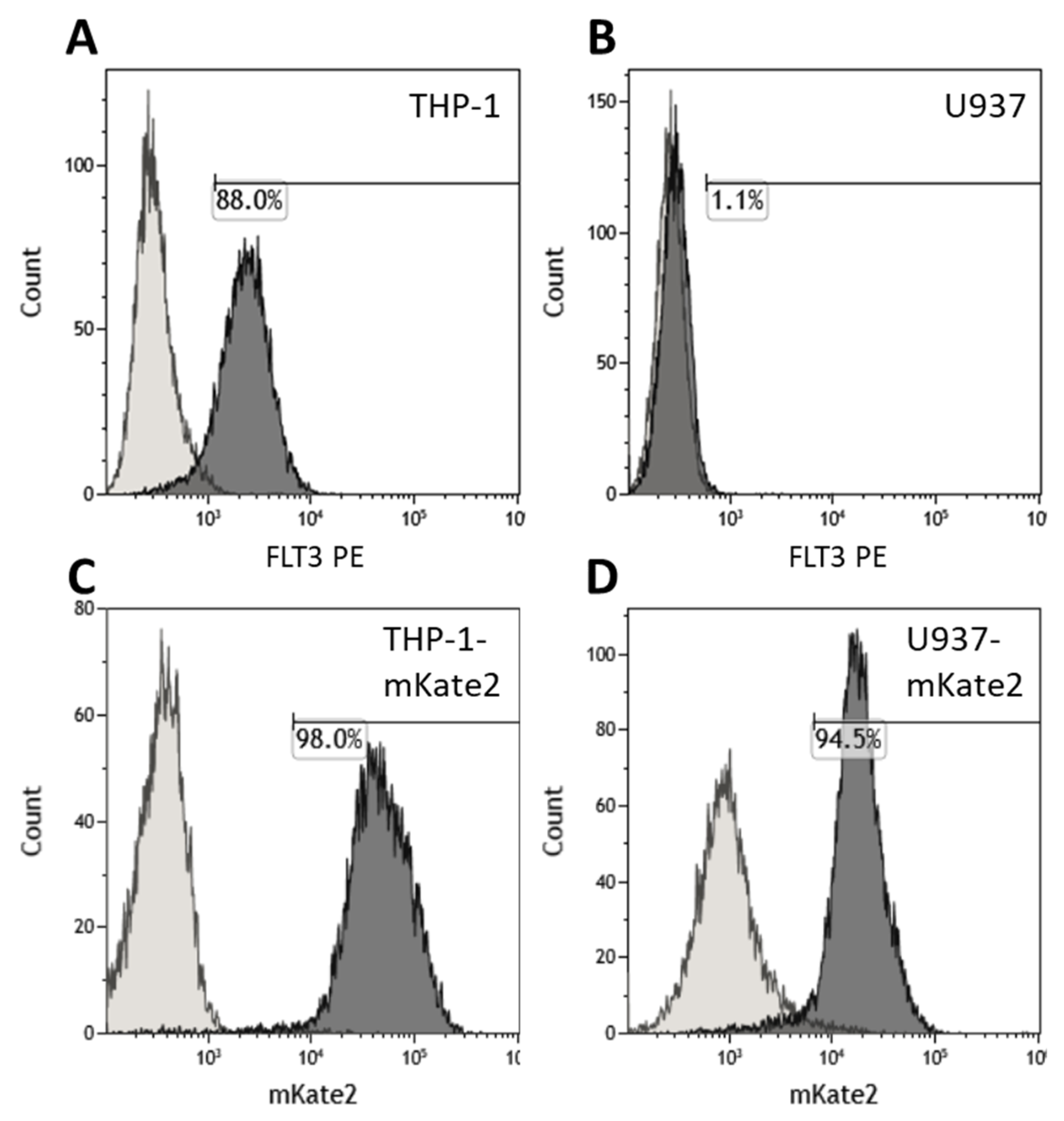
3.5. Flt3-CAR T Cells Recognize the Flt3Lg-Binding Site of Flt3 on the Surface of THP-1 Cells
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maschan, M.; Shelikhova, L.; Ilushina, M.; Shekhovtsova, Z.; Khismatullina, R.; Kurnikova, E.; Pershin, D.; Balashov, D.; Kalinina, I.; Muzalevskii, Y.; et al. Outcome of αβ T cell-depleted transplantation in children with high-risk acute myeloid leukemia, grafted in remission. Bone Marrow Transplant. 2019, 55, 256–259. [Google Scholar] [CrossRef] [PubMed]
- DeMaria, P.J.; Bilusic, M. Cancer Vaccines. Hematol. Clin. N. Am. 2019, 33, 199–214. [Google Scholar] [CrossRef] [PubMed]
- De Bruijn, S.; Anguille, S.; Verlooy, J.; Smits, E.L.; Van Tendeloo, V.F.; De Laere, M.; Norga, K.; Berneman, Z.N.; Lion, E. Dendritic Cell-Based and Other Vaccination Strategies for Pediatric Cancer. Cancers 2019, 11, 1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousseau, R.F.; Biagi, E.; Dutour, A.; Yvon, E.S.; Brown, M.P.; Lin, T.; Mei, Z.; Grilley, B.; Popek, E.; Heslop, H.E.; et al. Immunotherapy of high-risk acute leukemia with a recipient (autologous) vaccine expressing transgenic human CD40L and IL-2 after chemotherapy and allogeneic stem cell transplantation. Blood 2006, 107, 1332–1341. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Zheng, J.-E.; Wang, N.; Cai, H.-H.; Zhai, L.-N.; Wu, Y.-H.; Wang, F.; Jin, R.-M.; Zhou, D.-F. Effects of dendritic cell-activated and cytokine-induced killer cell therapy on 22 children with acute myeloid leukemia after chemotherapy. Acta Acad. Med. Wuhan 2015, 35, 689–693. [Google Scholar] [CrossRef]
- Lee, J.-B.; Vasic, D.; Kang, H.; Fang, K.; Zhang, L. State-of-Art of Cellular Therapy for Acute Leukemia. Int. J. Mol. Sci. 2021, 22, 4590. [Google Scholar] [CrossRef] [PubMed]
- Casucci, M.; Di Robilant, B.N.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef]
- Kenderian, S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.D.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M.C.; et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015, 29, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Tasian, S.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor–modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, Y.; Li, S.; Liu, J.; Xing, Y.; Xing, H.; Tian, Z.; Tang, K.; Rao, Q.; Wang, M.; et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J. Hematol. Oncol. 2018, 11, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3−ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef]
- Amgen A Phase 1 Study Evaluating the Safety, Tolerability, and Efficacy of FLT3 Chimeric Antigen Receptor T-Cell (CAR-T) AMG 553 in Subjects With FLT3-Positive Relapsed/Refractory Acute Myeloid Leukemia. 2020. Available online: clinicaltrials.gov (accessed on 30 September 2021).
- Sommer, C.; Cheng, H.-Y.; Nguyen, D.; Dettling, D.; Yeung, Y.A.; Sutton, J.; Hamze, M.; Valton, J.; Smith, J.; Djuretic, I.; et al. Allogeneic FLT3 CAR T Cells with an Off-Switch Exhibit Potent Activity against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol. Ther. 2020, 28, 2237–2251. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, E.; Liang, R.; Sirochinsky, B.C.; Ben Jehuda, R.; Sandler, V. Preclinical Development of Anti-FLT3 CAR-T Therapy for the Treatment of Acute Myeloid Leukemia. Blood 2020, 136, 4–5. [Google Scholar] [CrossRef]
- Kazi, J.U.; Rönnstrand, L. FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-L.; Yuan, H.-L.; Yang, Y.-Q.; Wang, L.; Zou, R.-C. A brief review concerning Chimeric Antigen Receptors T cell therapy. J. Cancer 2020, 11, 5424–5431. [Google Scholar] [CrossRef] [PubMed]
- Shcherbo, D.; Murphy, C.S.; Ermakova, G.V.; Solovieva, E.A.; Chepurnykh, T.V.; Shcheglov, A.S.; Verkhusha, V.; Pletnev, V.Z.; Hazelwood, K.L.; Roche, P.M.; et al. Far-red fluorescent tags for protein imaging in living tissues. Biochem. J. 2009, 418, 567–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graddis, T.J.; Brasel, K.; Friend, D.; Srinivasan, S.; Wee, S.; Lyman, S.D.; March, C.J.; McGrew, J.T. Structure-Function Analysis of FLT3 Ligand-FLT3 Receptor Interactions Using a Rapid Functional Screen. J. Biol. Chem. 1998, 273, 17626–17633. [Google Scholar] [CrossRef] [Green Version]
- Drexler, H.G. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia 1996, 10, 588–599. [Google Scholar]
- Lisovsky, M.; Estrov, Z.; Zhang, X.; Consoli, U.; Sanchez-Williams, G.; Snell, V.; Munker, R.; Goodacre, A.; Savchenko, V.G.; Andreeff, M. Flt3 ligand stimulates proliferation and inhibits apoptosis of acute myeloid leukemia cells: Regulation of Bcl-2 and Bax. Blood 1996, 88, 3987–3997. [Google Scholar] [CrossRef] [Green Version]
- Mardiana, S.; Gill, S. CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 697. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Petrov, I.; Suntsova, M.; Mutorova, O.; Sorokin, M.; Garazha, A.; Ilnitskaya, E.; Spirin, P.; Larin, S.; Kovalchuk, O.; Prassolov, V.; et al. Molecular pathway activation features of pediatric acute myeloid leukemia (AML) and acute lymphoblast leukemia (ALL) cells. Aging 2016, 8, 2936–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, S.; Desplat, V.; Villacreces, A.; Guitart, A.V.; Milpied, N.; Pigneux, A.; Vigon, I.; Pasquet, J.-M.; Dumas, P.-Y. Targeting Tyrosine Kinases in Acute Myeloid Leukemia: Why, Who and How? Int. J. Mol. Sci. 2019, 20, 3429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcangeli, S.; Rotiroti, M.C.; Bardelli, M.; Simonelli, L.; Magnani, C.F.; Biondi, A.; Biagi, E.; Tettamanti, S.; Varani, L. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol. Ther. 2017, 25, 1933–1945. [Google Scholar] [CrossRef] [PubMed]
- Thokala, R.; Olivares, S.; Mi, T.; Maiti, S.; Deniger, D.; Huls, H.; Torikai, H.; Singh, H.; Champlin, R.E.; Laskowski, T.; et al. Redirecting Specificity of T cells Using the Sleeping Beauty System to Express Chimeric Antigen Receptors by Mix-and-Matching of VL and VH Domains Targeting CD123+ Tumors. PLoS ONE 2016, 11, e0159477. [Google Scholar] [CrossRef]
- Lynn, R.C.; Feng, Y.; Schutsky, K.; Poussin, M.; Kalota, A.; Dimitrov, D.S., Jr. High-affinity FRβ-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia 2016, 30, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Casucci, M.; Falcone, L.; Camisa, B.; Norelli, M.; Porcellini, S.; Stornaiuolo, A.; Ciceri, F.; Traversari, C.; Bordignon, C.; Bonini, M.C.; et al. Extracellular NGFR Spacers Allow Efficient Tracking and Enrichment of Fully Functional CAR-T Cells Co-Expressing a Suicide Gene. Front. Immunol. 2018, 9, 507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterlin, P.; Gaschet, J.; Guillaume, T.; Garnier, A.; Eveillard, M.; Le Bourgeois, A.; Cherel, M.; Debord, C.; Le Bris, Y.; Theisen, O.; et al. FLT3 ligand plasma levels in acute myeloid leukemia. Haematology 2019, 104, e240–e243. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiorova, V.; Mollaev, M.D.; Vikhreva, P.; Kulakovskaya, E.; Pershin, D.; Chudakov, D.M.; Kibardin, A.; Maschan, M.A.; Larin, S. Natural Flt3Lg-Based Chimeric Antigen Receptor (Flt3-CAR) T Cells Successfully Target Flt3 on AML Cell Lines. Vaccines 2021, 9, 1238. https://doi.org/10.3390/vaccines9111238
Maiorova V, Mollaev MD, Vikhreva P, Kulakovskaya E, Pershin D, Chudakov DM, Kibardin A, Maschan MA, Larin S. Natural Flt3Lg-Based Chimeric Antigen Receptor (Flt3-CAR) T Cells Successfully Target Flt3 on AML Cell Lines. Vaccines. 2021; 9(11):1238. https://doi.org/10.3390/vaccines9111238
Chicago/Turabian StyleMaiorova, Varvara, Murad D. Mollaev, Polina Vikhreva, Elena Kulakovskaya, Dmitry Pershin, Dmitriy M. Chudakov, Alexey Kibardin, Michael A. Maschan, and Sergey Larin. 2021. "Natural Flt3Lg-Based Chimeric Antigen Receptor (Flt3-CAR) T Cells Successfully Target Flt3 on AML Cell Lines" Vaccines 9, no. 11: 1238. https://doi.org/10.3390/vaccines9111238
APA StyleMaiorova, V., Mollaev, M. D., Vikhreva, P., Kulakovskaya, E., Pershin, D., Chudakov, D. M., Kibardin, A., Maschan, M. A., & Larin, S. (2021). Natural Flt3Lg-Based Chimeric Antigen Receptor (Flt3-CAR) T Cells Successfully Target Flt3 on AML Cell Lines. Vaccines, 9(11), 1238. https://doi.org/10.3390/vaccines9111238