
Multiple Sclerosis and SARS-CoV-2 Vaccination: Considerations for Immune-Depleting Therapies


Abstract
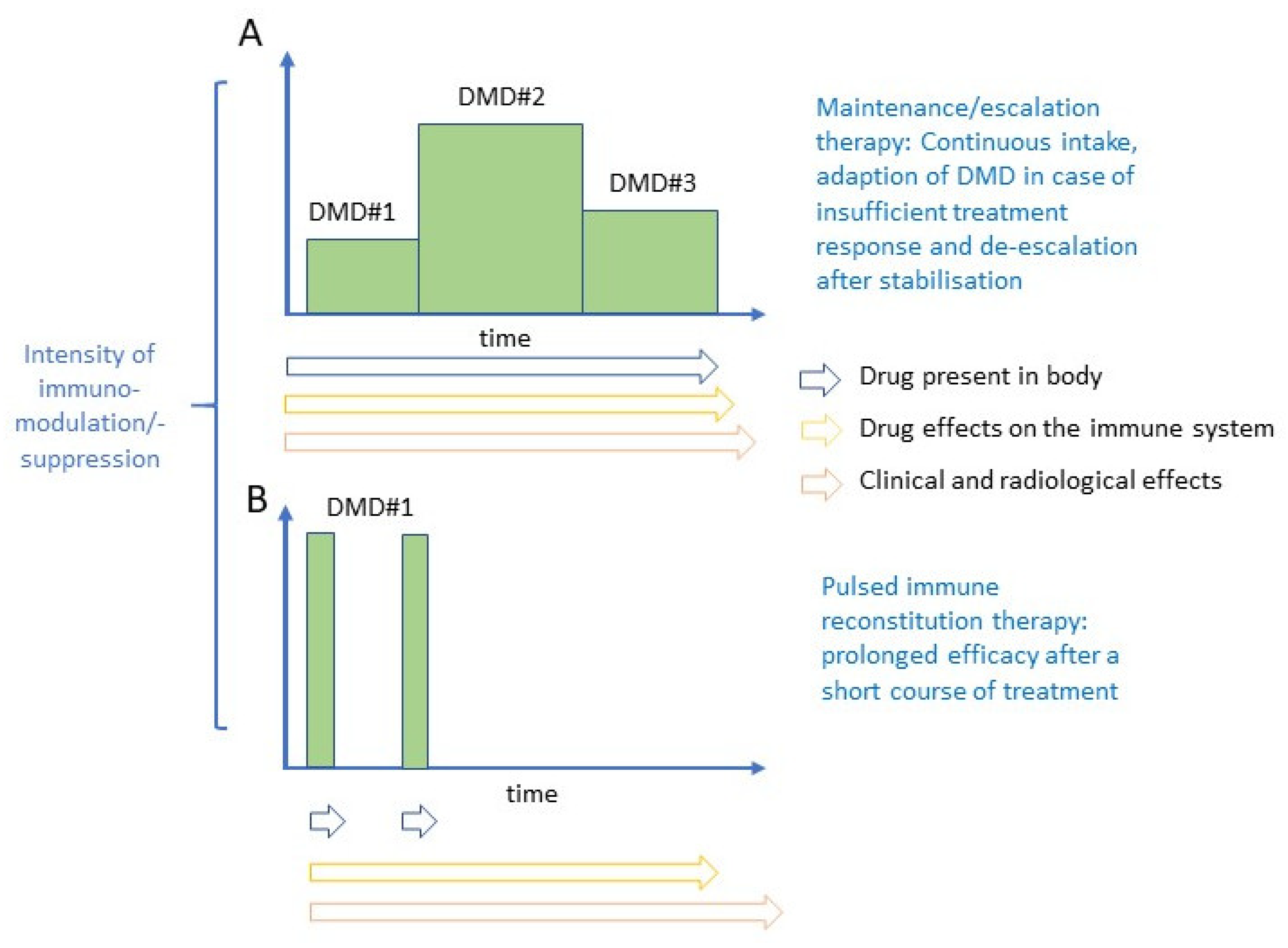
:1. Introduction
2. Immunological Consequences of Immune-Depleting Therapies Approved for the Treatment of MS and Infectious Complications
2.1. Alemtuzumab
2.2. Cladribine
2.3. Ocrelizumab
3. The Host Immune Response to SARS-CoV-2 Infection
4. Disease-Modifying Drugs and COVID-19: Restriction for Immune-Depleting Therapies?
- Temporarily delay (between 6 and 12 months depending on the DMD re-dosing of alemtuzumab, ocrelizumab and cladribine. This decision should be made according to individual factors such as disease severity and activity.
- For anti-CD20 DMTs, it is recommended to delay the next dose even beyond 6 months if CD19+ and CD20+ lymphocyte counts are severely decreased at the time the next dose is due.
5. Considerations for Vaccination in the Context of Immune-Depleting Therapies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- von Oertzen, T.J.; Macerollo, A.; Leone, M.A.; Beghi, E.; Crean, M.; Oztuk, S.; Bassetti, C.; Twardzik, A.; Bereczki, D.; Di Liberto, G.; et al. EAN consensus statement for management of patients with neurological diseases during the COVID-19 pandemic. Eur. J. Neurol. 2021, 28, 7–14. [Google Scholar] [CrossRef]
- Hauer, L.; Perneczky, J.; Sellner, J. A global view of comorbidity in multiple sclerosis: A systematic review with a focus on regional differences, methodology, and clinical implications. J. Neurol. 2020, 1–12. [Google Scholar] [CrossRef]
- Chaudhry, F.; Bulka, H.; Rathnam, A.S.; Said, O.M.; Lin, J.; Lorigan, H.; Bernitsas, E.; Rube, J.; Korzeniewski, S.J.; Memon, A.B.; et al. COVID-19 in multiple sclerosis patients and risk factors for severe infection. J. Neurol. Sci. 2020, 418, 117147. [Google Scholar] [CrossRef]
- Karamyan, A.; Dunser, M.W.; Wiebe, D.J.; Pilz, G.; Wipfler, P.; Chroust, V.; Novak, H.F.; Hauer, L.; Trinka, E.; Sellner, J. Critical Illness in Patients with Multiple Sclerosis: A Matched Case-Control Study. PLoS ONE 2016, 11, e0155795. [Google Scholar] [CrossRef]
- Bsteh, G.; Bitschnau, C.; Hegen, H.; Auer, M.; Di Pauli, F.; Rommer, P.; Deisenhammer, F.; Berger, T. Multiple sclerosis and COVID-19: How many are at risk? Eur. J. Neurol. 2020. [Google Scholar] [CrossRef]
- Louapre, C.; Collongues, N.; Stankoff, B.; Giannesini, C.; Papeix, C.; Bensa, C.; Deschamps, R.; Creange, A.; Wahab, A.; Pelletier, J.; et al. Clinical Characteristics and Outcomes in Patients With Coronavirus Disease 2019 and Multiple Sclerosis. JAMA Neurol. 2020, 77, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, F.; Laurent, S.; Fink, G.R.; Barnett, M.H.; Hartung, H.P.; Warnke, C. Effects of disease-modifying therapy on peripheral leukocytes in patients with multiple sclerosis. J. Neurol. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruck, T.; Bittner, S.; Wiendl, H.; Meuth, S.G. Alemtuzumab in Multiple Sclerosis: Mechanism of Action and Beyond. Int. J. Mol. Sci. 2015, 16, 16414–16439. [Google Scholar] [CrossRef] [PubMed]
- Hale, G. The CD52 antigen and development of the CAMPATH antibodies. Cytotherapy 2001, 3, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Ginaldi, L.; De Martinis, M.; Matutes, E.; Farahat, N.; Morilla, R.; Dyer, M.J.; Catovsky, D. Levels of expression of CD52 in normal and leukemic B and T cells: Correlation with in vivo therapeutic responses to Campath-1H. Leuk. Res. 1998, 22, 185–191. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Sellebjerg, F. Pulsed immune reconstitution therapy in multiple sclerosis. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419836913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.P.; Sancho, J.; Campos-Rivera, J.; Boutin, P.M.; Severy, P.B.; Weeden, T.; Shankara, S.; Roberts, B.L.; Kaplan, J.M. Human peripheral blood mononuclear cells exhibit heterogeneous CD52 expression levels and show differential sensitivity to alemtuzumab mediated cytolysis. PLoS ONE 2012, 7, e39416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coles, A.J.; Cox, A.; Le Page, E.; Jones, J.; Trip, S.A.; Deans, J.; Seaman, S.; Miller, D.H.; Hale, G.; Waldmann, H.; et al. The window of therapeutic opportunity in multiple sclerosis: Evidence from monoclonal antibody therapy. J. Neurol. 2006, 253, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.A.; Jones, J.L.; Cox, A.L.; Compston, D.A.; Coles, A.J. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J. Clin. Immunol. 2010, 30, 99–105. [Google Scholar] [CrossRef]
- Baker, D.; Herrod, S.S.; Alvarez-Gonzalez, C.; Giovannoni, G.; Schmierer, K. Interpreting Lymphocyte Reconstitution Data From the Pivotal Phase 3 Trials of Alemtuzumab. JAMA Neurol. 2017, 74, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.A.; Ruppert, A.S.; Zhao, W.; Lin, T.S.; Rai, K.; Peterson, B.; Larson, R.A.; Marcucci, G.; Heerema, N.A.; Byrd, J.C. Patients with chronic lymphocytic leukemia with high-risk genomic features have inferior outcome on successive Cancer and Leukemia Group B trials with alemtuzumab consolidation: Subgroup analysis from CALGB 19901 and CALGB 10101. Leuk. Lymphoma 2013, 54, 2654–2659. [Google Scholar] [CrossRef] [Green Version]
- Moser, T.; Akgun, K.; Proschmann, U.; Sellner, J.; Ziemssen, T. The role of TH17 cells in multiple sclerosis: Therapeutic implications. Autoimmun. Rev. 2020, 19, 102647. [Google Scholar] [CrossRef]
- Gross, C.C.; Ahmetspahic, D.; Ruck, T.; Schulte-Mecklenbeck, A.; Schwarte, K.; Jorgens, S.; Scheu, S.; Windhagen, S.; Graefe, B.; Melzer, N.; et al. Alemtuzumab treatment alters circulating innate immune cells in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e289. [Google Scholar] [CrossRef] [Green Version]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Wiendl, H.; Marcenaro, E.; Kerlero de Rosbo, N.; Uccelli, A.; Laroni, A. Regulatory Functions of Natural Killer Cells in Multiple Sclerosis. Front. Immunol. 2016, 7, 606. [Google Scholar] [CrossRef] [Green Version]
- Investigators, C.T.; Coles, A.J.; Compston, D.A.; Selmaj, K.W.; Lake, S.L.; Moran, S.; Margolin, D.H.; Norris, K.; Tandon, P.K. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N. Engl. J. Med. 2008, 359, 1786–1801. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.A.; Coles, A.J.; Arnold, D.L.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet 2012, 380, 1819–1828. [Google Scholar] [CrossRef]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Epstein, D.J.; Dunn, J.; Deresinski, S. Infectious Complications of Multiple Sclerosis Therapies: Implications for Screening, Prophylaxis, and Management. Open Forum Infect. Dis. 2018, 5, ofy174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappulo, E.; Buonomo, A.R.; Sacca, F.; Russo, C.V.; Scotto, R.; Scalia, G.; Nozzolillo, A.; Lanzillo, R.; Tosone, G.; Gentile, I. Incidence and Predictive Risk Factors of Infective Events in Patients with Multiple Sclerosis Treated With Agents Targeting CD20 and CD52 Surface Antigens. Open Forum Infect. Dis. 2019, 6, ofz445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzitelli, M.; Barone, S.; Greco, G.; Serapide, F.; Valentino, P.; Giancotti, A.; Costa, C.; Pisani, V.; Quirino, A.; Liberto, M.C.; et al. Listeria infection after treatment with alemtuzumab: A case report and literature review. Would antibiotic prophylaxis be considered? Infez. Med. 2020, 28, 258–262. [Google Scholar]
- Beutler, E. Cladribine (2-chlorodeoxyadenosine). Lancet 1992, 340, 952–956. [Google Scholar] [CrossRef]
- Leist, T.P.; Weissert, R. Cladribine: Mode of action and implications for treatment of multiple sclerosis. Clin. Neuropharmacol. 2011, 34, 28–35. [Google Scholar] [CrossRef]
- Ceronie, B.; Jacobs, B.M.; Baker, D.; Dubuisson, N.; Mao, Z.; Ammoscato, F.; Lock, H.; Longhurst, H.J.; Giovannoni, G.; Schmierer, K. Cladribine treatment of multiple sclerosis is associated with depletion of memory B cells. J. Neurol. 2018, 265, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Warnke, C.; Wiendl, H.; Hartung, H.P.; Stuve, O.; Kieseier, B.C. Identification of targets and new developments in the treatment of multiple sclerosis--focus on cladribine. Drug Des. Dev. Ther. 2010, 4, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Stuve, O.; Soelberg Soerensen, P.; Leist, T.; Giovannoni, G.; Hyvert, Y.; Damian, D.; Dangond, F.; Boschert, U. Effects of cladribine tablets on lymphocyte subsets in patients with multiple sclerosis: An extended analysis of surface markers. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419854986. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.; Herrod, S.S.; Alvarez-Gonzalez, C.; Zalewski, L.; Albor, C.; Schmierer, K. Both cladribine and alemtuzumab may effect MS via B-cell depletion. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, T.; Schwenker, K.; Seiberl, M.; Feige, J.; Akgun, K.; Haschke-Becher, E.; Ziemssen, T.; Sellner, J. Long-term peripheral immune cell profiling reveals further targets of oral cladribine in MS. Ann. Clin. Transl. Neurol. 2020, 7, 2199–2212. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Cook, S.; Giovannoni, G.; Rieckmann, P.; Sorensen, P.S.; Vermersch, P.; Galazka, A.; Nolting, A.; Hicking, C.; Dangond, F. Effect of cladribine tablets on lymphocyte reduction and repopulation dynamics in patients with relapsing multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 29, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Sellner, J.; Rommer, P.S. Immunological consequences of “immune reconstitution therapy” in multiple sclerosis: A systematic review. Autoimmun. Rev. 2020, 19, 102492. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, G.; Soelberg Sorensen, P.; Cook, S.; Rammohan, K.; Rieckmann, P.; Comi, G.; Dangond, F.; Adeniji, A.K.; Vermersch, P. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: Results from the randomized extension trial of the CLARITY study. Mult. Scler. 2018, 24, 1594–1604. [Google Scholar] [CrossRef] [Green Version]
- Schuh, E.; Berer, K.; Mulazzani, M.; Feil, K.; Meinl, I.; Lahm, H.; Krane, M.; Lange, R.; Pfannes, K.; Subklewe, M.; et al. Features of Human CD3+CD20+ T Cells. J. Immunol. 2016, 197, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Bar-Or, A.; Calabresi, P.A.; Arnold, D.; Markowitz, C.; Shafer, S.; Kasper, L.H.; Waubant, E.; Gazda, S.; Fox, R.J.; Panzara, M.; et al. Rituximab in relapsing-remitting multiple sclerosis: A 72-week, open-label, phase I trial. Ann. Neurol. 2008, 63, 395–400. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannon, G.; Hartung, S.P.; Hem-mer, B.; et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.; Amor, S.; Kang, A.S.; Schmierer, K.; Giovannoni, G. The underpinning biology relating to multiple sclerosis disease modifying treatments during the COVID-19 pandemic. Mult. Scler. Relat. Disord. 2020, 43, 102174. [Google Scholar] [CrossRef] [PubMed]
- Gingele, S.; Jacobus, T.L.; Konen, F.F.; Hummert, M.W.; Suhs, K.W.; Schwenkenbecher, P.; Ahlbrecht, J.; Mohn, N.; Muschen, L.H.; Bonig, L.; et al. Ocrelizumab Depletes CD20(+) T Cells in Multiple Sclerosis Patients. Cells 2018, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Huang, B.; Wu, M.; Zhong, A.; Li, L.; Cai, Y.; Wang, Z.; Wu, L.; Zhu, M.; Li, J.; et al. Dynamic changes in anti-SARS-CoV-2 antibodies during SARS-CoV-2 infection and recovery from COVID-19. Nat. Commun. 2020, 11, 6044. [Google Scholar] [CrossRef] [PubMed]
- Findling, O.; Sellner, J. Second-generation immunotherapeutics in multiple sclerosis: Can we discard their precursors? Drug Discov. Today 2020. [Google Scholar] [CrossRef]
- Zheng, C.; Kar, I.; Chen, C.K.; Sau, C.; Woodson, S.; Serra, A.; Abboud, H. Multiple Sclerosis Disease-Modifying Therapy and the COVID-19 Pandemic: Implications on the Risk of Infection and Future Vaccination. CNS Drugs 2020, 34, 879–896. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.S.; Rosenbult, C.L.; Tremlett, H. Safety profile of ocrelizumab for the treatment of multiple sclerosis: A systematic review. Expert Opin. Drug. Saf. 2020, 19, 1069–1094. [Google Scholar] [CrossRef]
- Munro, N.; Scordo, K.A.; Richmond, M.M. COVID-19: An Immunopathologic Assault. AACN Adv. Crit. Care 2020, 31, 268–280. [Google Scholar] [CrossRef]
- Sellner, J. Of mice and men: COVID-19 challenges translational neuroscience. Eur. J. Neurol. 2020, 27, 1762–1763. [Google Scholar] [CrossRef]
- Heming, M.; Li, X.; Rauber, S.; Mausberg, A.K.; Borsch, A.L.; Hartlehnert, M.; Singhal, A.; Lu, I.N.; Fleischer, M.; Szepanowski, F.; et al. Neurological Manifestations of COVID-19 Feature T Cell Exhaustion and Dedifferentiated Monocytes in Cerebrospinal Fluid. Immunity 2020. [Google Scholar] [CrossRef]
- Seyran, M.; Takayama, K.; Uversky, V.N.; Lundstrom, K.; Palu, G.; Sherchan, S.P.; Attrish, D.; Rezaei, N.; Aljabali, A.A.A.; Ghosh, S.; et al. The structural basis of accelerated host cell entry by SARS-CoV-2dagger. FEBS J. 2020. [Google Scholar] [CrossRef]
- Saksena, S.; Chattopadhyay, P. Illuminating the immunopathology of SARS-CoV-2. Cytometry B Clin. Cytom. 2021. [Google Scholar] [CrossRef] [PubMed]
- Laing, A.G.; Lorenc, A.; Del Molino Del Barrio, I.; Das, A.; Fish, M.; Monin, L.; Munoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Schmitz, K.S.; Raadsen, M.P.; Grifoni, A.; Okba, N.M.A.; Endeman, H.; van den Akker, J.P.C.; Molenkamp, R.; Koopmans, M.P.G.; van Gorp, E.C.M.; et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef] [PubMed]
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Thevarajan, I.; Nguyen, T.H.O.; Koutsakos, M.; Druce, J.; Caly, L.; van de Sandt, C.E.; Jia, X.; Nicholson, S.; Catton, M.; Cowie, B.; et al. Breadth of concomitant immune responses prior to patient recovery: A case report of non-severe COVID-19. Nat. Med. 2020, 26, 453–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodruff, M.; Ramonell, R.; Cashman, K.; Nguyen, D.; Ley, A.; Kyu, S.; Saini, A.; Haddad, N.; Chen, W.; Howell, J.C.; et al. Critically ill SARS-CoV-2 patients display lupus-like hallmarks of extrafollicular B cell activation. medRxiv 2020. [Google Scholar] [CrossRef]
- Rydyznski Moderbacher, C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.e19. [Google Scholar] [CrossRef]
- Zrzavy, T.; Wimmer, I.; Rommer, P.S.; Berger, T. Immunology of COVID-19 and disease-modifying therapies: The good, the bad and the unknown. Eur. J. Neurol. 2020. [Google Scholar] [CrossRef]
- Thakolwiboon, S.; Zhao-Fleming, H.; Pan, J.; Scott, J.K.; Shoji, E.; Sohn, G.; Avila, M. Disease-Modifying Therapies During the COVID-19 Outbreak: A Narrative Review of International and National Recommendations. Int. J. MS Care 2020, 22, 151–157. [Google Scholar] [CrossRef]
- Giovannoni, G.; Hawkes, C.; Lechner-Scott, J.; Levy, M.; Waubant, E.; Gold, J. The COVID-19 pandemic and the use of MS disease-modifying therapies. Mult. Scler. Relat. Disord. 2020, 39, 102073. [Google Scholar] [CrossRef] [PubMed]
- Zabalza, A.; Cárdenas-Robledo, S.; Tagliani, P.; Arrambide, S.; Otero-Romero, S. COVID-19 in MS patients: Susceptibility, severity risk factors and serological response. Eur. J. Neurol. 2020, in press. [Google Scholar]
- Sellner, J.; Jenkins, T.; von Oertzen, T.; Bassetti, C.L.; Beghi, E.; Bereczki, D.; Bodini, B.; Cavallieri, F.; Di Liberto, G.; Helbok, R.; et al. Primary prevention of COVID-19: Advocacy for vaccination from a neurological perspective. Eur. J. Neurol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Bar-Or, A.; Calkwood, J.C.; Chognot, C.; Evershed, J.; Fox, E.J.; Herman, A.; Manfrini, M.; McNamara, J.; Robertson, D.S.; Stokmaier, D.; et al. Effect of ocrelizumab on vaccine responses in patients with multiple sclerosis: The VELOCE study. Neurology 2020, 95, e1999–e2008. [Google Scholar] [CrossRef]
- Ciotti, J.R.; Valtcheva, M.V.; Cross, A.H. Effects of MS disease-modifying therapies on responses to vaccinations: A review. Mult. Scler. Relat. Disord. 2020, 45, 102439. [Google Scholar] [CrossRef]
- Rodda, L.B.; Netland, J.; Shehata, L.; Pruner, K.B.; Morawski, P.A.; Thouvenel, C.D.; Takehara, K.K.; Eggenberger, J.; Hemann, E.A.; Waterman, H.R.; et al. Functional SARS-CoV-2-Specific Immune Memory Persists after Mild COVID-19. Cell 2020. [Google Scholar] [CrossRef]
- Edridge, A.W.D.; Kaczorowska, J.; Hoste, A.C.R.; Bakker, M.; Klein, M.; Loens, K.; Jebbink, M.F.; Matser, A.; Kinsella, C.M.; Rueda, P.; et al. Seasonal coronavirus protective immunity is short-lasting. Nat. Med. 2020, 26, 1691–1693. [Google Scholar] [CrossRef]
- Tillett, R.L.; Sevinsky, J.R.; Hartley, P.D.; Kerwin, H.; Crawford, N.; Gorzalski, A.; Laverdure, C.; Verma, S.C.; Rossetto, C.C.; Jackson, D.; et al. Genomic evidence for reinfection with SARS-CoV-2: A case study. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Zrzavy, T.; Kollaritsch, H.; Rommer, P.S.; Boxberger, N.; Loebermann, M.; Wimmer, I.; Winkelmann, A.; Zettl, U.K. Vaccination in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2019, 10, 1883. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Mecham, R.P.; Mann, D.L. RNA Vaccines for COVID-19: Five Things Every Cardiologist Should Know. JACC Basic Transl. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A. Immunologic basis of vaccine vectors. Immunity 2010, 33, 504–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Alemtuzumab (Lemtrada®) | Cladribine (Mavenclad®) | Ocrelizumab (Ocrevus®) | |
|---|---|---|---|
| Indication | RRMS | RRMS | RRMS/PPMS |
| Route of administration | i.v. | oral | i.v. |
| Dosing/Interval | A total of 8 treatment days separated into two annual pulsed treatment periods: 5 × 12 mg (1st year) and 3 × 12 mg (2nd year) | Maximum 20 treatment days separated into two annual pulsed treatment periods: total of 3.5 mg/kg body weight (given as 1.75 mg/kg per year for each treatment period) | Induction with 300 mg at day 0 and 14 each, followed by maintenance dose of 600 mg every six months |
| Degree of lymphophenia # | Grade 3 and 4: 99.9% | Grade 3: 25.6%, Grade 4: 0.7% | Grade 1 and 2: majority, Grade 3: 1% |
| Lymphocyte recovery # | Total lymphocytes: 6 At 6 and 12 months in 40 and 80% of patients after each treatment cycle, respectively. | Recovery of lymphocytes at the end of each treatment year: 86% | After 2.5 years: 90% of patients had recovered CD19+ B cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sellner, J.; Rommer, P.S. Multiple Sclerosis and SARS-CoV-2 Vaccination: Considerations for Immune-Depleting Therapies. Vaccines 2021, 9, 99. https://doi.org/10.3390/vaccines9020099
Sellner J, Rommer PS. Multiple Sclerosis and SARS-CoV-2 Vaccination: Considerations for Immune-Depleting Therapies. Vaccines. 2021; 9(2):99. https://doi.org/10.3390/vaccines9020099
Chicago/Turabian StyleSellner, Johann, and Paulus S. Rommer. 2021. "Multiple Sclerosis and SARS-CoV-2 Vaccination: Considerations for Immune-Depleting Therapies" Vaccines 9, no. 2: 99. https://doi.org/10.3390/vaccines9020099
APA StyleSellner, J., & Rommer, P. S. (2021). Multiple Sclerosis and SARS-CoV-2 Vaccination: Considerations for Immune-Depleting Therapies. Vaccines, 9(2), 99. https://doi.org/10.3390/vaccines9020099