
Cancer Vaccines: Antigen Selection Strategy

 ,
,  and
and
Abstract
:1. Introduction
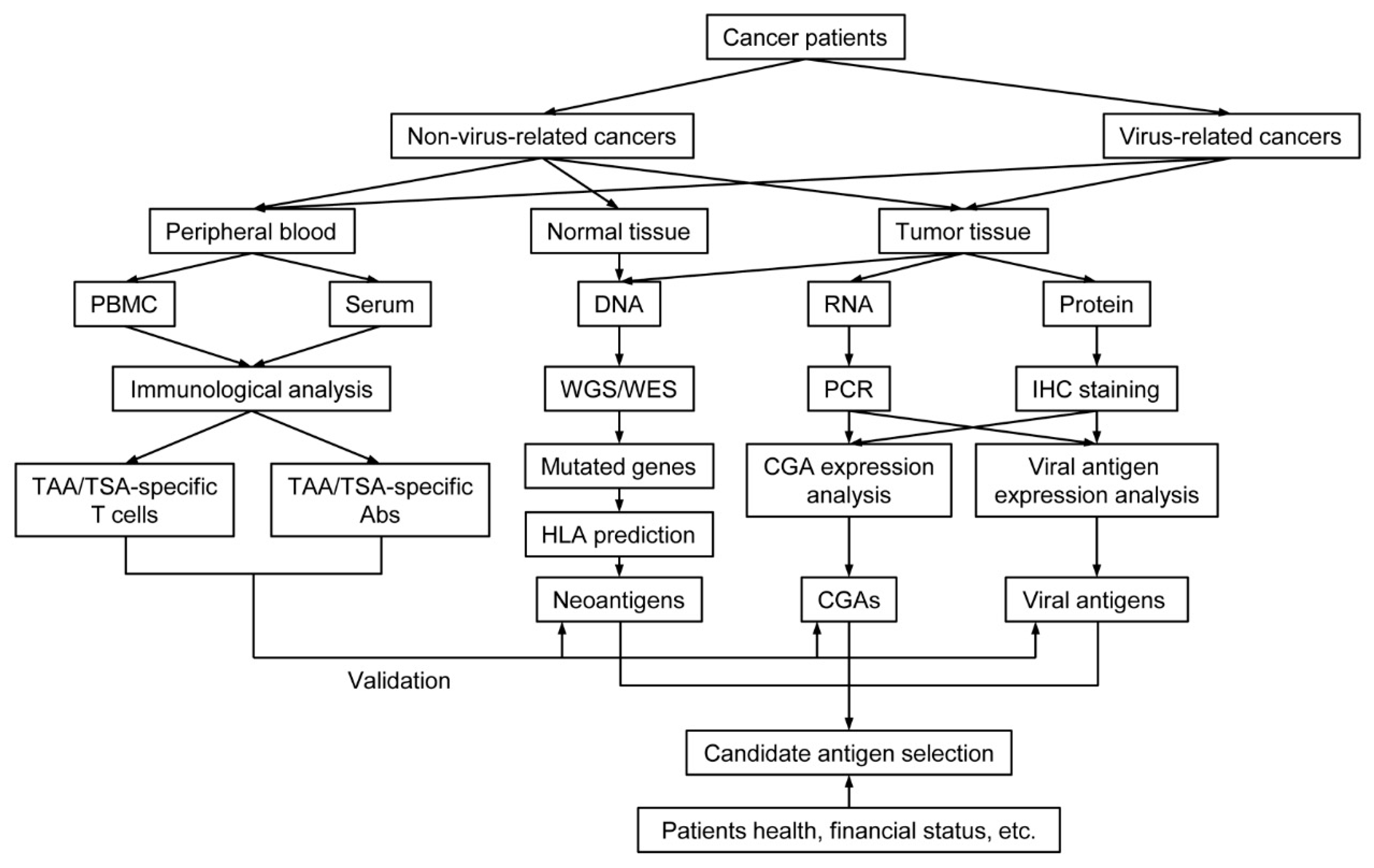
2. Tumor-Specific Antigens
2.1. Neoantigens
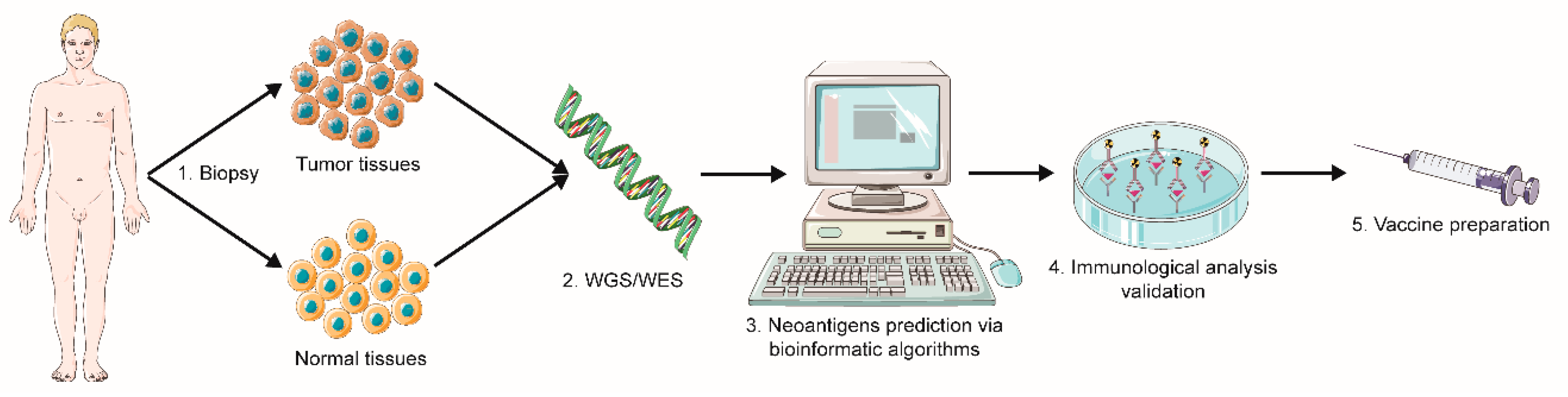
2.1.1. Sequencing and Prediction of Neoantigens
2.1.2. Neoantigen Administration Strategies
2.1.3. Clinical Progress
2.1.4. Combination with Other Therapies
2.2. Viral Antigens
2.2.1. Human Papillomavirus and Cervical Carcinoma
2.2.2. Hepatitis Viruses and Hepatocellular Carcinoma
2.2.3. Epstein–Barr Virus and Nasopharyngeal Carcinoma
2.2.4. Clinical Advance of Viral Antigen-Based Cancer Vaccines
3. Tumor-Associated Antigens
3.1. Cancer-Germline Antigens
3.1.1. Melanoma Antigen Family A3
3.1.2. “New York Esophageal Squamous Cell Carcinoma-1” Antigen
3.1.3. Preferentially Expressed Antigen in Melanoma
3.2. Clinical Advance in Cancer-Germline Antigen-Based Vaccination
3.3. Combination Therapy
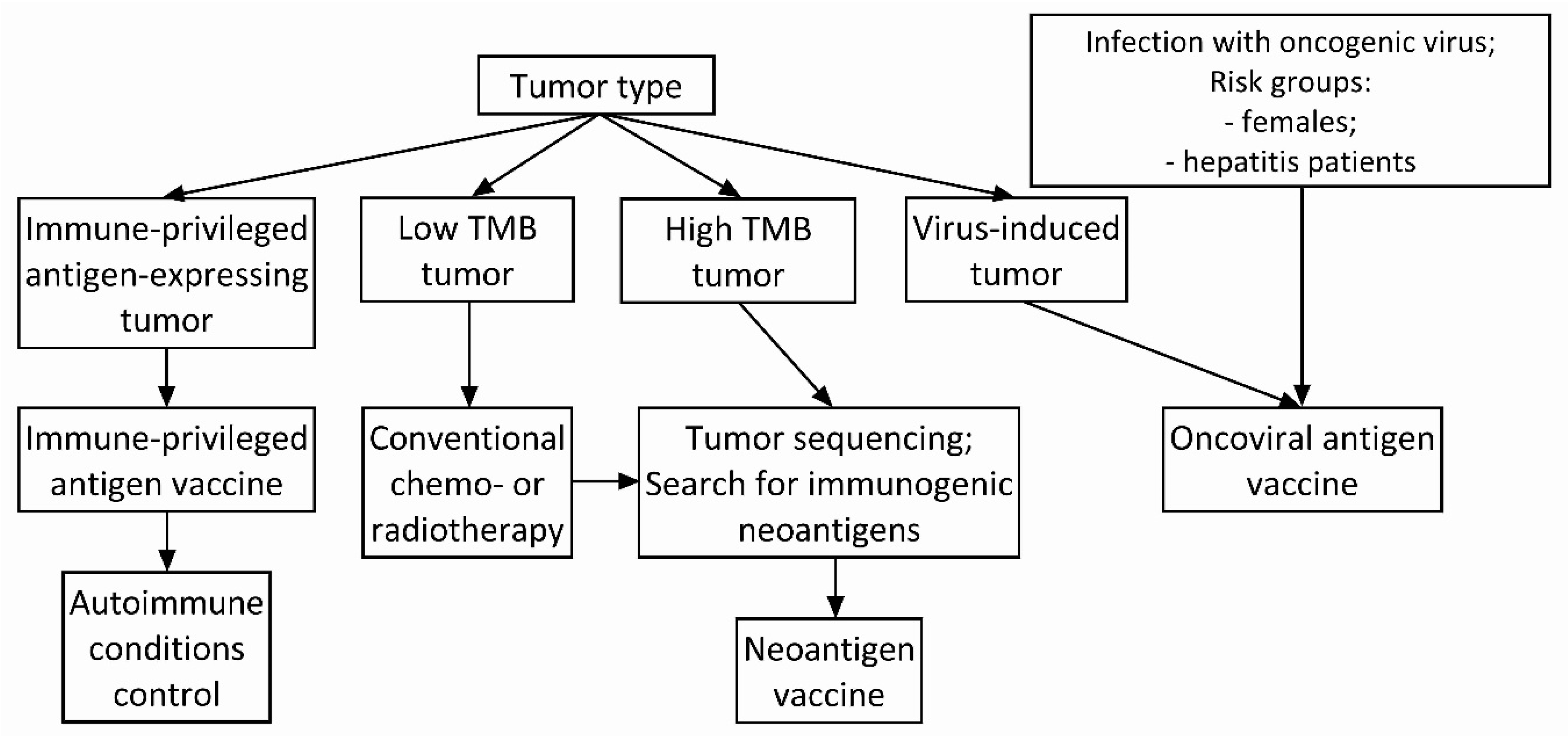
4. Antigen Selection Strategy for Cancer Vaccine
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, M.Y.; Jeon, J.W.; Sievers, C.; Allen, C.T. Antigen processing and presentation in cancer immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Segal, N.H.; Parsons, D.W.; Peggs, K.S.; Velculescu, V.; Kinzler, K.W.; Vogelstein, B.; Allison, J.P. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008, 68, 889–892. [Google Scholar] [CrossRef] [Green Version]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- De Plaen, E.; Lurquin, C.; Van Pel, A.; Mariamé, B.; Szikora, J.P.; Wölfel, T.; Sibille, C.; Chomez, P.; Boon, T. Immunogenic (tum-) variants of mouse tumor P815: Cloning of the gene of tum- antigen P91A and identification of the tum- mutation. Proc. Natl. Acad. Sci. USA 1988, 85, 2274–2278. [Google Scholar] [CrossRef] [Green Version]
- Sibille, C.; Chomez, P.; Wildmann, C.; Van Pel, A.; De Plaen, E.; Maryanski, J.L.; de Bergeyck, V.; Boon, T. Structure of the gene of tum- transplantation antigen P198: A point mutation generates a new antigenic peptide. J. Exp. Med. 1990, 172, 35–45. [Google Scholar] [CrossRef]
- Monach, P.A.; Meredith, S.C.; Siegel, C.T.; Schreiber, H. A unique tumor antigen produced by a single amino acid substitution. Immunity 1995, 2, 45–59. [Google Scholar] [CrossRef] [Green Version]
- Coulie, P.G.; Lehmann, F.; Lethe, B.; Herman, J.; Lurquin, C.; Andrawiss, M.; Boon, T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA 1995, 92, 7976–7980. [Google Scholar] [CrossRef] [Green Version]
- Wolfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wolfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Meyer zum Buschenfelde, K.H.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Besser, H.; Yunger, S.; Merhavi-Shoham, E.; Cohen, C.J.; Louzoun, Y. Level of neo-epitope predecessor and mutation type determine T cell activation of MHC binding peptides. J. Immunother. Cancer 2019, 7, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, J.S.; Chang, A.R.; Wick, D.A.; Sedgwick, C.G.; Zong, Z.; Mungall, A.J.; Martin, S.D.; Kinloch, N.N.; Ott-Langer, S.; Brumme, Z.L.; et al. Mapping the human T cell repertoire to recurrent driver mutations in MYD88 and EZH2 in lymphoma. Oncoimmunology 2017, 6, e1321184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleksic, M.; Liddy, N.; Molloy, P.E.; Pumphrey, N.; Vuidepot, A.; Chang, K.M.; Jakobsen, B.K. Different affinity windows for virus and cancer-specific T-cell receptors: Implications for therapeutic strategies. Eur. J. Immunol. 2012, 42, 3174–3179. [Google Scholar] [CrossRef]
- Stone, J.D.; Harris, D.T.; Kranz, D.M. TCR affinity for p/MHC formed by tumor antigens that are self-proteins: Impact on efficacy and toxicity. Curr. Opin. Immunol. 2015, 33, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Luo, H.; Kong, Y.; Lai, W.F.; Cui, L.; Zhu, X. Cancer neoantigen: Boosting immunotherapy. Biomed. Pharmacother. 2020, 131, 110640. [Google Scholar] [CrossRef]
- Karpanen, T.; Olweus, J. The Potential of Donor T-Cell Repertoires in Neoantigen-Targeted Cancer Immunotherapy. Front. Immunol. 2017, 8, 1718. [Google Scholar] [CrossRef] [Green Version]
- Van Buuren, M.M.; Calis, J.J.; Schumacher, T.N. High sensitivity of cancer exome-based CD8 T cell neo-antigen identification. Oncoimmunology 2014, 3, e28836. [Google Scholar] [CrossRef]
- Gopanenko, A.V.; Kosobokova, E.N.; Kosorukov, V.S. Main Strategies for the Identification of Neoantigens. Cancers 2020, 12, 2879. [Google Scholar] [CrossRef]
- Nakagawa, H.; Fujita, M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018, 109, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Investig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef] [PubMed]
- Sneddon, S.; Rive, C.M.; Ma, S.; Dick, I.M.; Allcock, R.J.N.; Brown, S.D.; Holt, R.A.; Watson, M.; Leary, S.; Lee, Y.C.G.; et al. Identification of a CD8+ T-cell response to a predicted neoantigen in malignant mesothelioma. Oncoimmunology 2020, 9, 1684713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hos, B.J.; Camps, M.G.M.; van den Bulk, J.; Tondini, E.; van den Ende, T.C.; Ruano, D.; Franken, K.; Janssen, G.M.C.; Ru, A.; Filippov, D.V.; et al. Identification of a neo-epitope dominating endogenous CD8 T cell responses to MC-38 colorectal cancer. Oncoimmunology 2019, 9, 1673125. [Google Scholar] [CrossRef] [Green Version]
- Bjerregaard, A.M.; Nielsen, M.; Hadrup, S.R.; Szallasi, Z.; Eklund, A.C. MuPeXI: Prediction of neo-epitopes from tumor sequencing data. Cancer Immunol. Immunother. CII 2017, 66, 1123–1130. [Google Scholar] [CrossRef]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szolek, A.; Schubert, B.; Mohr, C.; Sturm, M.; Feldhahn, M.; Kohlbacher, O. OptiType: Precision HLA typing from next-generation sequencing data. Bioinformatics 2014, 30, 3310–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bais, P.; Namburi, S.; Gatti, D.M.; Zhang, X.; Chuang, J.H. CloudNeo: A cloud pipeline for identifying patient-specific tumor neoantigens. Bioinformatics 2017, 33, 3110–3112. [Google Scholar] [CrossRef] [Green Version]
- Schenck, R.O.; Lakatos, E.; Gatenbee, C.; Graham, T.A.; Anderson, A.R.A. NeoPredPipe: High-throughput neoantigen prediction and recognition potential pipeline. BMC Bioinform. 2019, 20, 264. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Fulton, K.M.; Twine, S.M.; Li, J. Identification of mhc peptides using mass spectrometry for neoantigen discovery and cancer vaccine developmeNT. Mass Spectrom. Rev. 2019. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, Y.; Zhang, Q.; Liu, W. Application of mass spectrometry-based MHC immunopeptidome profiling in neoantigen identification for tumor immunotherapy. Biomed. Pharmacother. 2019, 120, 109542. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Goedegebuure, S.P.; Gillanders, W.E. Preclinical and clinical development of neoantigen vaccines. Ann. Oncol. 2017, 28, xii11–xii17. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, J.; Wang, L.; Liu, B. Personalized neoantigen vaccination with synthetic long peptides: Recent advances and future perspectives. Theranostics 2020, 10, 6011–6023. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Morisaki, T.; Hikichi, T.; Onishi, H.; Morisaki, T.; Kubo, M.; Hirano, T.; Yoshimura, S.; Kiyotani, K.; Nakamura, Y. Intranodal Administration of Neoantigen Peptide-loaded Dendritic Cell Vaccine Elicits Epitope-specific T Cell Responses and Clinical Effects in a Patient with Chemorefractory Ovarian Cancer with Malignant Ascites. Immunol. Investig. 2020, 1–18. [Google Scholar] [CrossRef]
- Zhang, R.; Yuan, F.; Shu, Y.; Tian, Y.; Zhou, B.; Yi, L.; Zhang, X.; Ding, Z.; Xu, H.; Yang, L. Personalized neoantigen-pulsed dendritic cell vaccines show superior immunogenicity to neoantigen-adjuvant vaccines in mouse tumor models. Cancer Immunol. Immunother. CII 2020, 69, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Duperret, E.K.; Perales-Puchalt, A.; Stoltz, R.; Hiranjith, G.H.; Mandloi, N.; Barlow, J.; Chaudhuri, A.; Sardesai, N.Y.; Weiner, D.B. A Synthetic DNA, Multi-Neoantigen Vaccine Drives Predominately MHC Class I CD8(+) T-cell Responses, Impacting Tumor Challenge. Cancer Immunol. Res. 2019, 7, 174–182. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Salomon, N.; Vascotto, F.; Selmi, A.; Vormehr, M.; Quinkhardt, J.; Bukur, T.; Schrörs, B.; Löewer, M.; Diken, M.; Türeci, Ö.; et al. A liposomal RNA vaccine inducing neoantigen-specific CD4(+) T cells augments the antitumor activity of local radiotherapy in mice. Oncoimmunology 2020, 9, 1771925. [Google Scholar] [CrossRef]
- Zhu, G.; Zhang, F.; Ni, Q.; Niu, G.; Chen, X. Efficient Nanovaccine Delivery in Cancer Immunotherapy. ACS Nano 2017, 11, 2387–2392. [Google Scholar] [CrossRef]
- Reuven, E.M.; Leviatan Ben-Arye, S.; Yu, H.; Duchi, R.; Perota, A.; Conchon, S.; Bachar Abramovitch, S.; Soulillou, J.P.; Galli, C.; Chen, X.; et al. Biomimetic Glyconanoparticle Vaccine for Cancer Immunotherapy. ACS Nano 2019, 13, 2936–2947. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, M.O.; Vogel, M.; Riether, C.; Muller, J.; Salatino, S.; Ternette, N.; Gomes, A.C.; Cabral-Miranda, G.; El-Turabi, A.; Ruedl, C.; et al. Targeting Mutated Plus Germline Epitopes Confers Pre-clinical Efficacy of an Instantly Formulated Cancer Nano-Vaccine. Front. Immunol. 2019, 10, 1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurisicchio, L.; Salvatori, E.; Lione, L.; Bandini, S.; Pallocca, M.; Maggio, R.; Fanciulli, M.; De Nicola, F.; Goeman, F.; Ciliberto, G.; et al. Poly-specific neoantigen-targeted cancer vaccines delay patient derived tumor growth. J. Exp. Clin. Cancer Res. 2019, 38, 78. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lin, Z.; Wan, Y.; Cai, H.; Deng, L.; Li, R. The Immunogenicity and Anti-tumor Efficacy of a Rationally Designed Neoantigen Vaccine for B16F10 Mouse Melanoma. Front. Immunol. 2019, 10, 2472. [Google Scholar] [CrossRef] [Green Version]
- Scheetz, L.; Kadiyala, P.; Sun, X.; Son, S.; Hassani Najafabadi, A.; Aikins, M.; Lowenstein, P.R.; Schwendeman, A.; Castro, M.G.; Moon, J.J. Synthetic High-density Lipoprotein Nanodiscs for Personalized Immunotherapy Against Gliomas. Clin. Cancer Res. 2020, 26, 4369–4380. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Kinkead, H.L.; Hopkins, A.; Lutz, E.; Wu, A.A.; Yarchoan, M.; Cruz, K.; Woolman, S.; Vithayathil, T.; Glickman, L.H.; Ndubaku, C.O.; et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Forghanifard, M.M.; Gholamin, M.; Moaven, O.; Farshchian, M.; Ghahraman, M.; Aledavood, A.; Abbaszadegan, M.R. Neoantigen in esophageal squamous cell carcinoma for dendritic cell-based cancer vaccine development. Med. Oncol. 2014, 31, 191. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, A.M.; Lesterhuis, W.J.; Nowak, A.K.; Lake, R.A. Chemotherapy and immunotherapy: Mapping the road ahead. Curr. Opin. Immunol. 2016, 39, 23–29. [Google Scholar] [CrossRef]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018, 419, 210–221. [Google Scholar] [CrossRef]
- Boccardo, E.; Villa, L.L. Viral origins of human cancer. Curr. Med. Chem. 2007, 14, 2526–2539. [Google Scholar] [CrossRef]
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. (Eds.) Retroviruses. In Retroviruses; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Valdespino, V.; Gorodezky, C.; Ortiz, V.; Kaufmann, A.M.; Roman-Basaure, E.; Vazquez, A.; Berumen, J. HPV16-specific cytotoxic T lymphocyte responses are detected in all HPV16-positive cervical cancer patients. Gynecol. Oncol. 2005, 96, 92–102. [Google Scholar] [CrossRef]
- Drury, S.E.; Gough, R.E.; McArthur, S.; Jessop, M. Detection of herpesvirus-like and papillomavirus-like particles associated with diseases of tortoises. Vet. Rec. 1998, 143, 639. [Google Scholar]
- Mühr, L.S.A.; Eklund, C.; Dillner, J. Towards quality and order in human papillomavirus research. Virology 2018, 519, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Lorincz, A.; Muñoz, N.; Meijer, C.J.; Shah, K.V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, G.; Kreimer, A.R.; Viscidi, R.; Pawlita, M.; Fakhry, C.; Koch, W.M.; Westra, W.H.; Gillison, M.L. Case-control study of human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007, 356, 1944–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Pan, W.; Jin, L.; Huang, W.; Li, Y.; Wu, D.; Gao, C.; Ma, D.; Liao, S. Human papillomavirus vaccine against cervical cancer: Opportunity and challenge. Cancer Lett. 2020, 471, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Dyson, N.; Howley, P.M.; Münger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef]
- Van der Burg, S.H.; Melief, C.J. Therapeutic vaccination against human papilloma virus induced malignancies. Curr. Opin. Immunol. 2011, 23, 252–257. [Google Scholar] [CrossRef]
- Morrow, M.P.; Yan, J.; Sardesai, N.Y. Human papillomavirus therapeutic vaccines: Targeting viral antigens as immunotherapy for precancerous disease and cancer. Expert Rev. Vaccines 2013, 12, 271–283. [Google Scholar] [CrossRef]
- Monnier-Benoit, S.; Mauny, F.; Riethmuller, D.; Guerrini, J.S.; Căpîlna, M.; Félix, S.; Seillès, E.; Mougin, C.; Prétet, J.L. Immunohistochemical analysis of CD4+ and CD8+ T-cell subsets in high risk human papillomavirus-associated pre-malignant and malignant lesions of the uterine cervix. Gynecol. Oncol. 2006, 102, 22–31. [Google Scholar] [CrossRef]
- Kim, K.H.; Greenfield, W.W.; Cannon, M.J.; Coleman, H.N.; Spencer, H.J.; Nakagawa, M. CD4+ T-cell response against human papillomavirus type 16 E6 protein is associated with a favorable clinical trend. Cancer Immunol. Immunother. CII 2012, 61, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.J.; Jin, H.T.; Hur, S.Y.; Yang, H.G.; Seo, Y.B.; Hong, S.R.; Lee, C.W.; Kim, S.; Woo, J.W.; Park, K.S.; et al. Clearance of persistent HPV infection and cervical lesion by therapeutic DNA vaccine in CIN3 patients. Nat. Commun. 2014, 5, 5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Oosterhuis, K.; Wunderlich, K.; Bunnik, E.M.; Bhaggoe, M.; Boedhoe, S.; Karia, S.; Steenbergen, R.D.M.; Bosch, L.; Serroyen, J.; et al. Development of a replication-deficient adenoviral vector-based vaccine candidate for the interception of HPV16- and HPV18-induced infections and disease. Int. J. cancer 2017, 141, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallecha, A.; French, C.; Petit, R.; Singh, R.; Amin, A.; Rothman, J. Lm-LLO-Based Immunotherapies and HPV-Associated Disease. J. Oncol. 2012, 2012, 542851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahma, O.E.; Herrin, V.E.; Ibrahim, R.A.; Toubaji, A.; Bernstein, S.; Dakheel, O.; Steinberg, S.M.; Abu Eid, R.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Pre-immature dendritic cells (PIDC) pulsed with HPV16 E6 or E7 peptide are capable of eliciting specific immune response in patients with advanced cervical cancer. J. Transl. Med. 2014, 12, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenter, G.G.; Welters, M.J.; Valentijn, A.R.; Lowik, M.J.; Berends-van der Meer, D.M.; Vloon, A.P.; Essahsah, F.; Fathers, L.M.; Offringa, R.; Drijfhout, J.W.; et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 2009, 361, 1838–1847. [Google Scholar] [CrossRef] [Green Version]
- Stöppler, M.C.; Straight, S.W.; Tsao, G.; Schlegel, R.; McCance, D.J. The E5 gene of HPV-16 enhances keratinocyte immortalization by full-length DNA. Virology 1996, 223, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Straight, S.W.; Herman, B.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J. Virol. 1995, 69, 3185–3192. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.K.; Kim, H.S.; Kim, S.H.; Oh, J.M.; Han, J.Y.; Lim, J.M.; Juhnn, Y.S.; Song, Y.S. Human papillomavirus type 16 E5 oncoprotein as a new target for cervical cancer treatment. Biochem. Pharmacol. 2010, 80, 1930–1935. [Google Scholar] [CrossRef]
- Liao, S.J.; Deng, D.R.; Zeng, D.; Zhang, L.; Hu, X.J.; Zhang, W.N.; Li, L.; Jiang, X.F.; Wang, C.Y.; Zhou, J.F.; et al. HPV16 E5 peptide vaccine in treatment of cervical cancer in vitro and in vivo. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 735–742. [Google Scholar] [CrossRef]
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Primers 2018, 4, 18035. [Google Scholar] [CrossRef]
- Xu, H.Z.; Liu, Y.P.; Guleng, B.; Ren, J.L. Hepatitis B Virus-Related Hepatocellular Carcinoma: Pathogenic Mechanisms and Novel Therapeutic Interventions. Gastrointest. Tumors 2014, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Gong, J.; Tian, D.; Wang, Z. Hepatitis B Virus X Protein Induces SATB1 Expression Through Activation of ERK and p38MAPK Pathways to Suppress Anoikis. Dig. Dis. Sci. 2019, 64, 3203–3214. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Wu, D.W.; Shen, C.J.; Cheng, Y.M.; Wu, C.C.; Lee, H. Hepatitis B virus X protein promotes tumor invasion and poor prognosis in hepatocellular carcinoma via phosphorylation of paxillin at Serine 178 by activation of the c-Jun NH2-terminal kinase. Am. J. Cancer Res. 2020, 10, 275–283. [Google Scholar] [PubMed]
- Lupberger, J.; Hildt, E. Hepatitis B virus-induced oncogenesis. World J. Gastroenterol. 2007, 13, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, F.X.; Wang, F.; Lu, Y.M.; Li, K.; Wang, K.H.; He, X.W.; Sun, S.H. Multiepitope peptide-loaded virus-like particles as a vaccine against hepatitis B virus-related hepatocellular carcinoma. Hepatology 2009, 49, 1492–1502. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, D.; Li, S.; Gao, Y.; Jiang, R.; Deng, L.; Frankel, F.R.; Sun, B. Development of a Listeria monocytogenes-based vaccine against hepatocellular carcinoma. Oncogene 2012, 31, 2140–2152. [Google Scholar] [CrossRef] [Green Version]
- Rongrui, L.; Na, H.; Zongfang, L.; Fanpu, J.; Shiwen, J. Epigenetic mechanism involved in the HBV/HCV-related hepatocellular carcinoma tumorigenesis. Curr. Pharm. Des. 2014, 20, 1715–1725. [Google Scholar] [CrossRef]
- Navas, M.C.; Glaser, S.; Dhruv, H.; Celinski, S.; Alpini, G.; Meng, F. Hepatitis C Virus Infection and Cholangiocarcinoma: An Insight into Epidemiologic Evidences and Hypothetical Mechanisms of Oncogenesis. Am. J. Pathol. 2019, 189, 1122–1132. [Google Scholar] [CrossRef] [Green Version]
- Preciado, M.V.; Valva, P.; Escobar-Gutierrez, A.; Rahal, P.; Ruiz-Tovar, K.; Yamasaki, L.; Vazquez-Chacon, C.; Martinez-Guarneros, A.; Carpio-Pedroza, J.C.; Fonseca-Coronado, S.; et al. Hepatitis C virus molecular evolution: Transmission, disease progression and antiviral therapy. World J. Gastroenterol. 2014, 20, 15992–16013. [Google Scholar] [CrossRef]
- Tagliamonte, M.; Petrizzo, A.; Napolitano, M.; Luciano, A.; Arra, C.; Maiolino, P.; Izzo, F.; Tornesello, M.L.; Aurisicchio, L.; Ciliberto, G.; et al. Novel metronomic chemotherapy and cancer vaccine combinatorial strategy for hepatocellular carcinoma in a mouse model. Cancer Immunol. Immunother. CII 2015, 64, 1305–1314. [Google Scholar] [CrossRef]
- Nowalk, A.; Green, M. Epstein-Barr Virus. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teow, S.Y.; Yap, H.Y.; Peh, S.C. Epstein-Barr Virus as a Promising Immunotherapeutic Target for Nasopharyngeal Carcinoma Treatment. J. Pathog 2017, 2017, 7349268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duraiswamy, J.; Sherritt, M.; Thomson, S.; Tellam, J.; Cooper, L.; Connolly, G.; Bharadwaj, M.; Khanna, R. Therapeutic LMP1 polyepitope vaccine for EBV-associated Hodgkin disease and nasopharyngeal carcinoma. Blood 2003, 101, 3150–3156. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Zhang, Q.; Zhou, J.; Ma, D.; Xiao, X.; Wang, D.W. Recombinant adeno-associated virus encoding Epstein-Barr virus latent membrane proteins fused with heat shock protein as a potential vaccine for nasopharyngeal carcinoma. Mol. Cancer Ther. 2009, 8, 2754–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, G.S.; Haigh, T.A.; Gudgeon, N.H.; Phelps, R.J.; Lee, S.P.; Steven, N.M.; Rickinson, A.B. Dual stimulation of Epstein-Barr Virus (EBV)-specific CD4+- and CD8+-T-cell responses by a chimeric antigen construct: Potential therapeutic vaccine for EBV-positive nasopharyngeal carcinoma. J. Virol. 2004, 78, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutzky, V.P.; Corban, M.; Heslop, L.; Morrison, L.E.; Crooks, P.; Hall, D.F.; Coman, W.B.; Thomson, S.A.; Moss, D.J. Novel approach to the formulation of an Epstein-Barr virus antigen-based nasopharyngeal carcinoma vaccine. J. Virol. 2010, 84, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Hancock, G.; Hellner, K.; Dorrell, L. Therapeutic HPV vaccines. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Castellsague, X.; Garland, S.M. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine 2012, 30 (Suppl. 5), F123–F138. [Google Scholar] [CrossRef] [Green Version]
- Maciag, P.C.; Radulovic, S.; Rothman, J. The first clinical use of a live-attenuated Listeria monocytogenes vaccine: A Phase I safety study of Lm-LLO-E7 in patients with advanced carcinoma of the cervix. Vaccine 2009, 27, 3975–3983. [Google Scholar] [CrossRef]
- Welters, M.J.; Kenter, G.G.; Piersma, S.J.; Vloon, A.P.; Lowik, M.J.; Berends-van der Meer, D.M.; Drijfhout, J.W.; Valentijn, A.R.; Wafelman, A.R.; Oostendorp, J.; et al. Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin. Cancer Res. 2008, 14, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, C.; Cohen, R.B.; Morrow, M.P.; Kraynyak, K.A.; Sylvester, A.J.; Knoblock, D.M.; Bauml, J.M.; Weinstein, G.S.; Lin, A.; Boyer, J.; et al. Immunotherapy Targeting HPV16/18 Generates Potent Immune Responses in HPV-Associated Head and Neck Cancer. Clin. Cancer Res. 2019, 25, 110–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.L.; Lo, W.F.; Lee, T.H.; Ren, Y.; Hwang, S.L.; Cheng, Y.F.; Chen, C.L.; Chang, Y.S.; Lee, S.P.; Rickinson, A.B.; et al. Immunization with Epstein-Barr Virus (EBV) peptide-pulsed dendritic cells induces functional CD8+ T-cell immunity and may lead to tumor regression in patients with EBV-positive nasopharyngeal carcinoma. Cancer Res. 2002, 62, 6952–6958. [Google Scholar] [PubMed]
- Chia, W.K.; Wang, W.W.; Teo, M.; Tai, W.M.; Lim, W.T.; Tan, E.H.; Leong, S.S.; Sun, L.; Chen, J.J.; Gottschalk, S.; et al. A phase II study evaluating the safety and efficacy of an adenovirus-DeltaLMP1-LMP2 transduced dendritic cell vaccine in patients with advanced metastatic nasopharyngeal carcinoma. Ann. Oncol. 2012, 23, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.S.; Jia, H.; Harrington, K.; Lee, L.W.; Turner, J.; Ladell, K.; Price, D.A.; Tanday, M.; Matthews, J.; Roberts, C.; et al. A recombinant modified vaccinia ankara vaccine encoding Epstein-Barr Virus (EBV) target antigens: A phase I trial in UK patients with EBV-positive cancer. Clin. Cancer Res. 2014, 20, 5009–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, E.P.; Taylor, G.S.; Jia, H.; Ma, B.B.; Chan, S.L.; Ho, R.; Wong, W.L.; Wilson, S.; Johnson, B.F.; Edwards, C.; et al. Phase I trial of recombinant modified vaccinia ankara encoding Epstein-Barr viral tumor antigens in nasopharyngeal carcinoma patients. Cancer Res. 2013, 73, 1676–1688. [Google Scholar] [CrossRef] [Green Version]
- Yutani, S.; Ueshima, K.; Abe, K.; Ishiguro, A.; Eguchi, J.; Matsueda, S.; Komatsu, N.; Shichijo, S.; Yamada, A.; Itoh, K.; et al. Phase II Study of Personalized Peptide Vaccination with Both a Hepatitis C Virus-Derived Peptide and Peptides from Tumor-Associated Antigens for the Treatment of HCV-Positive Advanced Hepatocellular Carcinoma Patients. J. Immunol. Res. 2015, 2015, 473909. [Google Scholar] [CrossRef] [Green Version]
- Geall, A.J.; Ulmer, J.B. Introduction to RNA-based vaccines and therapeutics. Expert Rev. Vaccines 2015, 14, 151–152. [Google Scholar] [CrossRef] [Green Version]
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Pinzani, P.; Lind, K.; Malentacchi, F.; Nesi, G.; Salvianti, F.; Villari, D.; Kubista, M.; Pazzagli, M.; Orlando, C. Prostate-specific antigen mRNA and protein levels in laser microdissected cells of human prostate measured by real-time reverse transcriptase-quantitative polymerase chain reaction and immuno-quantitative polymerase chain reaction. Hum. Pathol. 2008, 39, 1474–1482. [Google Scholar] [CrossRef]
- Liu, C.C.; Yang, H.; Zhang, R.; Zhao, J.J.; Hao, D.J. Tumour-associated antigens and their anti-cancer applications. Eur. J. Cancer Care 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.K.; Bright, J.D.; Byrne, J.A. Overexpressed oncogenic tumor-self antigens. Hum. Vaccines Immunother. 2014, 10, 3297–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haen, S.P.; Rammensee, H.G. The repertoire of human tumor-associated epitopes—Identification and selection of antigens and their application in clinical trials. Curr. Opin. Immunol. 2013, 25, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Bart, J.; Groen, H.J.; van der Graaf, W.T.; Hollema, H.; Hendrikse, N.H.; Vaalburg, W.; Sleijfer, D.T.; de Vries, E.G. An oncological view on the blood-testis barrier. Lancet. Oncol. 2002, 3, 357–363. [Google Scholar] [CrossRef]
- Gjerstorff, M.F.; Kock, K.; Nielsen, O.; Ditzel, H.J. MAGE-A1, GAGE and NY-ESO-1 cancer/testis antigen expression during human gonadal development. Hum. Reprod. 2007, 22, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Fiszer, D.; Kurpisz, M. Major histocompatibility complex expression on human, male germ cells: A review. Am. J. Reprod. Immunol. 1998, 40, 172–176. [Google Scholar] [CrossRef]
- Fijak, M.; Meinhardt, A. The testis in immune privilege. Immunol. Rev. 2006, 213, 66–81. [Google Scholar] [CrossRef]
- Fratta, E.; Coral, S.; Covre, A.; Parisi, G.; Colizzi, F.; Danielli, R.; Nicolay, H.J.; Sigalotti, L.; Maio, M. The biology of cancer testis antigens: Putative function, regulation and therapeutic potential. Mol. Oncol. 2011, 5, 164–182. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Caballero, O.L.; Yung, W.K.; Weinstein, J.N.; Riggins, G.J.; Strausberg, R.L.; Zhao, Q. Tumor subtype-specific cancer-testis antigens as potential biomarkers and immunotherapeutic targets for cancers. Cancer Immunol. Res. 2014, 2, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Simpson, A.J.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef]
- Van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.G.; Sakabe, N.J.; deoliveira, A.R.; Silva, M.C.; Mundstein, A.S.; Cohen, T.; Chen, Y.T.; Chua, R.; Gurung, S.; Gnjatic, S.; et al. CTdatabase: A knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 2009, 37, D816–D819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomez, P.; De Backer, O.; Bertrand, M.; De Plaen, E.; Boon, T.; Lucas, S. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res. 2001, 61, 5544–5551. [Google Scholar]
- Sang, M.; Lian, Y.; Zhou, X.; Shan, B. MAGE-A family: Attractive targets for cancer immunotherapy. Vaccine 2011, 29, 8496–8500. [Google Scholar] [CrossRef] [PubMed]
- Esfandiary, A.; Ghafouri-Fard, S. MAGE-A3: An immunogenic target used in clinical practice. Immunotherapy 2015, 7, 683–704. [Google Scholar] [CrossRef]
- Gaugler, B.; Van den Eynde, B.; van der Bruggen, P.; Romero, P.; Gaforio, J.J.; De Plaen, E.; Lethé, B.; Brasseur, F.; Boon, T. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J. Exp. Med. 1994, 179, 921–930. [Google Scholar] [CrossRef]
- Van der Bruggen, P.; Bastin, J.; Gajewski, T.; Coulie, P.G.; Boël, P.; De Smet, C.; Traversari, C.; Townsend, A.; Boon, T. A peptide encoded by human gene MAGE-3 and presented by HLA-A2 induces cytolytic T lymphocytes that recognize tumor cells expressing MAGE-3. Eur. J. Immunol. 1994, 24, 3038–3043. [Google Scholar] [CrossRef]
- Oiso, M.; Eura, M.; Katsura, F.; Takiguchi, M.; Sobao, Y.; Masuyama, K.; Nakashima, M.; Itoh, K.; Ishikawa, T. A newly identified MAGE-3-derived epitope recognized by HLA-A24-restricted cytotoxic T lymphocytes. Int. J. Cancer 1999, 81, 387–394. [Google Scholar] [CrossRef]
- Tanzarella, S.; Russo, V.; Lionello, I.; Dalerba, P.; Rigatti, D.; Bordignon, C.; Traversari, C. Identification of a promiscuous T-cell epitope encoded by multiple members of the MAGE family. Cancer Res. 1999, 59, 2668–2674. [Google Scholar]
- Schultz, E.S.; Lethé, B.; Cambiaso, C.L.; Van Snick, J.; Chaux, P.; Corthals, J.; Heirman, C.; Thielemans, K.; Boon, T.; van der Bruggen, P. A MAGE-A3 peptide presented by HLA-DP4 is recognized on tumor cells by CD4+ cytolytic T lymphocytes. Cancer Res. 2000, 60, 6272–6275. [Google Scholar]
- Kobayashi, H.; Song, Y.; Hoon, D.S.; Appella, E.; Celis, E. Tumor-reactive T helper lymphocytes recognize a promiscuous MAGE-A3 epitope presented by various major histocompatibility complex class II alleles. Cancer Res. 2001, 61, 4773–4778. [Google Scholar] [PubMed]
- Duperret, E.K.; Liu, S.; Paik, M.; Trautz, A.; Stoltz, R.; Liu, X.; Ze, K.; Perales-Puchalt, A.; Reed, C.; Yan, J.; et al. A Designer Cross-reactive DNA Immunotherapeutic Vaccine that Targets Multiple MAGE-A Family Members Simultaneously for Cancer Therapy. Clin. Cancer Res. 2018, 24, 6015–6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Breckpot, K.; Brasseur, F.; Zhang, Y.; Van Der Bruggen, P.; Thielemans, K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J. Immunol. 2004, 172, 6649–6657. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Song, N.; Liu, Y.; Liu, Y.; Li, J.; Ding, J.; Tong, Z. Efficient induction of anti-tumor immune response in esophageal squamous cell carcinoma via dendritic cells expressing MAGE-A3 and CALR antigens. Cell. Immunol. 2015, 295, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, R.; Pisu, P.; D’Apice, L.; Pizzella, L.; Romano, C.; Cortese, G.; Giorgini, A.; Santoni, A.; Velotti, F.; De Berardinis, P. The use of filamentous bacteriophage fd to deliver MAGE-A10 or MAGE-A3 HLA-A2-restricted peptides and to induce strong antitumor CTL responses. J. Immunol. 2008, 180, 3719–3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Zheng, H.; Liu, Q.; Lu, X.; Deng, X.; Jiang, L.; Hou, B.; Fu, Y.; Zhu, F.; Ding, Y.; et al. Attenuated plasmodium sporozoite expressing MAGE-A3 induces antigen-specific CD8+ T cell response against lung cancer in mice. Cancer Biol. Med. 2019, 16, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchu, R.B.; Gruzdyn, O.; Potti, R.B.; Weaver, D.W.; Gruber, S.A. MAGE-A3 with cell-penetrating domain as an efficient therapeutic cancer vaccine. JAMA Surg. 2014, 149, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef]
- Hemminger, J.A.; Ewart Toland, A.; Scharschmidt, T.J.; Mayerson, J.L.; Kraybill, W.G.; Guttridge, D.C.; Iwenofu, O.H. The cancer-testis antigen NY-ESO-1 is highly expressed in myxoid and round cell subset of liposarcomas. Mod. Pathol. 2013, 26, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Lee, S.; Lee, C.H.; Lee, M.K.; Kim, Y.D.; Shin, D.H.; Choi, K.U.; Kim, J.Y.; Park, D.Y.; Sol, M.Y. Expression of cancer-testis antigens MAGE-A3/6 and NY-ESO-1 in non-small-cell lung carcinomas and their relationship with immune cell infiltration. Lung 2009, 187, 401–411. [Google Scholar] [CrossRef]
- Aung, P.P.; Liu, Y.C.; Ballester, L.Y.; Robbins, P.F.; Rosenberg, S.A.; Lee, C.C. Expression of New York esophageal squamous cell carcinoma-1 in primary and metastatic melanoma. Hum. Pathol. 2014, 45, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fosså, A.; Berner, A.; Fosså, S.D.; Hernes, E.; Gaudernack, G.; Smeland, E.B. NY-ESO-1 protein expression and humoral immune responses in prostate cancer. Prostate 2004, 59, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Scanlan, M.J.; Sahin, U.; Türeci, O.; Gure, A.O.; Tsang, S.; Williamson, B.; Stockert, E.; Pfreundschuh, M.; Old, L.J. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc. Natl. Acad. Sci. USA 1997, 94, 1914–1918. [Google Scholar] [CrossRef] [Green Version]
- Jäger, E.; Chen, Y.T.; Drijfhout, J.W.; Karbach, J.; Ringhoffer, M.; Jäger, D.; Arand, M.; Wada, H.; Noguchi, Y.; Stockert, E.; et al. Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: Definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J. Exp. Med. 1998, 187, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Dunbar, P.R.; Gileadi, U.; Jäger, E.; Gnjatic, S.; Nagata, Y.; Stockert, E.; Panicali, D.L.; Chen, Y.T.; Knuth, A.; et al. Identification of NY-ESO-1 peptide analogues capable of improved stimulation of tumor-reactive CTL. J. Immunol. 2000, 165, 948–955. [Google Scholar] [CrossRef]
- Zarour, H.M.; Storkus, W.J.; Brusic, V.; Williams, E.; Kirkwood, J.M. NY-ESO-1 encodes DRB1*0401-restricted epitopes recognized by melanoma-reactive CD4+ T cells. Cancer Res. 2000, 60, 4946–4952. [Google Scholar]
- Chen, Y.; Huang, A.; Gao, M.; Yan, Y.; Zhang, W. Potential therapeutic value of dendritic cells loaded with NY-ESO-1 protein for the immunotherapy of advanced hepatocellular carcinoma. Int. J. Mol. Med. 2013, 32, 1366–1372. [Google Scholar] [CrossRef]
- Campos-Perez, J.; Rice, J.; Escors, D.; Collins, M.; Paterson, A.; Savelyeva, N.; Stevenson, F.K. DNA fusion vaccine designs to induce tumor-lytic CD8+ T-cell attack via the immunodominant cysteine-containing epitope of NY-ESO 1. Int. J. Cancer 2013, 133, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Delaunay, T.; Violland, M.; Boisgerault, N.; Dutoit, S.; Vignard, V.; Münz, C.; Gannage, M.; Dréno, B.; Vaivode, K.; Pjanova, D.; et al. Oncolytic viruses sensitize human tumor cells for NY-ESO-1 tumor antigen recognition by CD4+ effector T cells. Oncoimmunology 2018, 7, e1407897. [Google Scholar] [CrossRef]
- Li, M.; Shi, H.; Mu, Y.; Luo, Z.; Zhang, H.; Wan, Y.; Zhang, D.; Lu, L.; Men, K.; Tian, Y.; et al. Effective inhibition of melanoma tumorigenesis and growth via a new complex vaccine based on NY-ESO-1-alum-polysaccharide-HH2. Mol. Cancer 2014, 13, 179. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Li, M.; Yu, C.; Zhang, R.; Zhang, X.; Huang, R.; Lu, L.; Yuan, F.; Fan, Y.; Zhou, B.; et al. The novel complex combination of alum, CpG ODN and HH2 as adjuvant in cancer vaccine effectively suppresses tumor growth in vivo. Oncotarget 2017, 8, 45951–45964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, H.; Lethé, B.; Lehmann, F.; van Baren, N.; Baurain, J.F.; de Smet, C.; Chambost, H.; Vitale, M.; Moretta, A.; Boon, T.; et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 1997, 6, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Al-Khadairi, G.; Decock, J. Cancer Testis Antigens and Immunotherapy: Where Do We Stand in the Targeting of PRAME? Cancers 2019, 11, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, M.G.; Decatur, C.L.; Kurtenbach, S.; Gezgin, G.; van der Velden, P.A.; Jager, M.J.; Kozak, K.N.; Harbour, J.W. PRAME as an Independent Biomarker for Metastasis in Uveal Melanoma. Clin. Cancer Res. 2016, 22, 1234–1242. [Google Scholar] [CrossRef] [Green Version]
- Tan, P.; Zou, C.; Yong, B.; Han, J.; Zhang, L.; Su, Q.; Yin, J.; Wang, J.; Huang, G.; Peng, T.; et al. Expression and prognostic relevance of PRAME in primary osteosarcoma. Biochem. Biophys. Res. Commun. 2012, 419, 801–808. [Google Scholar] [CrossRef]
- Iura, K.; Kohashi, K.; Hotokebuchi, Y.; Ishii, T.; Maekawa, A.; Yamada, Y.; Yamamoto, H.; Iwamoto, Y.; Oda, Y. Cancer-testis antigens PRAME and NY-ESO-1 correlate with tumour grade and poor prognosis in myxoid liposarcoma. J. Pathol. Clin. Res. 2015, 1, 144–159. [Google Scholar] [CrossRef]
- Rezvani, K.; Yong, A.S.; Tawab, A.; Jafarpour, B.; Eniafe, R.; Mielke, S.; Savani, B.N.; Keyvanfar, K.; Li, Y.; Kurlander, R.; et al. Ex vivo characterization of polyclonal memory CD8+ T-cell responses to PRAME-specific peptides in patients with acute lymphoblastic leukemia and acute and chronic myeloid leukemia. Blood 2009, 113, 2245–2255. [Google Scholar] [CrossRef] [Green Version]
- Gérard, C.; Baudson, N.; Ory, T.; Segal, L.; Louahed, J. A Comprehensive Preclinical Model Evaluating the Recombinant PRAME Antigen Combined With the AS15 Immunostimulant to Fight Against PRAME-expressing Tumors. J. Immunother. 2015, 38, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Epping, M.T.; Wang, L.; Edel, M.J.; Carlée, L.; Hernandez, M.; Bernards, R. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell 2005, 122, 835–847. [Google Scholar] [CrossRef] [Green Version]
- Vansteenkiste, J.F.; Cho, B.C.; Vanakesa, T.; De Pas, T.; Zielinski, M.; Kim, M.S.; Jassem, J.; Yoshimura, M.; Dahabreh, J.; Nakayama, H.; et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016, 17, 822–835. [Google Scholar] [CrossRef]
- Dreno, B.; Thompson, J.F.; Smithers, B.M.; Santinami, M.; Jouary, T.; Gutzmer, R.; Levchenko, E.; Rutkowski, P.; Grob, J.-J.; Korovin, S.; et al. MAGE-A3 immunotherapeutic as adjuvant therapy for patients with resected, MAGE-A3-positive, stage III melanoma (DERMA): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 916–929. [Google Scholar] [CrossRef] [Green Version]
- Krishnadas, D.K.; Shusterman, S.; Bai, F.; Diller, L.; Sullivan, J.E.; Cheerva, A.C.; George, R.E.; Lucas, K.G. A phase I trial combining decitabine/dendritic cell vaccine targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol. Immunother. CII 2015, 64, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Gasser, O.; Sharples, K.J.; Barrow, C.; Williams, G.M.; Bauer, E.; Wood, C.E.; Mester, B.; Dzhelali, M.; Caygill, G.; Jones, J.; et al. A phase I vaccination study with dendritic cells loaded with NY-ESO-1 and alpha-galactosylceramide: Induction of polyfunctional T cells in high-risk melanoma patients. Cancer Immunol. Immunother. CII 2018, 67, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Westdorp, H.; Creemers, J.H.A.; van Oort, I.M.; Schreibelt, G.; Gorris, M.A.J.; Mehra, N.; Simons, M.; de Goede, A.L.; van Rossum, M.M.; Croockewit, A.J.; et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J. Immunother. Cancer 2019, 7, 302. [Google Scholar] [CrossRef]
- Pujol, J.L.; De Pas, T.; Rittmeyer, A.; Vallieres, E.; Kubisa, B.; Levchenko, E.; Wiesemann, S.; Masters, G.A.; Shen, R.; Tjulandin, S.A.; et al. Safety and Immunogenicity of the PRAME Cancer Immunotherapeutic in Patients with Resected Non-Small Cell Lung Cancer: A Phase I Dose Escalation Study. J. Thorac. Oncol. 2016, 11, 2208–2217. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.S.; Vogelzang, N.J.; Ernstoff, M.S.; Goodman, O.B.; Cranmer, L.D.; Marshall, J.L.; Miles, S.; Rosario, D.; Diamond, D.C.; Qiu, Z.; et al. A phase 1 study of a vaccine targeting preferentially expressed antigen in melanoma and prostate-specific membrane antigen in patients with advanced solid tumors. J. Immunother. 2011, 34, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Dalgleish, A.G. Rationale for combining immunotherapy with chemotherapy. Immunotherapy 2015, 7, 309–316. [Google Scholar] [CrossRef]
- Gordeeva, O. Cancer-testis antigens: Unique cancer stem cell biomarkers and targets for cancer therapy. Semin. Cancer Biol. 2018, 53, 75–89. [Google Scholar] [CrossRef]
- Fukuda, K.; Funakoshi, T.; Sakurai, T.; Nakamura, Y.; Mori, M.; Tanese, K.; Tanikawa, A.; Taguchi, J.; Fujita, T.; Okamoto, M.; et al. Peptide-pulsed dendritic cell vaccine in combination with carboplatin and paclitaxel chemotherapy for stage IV melanoma. Melanoma Res. 2017, 27, 326–334. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Li, P.; Song, M.; Qin, G.; Gao, Q.; Zhang, Z.; Yue, D.; Wang, D.; Nan, S.; et al. Dual TGF-β and PD-1 blockade synergistically enhances MAGE-A3-specific CD8(+) T cell response in esophageal squamous cell carcinoma. Int. J. Cancer 2018, 143, 2561–2574. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Gnjatic, S.; Li, H.; Powel, S.; Gallardo, H.F.; Ritter, E.; Ku, G.Y.; Jungbluth, A.A.; Segal, N.H.; Rasalan, T.S.; et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc. Natl. Acad. Sci. USA 2008, 105, 20410–20415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, L.I.; Galvao-Filho, B.; de Faria, P.C.; Junqueira, C.; Dutra, M.S.; Teixeira, S.M.; Rodrigues, M.M.; Ritter, G.; Bannard, O.; Fearon, D.T.; et al. Blockade of CTLA-4 promotes the development of effector CD8+ T lymphocytes and the therapeutic effect of vaccination with an attenuated protozoan expressing NY-ESO-1. Cancer Immunol. Immunother. CII 2015, 64, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Kudchadkar, R.R.; DeConti, R.C.; Thebeau, M.S.; Czupryn, M.P.; Tetteh, L.; Eysmans, C.; Richards, A.; Schell, M.J.; Fisher, K.J.; et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin. Cancer Res. 2015, 21, 712–720. [Google Scholar] [CrossRef] [Green Version]
- Sigalotti, L.; Coral, S.; Fratta, E.; Lamaj, E.; Danielli, R.; Di Giacomo, A.M.; Altomonte, M.; Maio, M. Epigenetic modulation of solid tumors as a novel approach for cancer immunotherapy. Semin. Oncol. 2005, 32, 473–478. [Google Scholar] [CrossRef]
- Lyko, F.; Brown, R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J. Natl. Cancer Inst. 2005, 97, 1498–1506. [Google Scholar] [CrossRef]
- Bao, L.; Dunham, K.; Lucas, K. MAGE-A1, MAGE-A3, and NY-ESO-1 can be upregulated on neuroblastoma cells to facilitate cytotoxic T lymphocyte-mediated tumor cell killing. Cancer Immunol. Immunother. CII 2011, 60, 1299–1307. [Google Scholar] [CrossRef]
- Odunsi, K.; Matsuzaki, J.; James, S.R.; Mhawech-Fauceglia, P.; Tsuji, T.; Miller, A.; Zhang, W.; Akers, S.N.; Griffiths, E.A.; Miliotto, A.; et al. Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunol. Res. 2014, 2, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Golovastova, M.O.; Bazhin, A.V.; Philippov, P.P. Cancer-retina antigens—A new group of tumor antigens. Biochem. Biochim. 2014, 79, 733–739. [Google Scholar] [CrossRef]
- Bazhin, A.V.; Schadendorf, D.; Willner, N.; De Smet, C.; Heinzelmann, A.; Tikhomirova, N.K.; Umansky, V.; Philippov, P.P.; Eichmuller, S.B. Photoreceptor proteins as cancer-retina antigens. Int. J. Cancer 2007, 120, 1268–1276. [Google Scholar] [CrossRef]
- Baldin, A.V.; Grishina, A.N.; Korolev, D.O.; Kuznetsova, E.B.; Golovastova, M.O.; Kalpinskiy, A.S.; Alekseev, B.Y.; Kaprin, A.D.; Zinchenko, D.V.; Savvateeva, L.V.; et al. Autoantibody against arrestin-1 as a potential biomarker of renal cell carcinoma. Biochimie 2019, 157, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Milam, A.H.; Saari, J.C.; Jacobson, S.G.; Lubinski, W.P.; Feun, L.G.; Alexander, K.R. Autoantibodies against retinal bipolar cells in cutaneous melanoma-associated retinopathy. Investig. Ophthalmol. Vis. Sci. 1993, 34, 91–100. [Google Scholar]
- Baldin, A.V.; Savvateeva, L.V.; Bazhin, A.V.; Zamyatnin, A.A., Jr. Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting. Cancers 2020, 12, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldin, A.V.; Zamyatnin, A.A., Jr.; Bazhin, A.V.; Xu, W.H.; Savvateeva, L.V. Advances in the Development of Anticancer HSP-based Vaccines. Curr. Med. Chem. 2019, 26, 427–445. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Intervention | Phase | Status as of October 2020 | Disease | Starting Date | Trial ID |
|---|---|---|---|---|---|
| Peptides | I | Recruiting | Pancrease carcinoma | April 2018 | NCT03558945 |
| Peptides | I | Recruiting | NSCLC | May 2020 | NCT04487093 |
| Peptides | Ⅰ | Not yet recruiting | Melanoma | September 2020 | NCT03929029 |
| Peptides | Ⅰ | Recruiting | NSCLC | May 2020 | NCT04397926 |
| Peptides | Ⅱ | Recruiting | Breast carcinoma | December 2018 | NCT03606967 |
| Peptides | Ⅰ | Recruiting | Renal carcinoma | March 2019 | NCT02950766 |
| Peptides | Ⅰ | Recruiting | Urothelial carcinoma | May 2019 | NCT03359239 |
| Peptides | Ⅰ | Not yet recruiting | Lymphocytic leukemia | September 2020 | NCT03219450 |
| DC | Ⅰ | Recruiting | Hepatocellular carcinoma | October 2018 | NCT03674073 |
| DC | I/II | Active | Colorectal carcinoma | October 2010 | NCT01885702 |
| DC | Ⅰ | Recruiting | NSCLC | August 2019 | NCT04078269 |
| DC | I/II | Unknown | Biliary tract carcinoma | September 2015 | NCT02632019 |
| DNA | Ⅰ | Recruiting | Breast carcinoma | August 2019 | NCT03199040 |
| DNA | Ⅰ | Active | Pancrease carcinoma | January 2018 | NCT03122106 |
| DNA | Ⅱ | Not yet recruiting | NSCLC | January 2021 | NCT04397003 |
| DNA | Ⅰ | Recruiting | Glioblastoma | July 2020 | NCT04015700 |
| RNA | Ⅰ/Ⅱ | Terminated | Solid tumors | May 2018 | NCT03480152 |
| RNA | N/A | Not yet recruiting | Esophageal carcinoma, NSCLC | May 2019 | NCT03908671 |
| RNA | N/A | Recruiting | Solid tumors | May 2018 | NCT03468244 |
| Cancer Antigen Type | Pros | Cons |
|---|---|---|
| Immune-privileged antigens |
|
|
| Neoantigens |
|
|
| Viral antigens |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Baldin, A.V.; Isayev, O.; Werner, J.; Zamyatnin, A.A., Jr.; Bazhin, A.V. Cancer Vaccines: Antigen Selection Strategy. Vaccines 2021, 9, 85. https://doi.org/10.3390/vaccines9020085
Zhao Y, Baldin AV, Isayev O, Werner J, Zamyatnin AA Jr., Bazhin AV. Cancer Vaccines: Antigen Selection Strategy. Vaccines. 2021; 9(2):85. https://doi.org/10.3390/vaccines9020085
Chicago/Turabian StyleZhao, Yue, Alexey V. Baldin, Orkhan Isayev, Jens Werner, Andrey A. Zamyatnin, Jr., and Alexandr V. Bazhin. 2021. "Cancer Vaccines: Antigen Selection Strategy" Vaccines 9, no. 2: 85. https://doi.org/10.3390/vaccines9020085
APA StyleZhao, Y., Baldin, A. V., Isayev, O., Werner, J., Zamyatnin, A. A., Jr., & Bazhin, A. V. (2021). Cancer Vaccines: Antigen Selection Strategy. Vaccines, 9(2), 85. https://doi.org/10.3390/vaccines9020085