
Transcriptome of Tumor-Infiltrating T Cells in Colorectal Cancer Patients Uncovered a Unique Gene Signature in CD4+ T Cells Associated with Poor Disease-Specific Survival

 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Sample Collection and Storage
2.3. Dissociation of Tissues
2.4. Cell Sorting
2.5. RNA Isolation and Amplification
2.6. Library Preparation
2.7. RNA-Sequencing Data Processing and Analyses
2.8. Data Alignment with the Cancer Genome Atlas (TCGA) Colorectal Cancer
2.9. TCGA Immune Estimations
3. Results
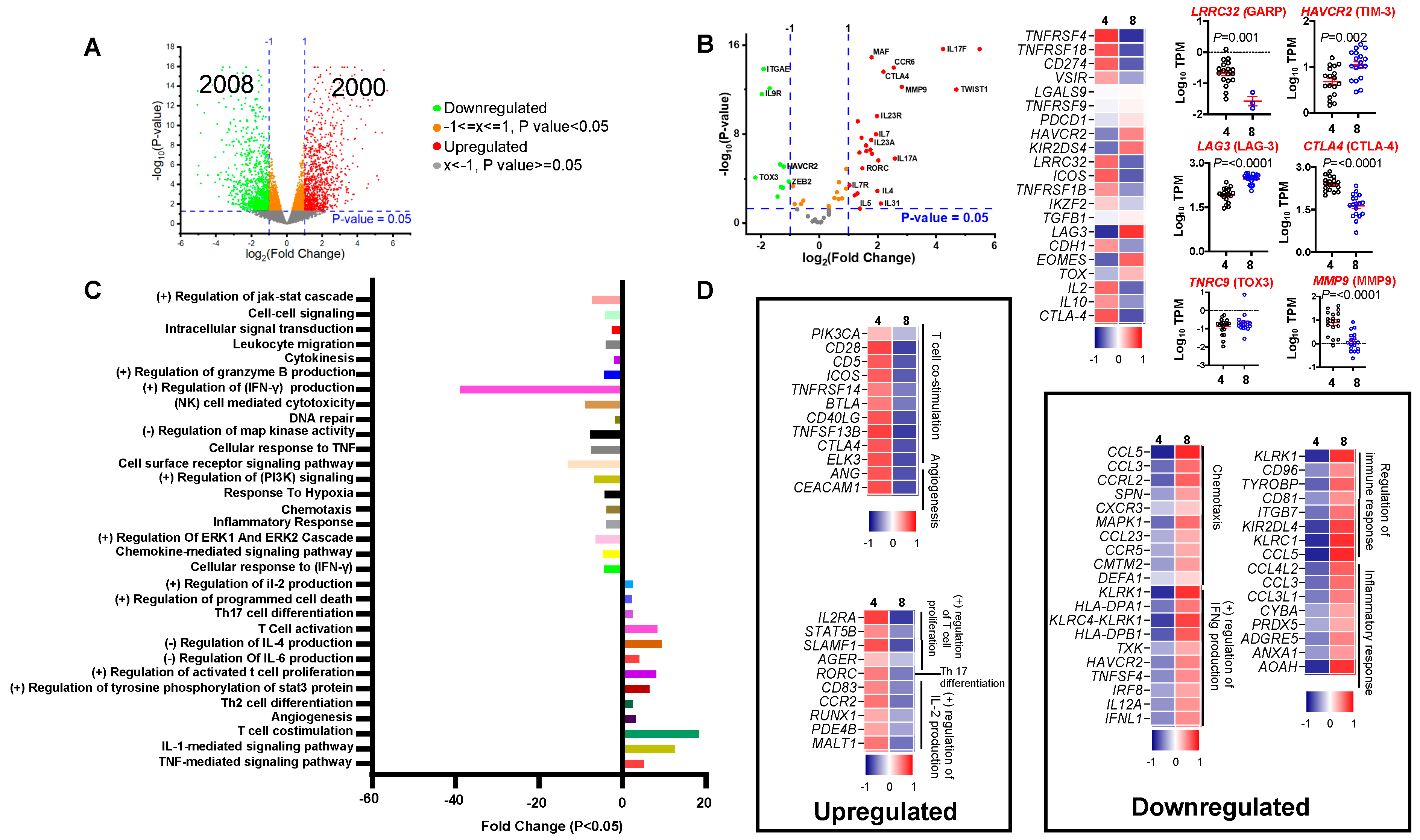
3.1. Comparing Transcriptomes of CD4+ and CD8+ Tumor-Infiltrating Lymphocytes in Colorectal Cancer Patients
3.2. Differential Expression of T Cell Related Genes
3.3. Functional Pathways Enriched in CD4+ TILs Compared to CD8+ TILs
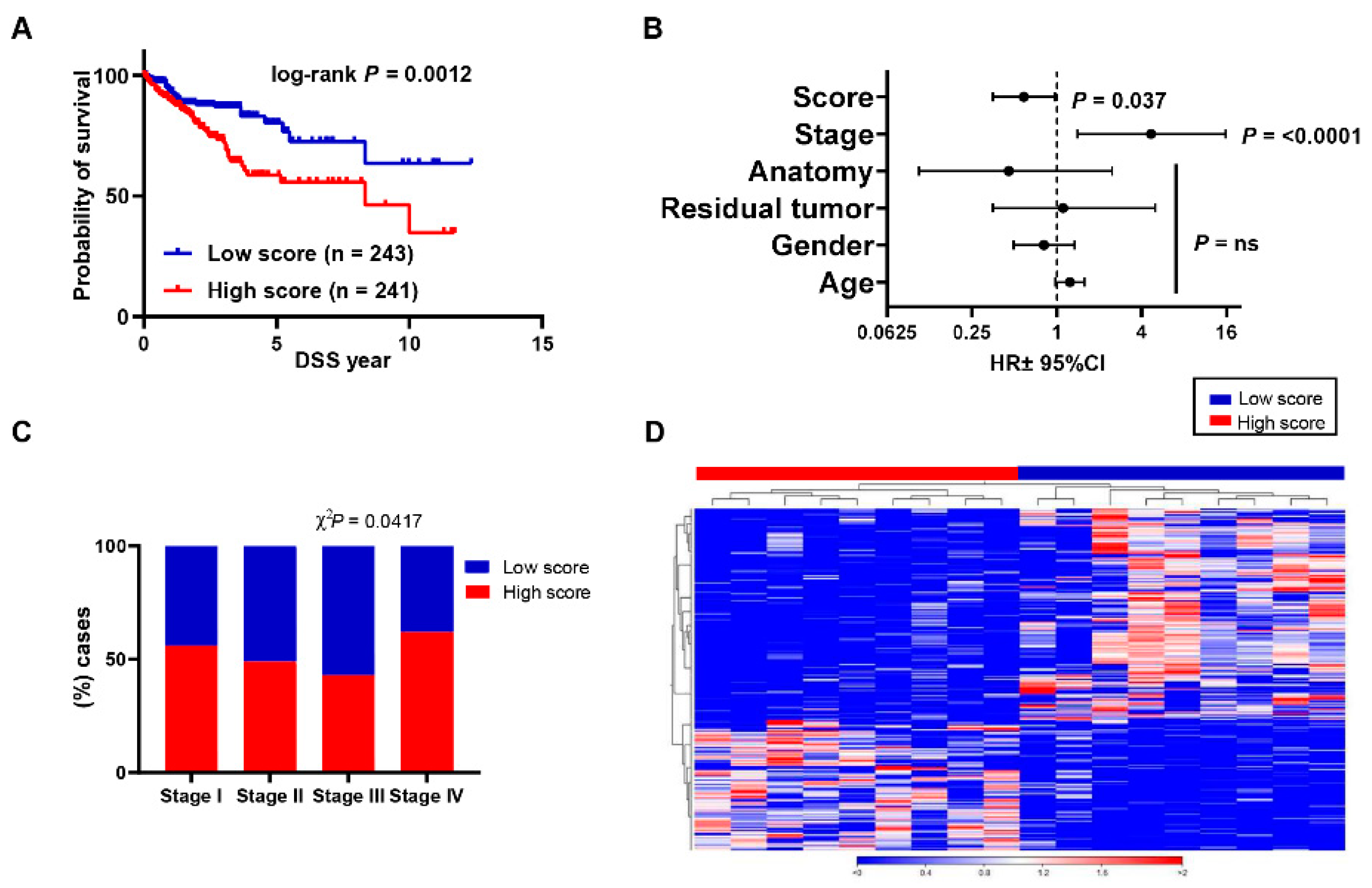
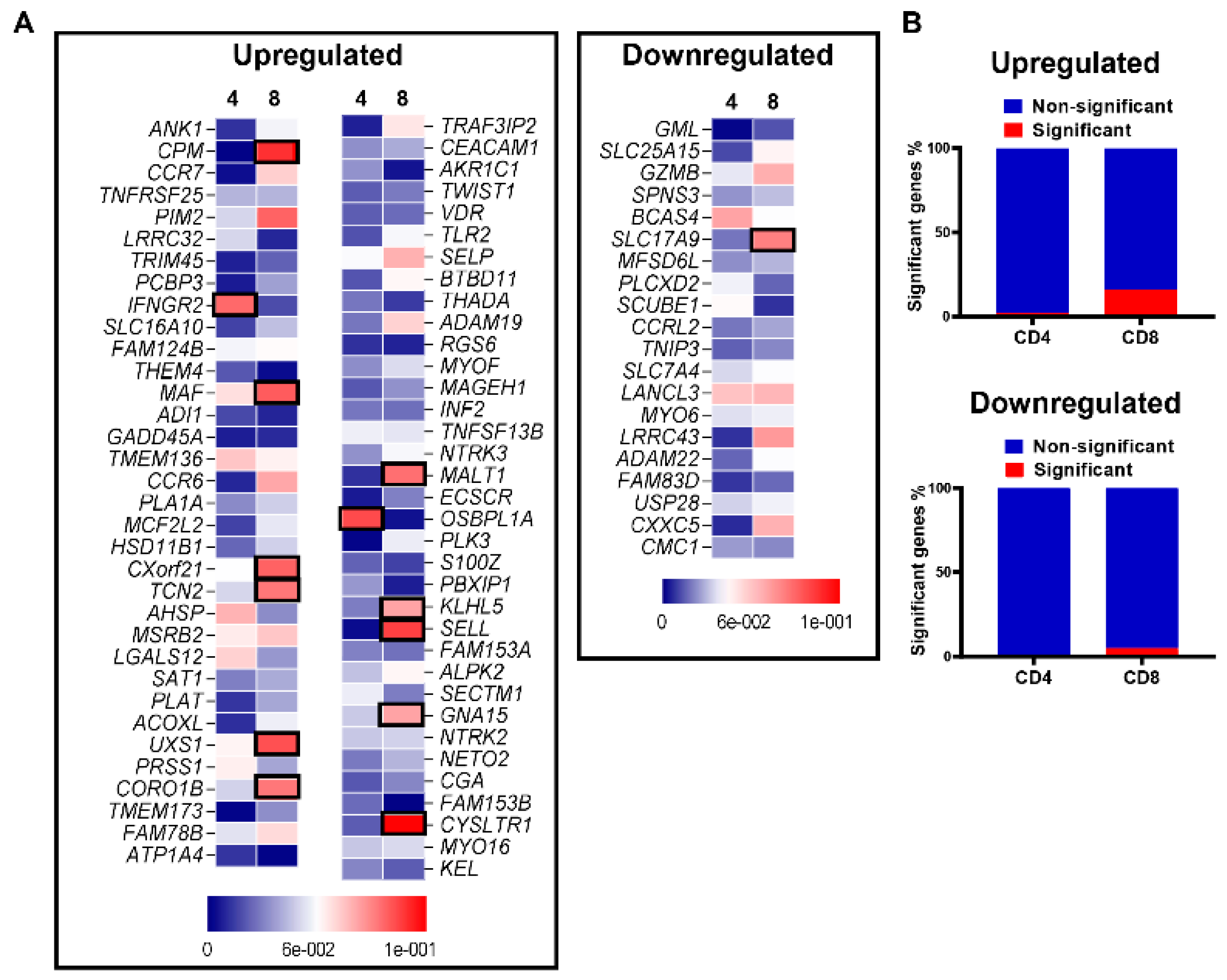
3.4. Aligning Gene Profile of CD4+ TILs from CRC Patients with TCGA Revealed a Distinct Gene Signature Associated with Poor Disease-Specific Survival
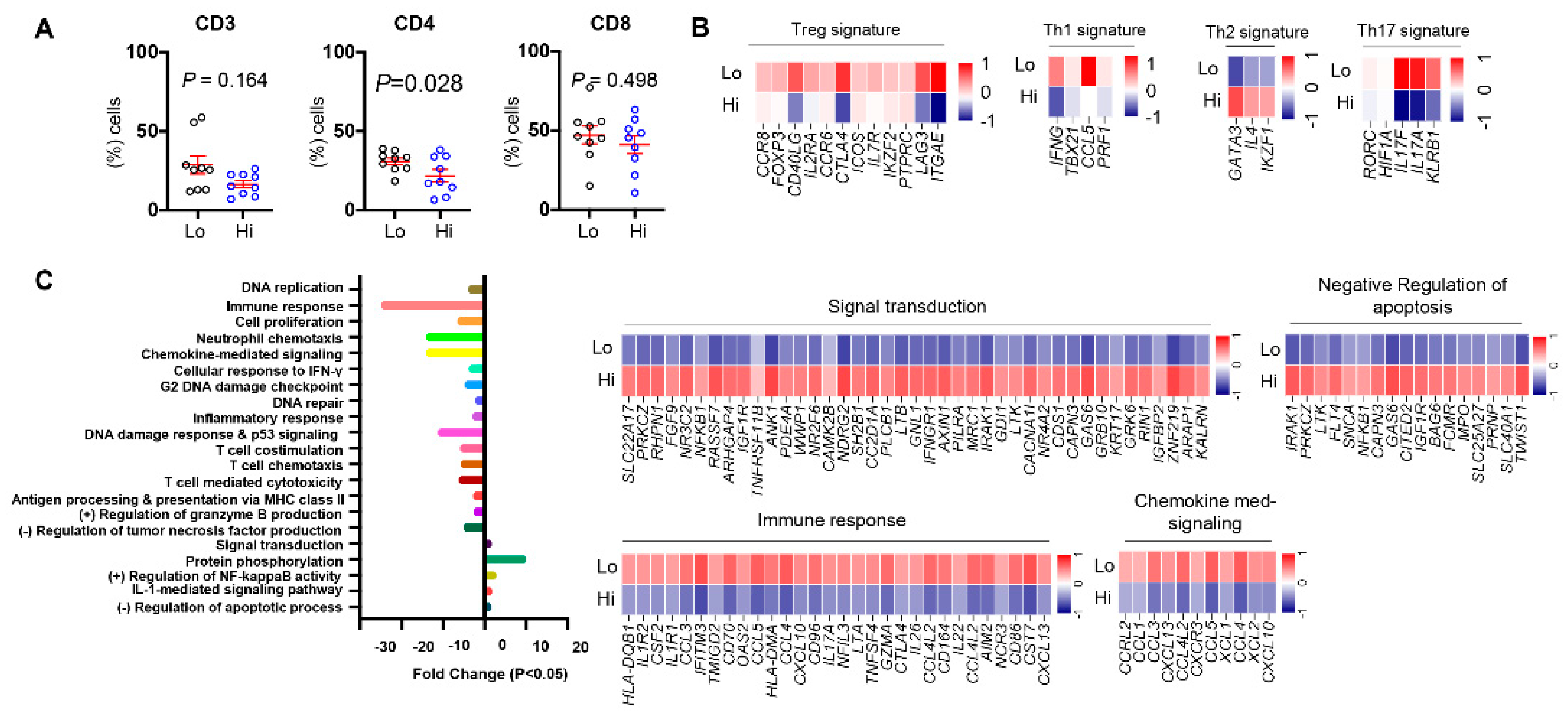
3.5. Differences in Tumor-Infiltrating T Cells between CRC Patients with High and Low ppScore
3.6. Differentially Expressed Genes and Associated Pathways between CRC Patients with High and Low ppScore
4. Discussion
4.1. Limitations and Future Directions
4.2. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chew, V.; Toh, H.C.; Abastado, J.P. Immune microenvironment in tumor progression: Characteristics and challenges for therapy. J. Oncol. 2012, 2012, 608406. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed Khaja, A.S.; Toor, S.M.; El Salhat, H.; Ali, B.R.; Elkord, E. Intratumoral FoxP3(+)Helios(+) Regulatory T Cells Upregulating Immunosuppressive Molecules Are Expanded in Human Colorectal Cancer. Front. Immunol. 2017, 8, 619. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Syed Khaja, A.S.; El Salhat, H.; Bekdache, O.; Kanbar, J.; Jaloudi, M.; Elkord, E. Increased Levels of Circulating and Tumor-Infiltrating Granulocytic Myeloid Cells in Colorectal Cancer Patients. Front. Immunol. 2016, 7, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Li, Z.; Wang, S. Tumor-infiltrating FoxP3(+) Tregs predict favorable outcome in colorectal cancer patients: A meta-analysis. Oncotarget 2017, 8, 75361–75371. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Fan, W.; Zhang, Z.; Wang, J.; Wang, P.; Li, Y.; Yu, M. The Clinicopathological and Prognostic Implications of FoxP3(+) Regulatory T Cells in Patients with Colorectal Cancer: A Meta-Analysis. Front. Physiol. 2017, 8, 950. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.C.; Mahalingam, J.; Chiang, J.M.; Su, P.J.; Chu, Y.Y.; Lai, H.Y.; Fang, J.H.; Huang, C.T.; Chiu, C.T.; Lin, C.Y. Activated but not resting regulatory T cells accumulated in tumor microenvironment and correlated with tumor progression in patients with colorectal cancer. Int. J. Cancer 2013, 132, 1341–1350. [Google Scholar] [CrossRef]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef]
- Reissfelder, C.; Stamova, S.; Gossmann, C.; Braun, M.; Bonertz, A.; Walliczek, U.; Grimm, M.; Rahbari, N.N.; Koch, M.; Saadati, M.; et al. Tumor-specific cytotoxic T lymphocyte activity determines colorectal cancer patient prognosis. J. Clin. Investig. 2015, 125, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Hazama, S.; Suzuki, N.; Yoshida, S.; Tomochika, S.; Nakagami, Y.; Matsui, H.; Shindo, Y.; Kanekiyo, S.; Tokumitsu, Y.; et al. Intratumoural-infiltrating CD4 + and FOXP3 + T cells as strong positive predictive markers for the prognosis of resectable colorectal cancer. Br. J. Cancer 2019, 121, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Pages, F.; Marincola, F.M.; Angell, H.K.; Thurin, M.; Lugli, A.; Zlobec, I.; Berger, A.; Bifulco, C.; Botti, G.; et al. Cancer classification using the Immunoscore: A worldwide task force. J. Transl. Med. 2012, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Sasidharan Nair, V.; Saleh, R.; Taha, R.Z.; Toor, S.M.; Murshed, K.; Ahmed, A.A.; Kurer, M.A.; Abu Nada, M.; Al Ejeh, F.; Elkord, E. Differential gene expression of tumor-infiltrating CD4(+) T cells in advanced versus early stage colorectal cancer and identification of a gene signature of poor prognosis. Oncoimmunology 2020, 9, 1825178. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Sasidharan Nair, V.; Toor, S.M.; Taha, R.Z.; Murshed, K.; Al-Dhaheri, M.; Khawar, M.; Petkar, M.A.; Abu Nada, M.; Al-Ejeh, F.; et al. Differential gene expression of tumor-infiltrating CD8(+) T cells in advanced versus early-stage colorectal cancer and identification of a gene signature of poor prognosis. J. Immunother Cancer 2020, 8. [Google Scholar] [CrossRef]
- Toor, S.M.; Murshed, K.; Al-Dhaheri, M.; Khawar, M.; Abu Nada, M.; Elkord, E. Immune Checkpoints in Circulating and Tumor-Infiltrating CD4(+) T Cell Subsets in Colorectal Cancer Patients. Front. Immunol. 2019, 10, 2936. [Google Scholar] [CrossRef] [PubMed]
- Toor, S.M.; Sasidharan Nair, V.; Pfister, G.; Elkord, E. Effect of pembrolizumab on CD4(+) CD25(+), CD4(+) LAP(+) and CD4(+) TIM-3(+) T cell subsets. Clin. Exp. Immunol. 2019, 196, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Vishnubalaji, R.; Sasidharan Nair, V.; Ouararhni, K.; Elkord, E.; Alajez, N.M. Integrated Transcriptome and Pathway Analyses Revealed Multiple Activated Pathways in Breast Cancer. Front. Oncol. 2019, 9, 910. [Google Scholar] [CrossRef] [Green Version]
- Malone, B.M.; Tan, F.; Bridges, S.M.; Peng, Z. Comparison of four ChIP-Seq analytical algorithms using rice endosperm H3K27 trimethylation profiling data. PLoS ONE 2011, 6, e25260. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Saleh, R.; Toor, S.M.; Taha, R.Z.; Ahmed, A.A.; Kurer, M.A.; Murshed, K.; Alajez, N.M.; Abu Nada, M.; Elkord, E. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin. Epigenetics 2020, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Huh, J.W.; Lee, J.H.; Kim, H.R. Prognostic significance of tumor-infiltrating lymphocytes for patients with colorectal cancer. Arch. Surg. 2012, 147, 366–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ropponen, K.M.; Eskelinen, M.J.; Lipponen, P.K.; Alhava, E.; Kosma, V.M. Prognostic value of tumour-infiltrating lymphocytes (TILs) in colorectal cancer. J. Pathol. 1997, 182, 318–324. [Google Scholar] [CrossRef]
- Scurr, M.; Gallimore, A.; Godkin, A. T cell subsets and colorectal cancer: Discerning the good from the bad. Cell Immunol. 2012, 279, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Fountzilas, E.; Kotoula, V.; Tikas, I.; Manousou, K.; Papadopoulou, K.; Poulios, C.; Karavasilis, V.; Efstratiou, I.; Pectasides, D.; Papaparaskeva, K.; et al. Prognostic significance of tumor genotypes and CD8+ infiltrates in stage I-III colorectal cancer. Oncotarget 2018, 9, 35623–35638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.S.; McCall, J.L.; Girardin, A.; Munro, F.M.; Black, M.A.; Kemp, R.A. Functional impairment of infiltrating T cells in human colorectal cancer. Oncoimmunology 2016, 5, e1234573. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, R.; Elkord, E. FoxP3+ T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett. 2020, 490, 174–185. [Google Scholar] [CrossRef] [PubMed]
- di Bari, M.G.; Lutsiak, M.E.; Takai, S.; Mostbock, S.; Farsaci, B.; Semnani, R.T.; Wakefield, L.M.; Schlom, J.; Sabzevari, H. TGF-beta modulates the functionality of tumor-infiltrating CD8+ T cells through effects on TCR signaling and Spred1 expression. Cancer Immunol. Immunother. 2009, 58, 1809–1818. [Google Scholar] [CrossRef] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin. Cancer Biol. 2020, 65, 13–27. [Google Scholar] [CrossRef]
- Gerstel, D.; Wegwitz, F.; Jannasch, K.; Ludewig, P.; Scheike, K.; Alves, F.; Beauchemin, N.; Deppert, W.; Wagener, C.; Horst, A.K. CEACAM1 creates a pro-angiogenic tumor microenvironment that supports tumor vessel maturation. Oncogene 2011, 30, 4275–4288. [Google Scholar] [CrossRef]
- Lavergne, E.; Combadiere, C.; Iga, M.; Boissonnas, A.; Bonduelle, O.; Maho, M.; Debre, P.; Combadiere, B. Intratumoral CC chemokine ligand 5 overexpression delays tumor growth and increases tumor cell infiltration. J. Immunol. 2004, 173, 3755–3762. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.M. The Jak-STAT pathway stimulated by interferon gamma. Sci. STKE 2004, 2004, tr8. [Google Scholar] [CrossRef]
- Sercan, O.; Stoycheva, D.; Hammerling, G.J.; Arnold, B.; Schuler, T. IFN-gamma receptor signaling regulates memory CD8+ T cell differentiation. J. Immunol. 2010, 184, 2855–2862. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, H.; Ido, M.; Nakanishi, K.; Nakano, T.; Kamiya, H.; Matsumine, A.; Uchida, A.; Mizutani, H.; de Beaucoudrey, L.; Vogt, G.; et al. Multiple cutaneous squamous cell carcinomas in a patient with interferon gamma receptor 2 (IFN gamma R2) deficiency. J. Med. Genet. 2010, 47, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bao, C.; Wang, L.; Han, R.; Beier, U.H.; Akimova, T.; Cole, P.A.; Dent, S.Y.R.; Hancock, W.W. Complementary Roles of GCN5 and PCAF in Foxp3+ T-Regulatory Cells. Cancers 2019, 11, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villagra, A.; Sotomayor, E.M.; Seto, E. Histone deacetylases and the immunological network: Implications in cancer and inflammation. Oncogene 2010, 29, 157–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Jiao, J.; Wang, L.; O’Brien, S.; Newick, K.; Wang, L.C.; Falkensammer, E.; Liu, Y.; Han, R.; Kapoor, V.; et al. HDAC5 controls the functions of Foxp3(+) T-regulatory and CD8(+) T cells. Int. J. Cancer 2016, 138, 2477–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ptaschinski, C.; Mukherjee, S.; Moore, M.L.; Albert, M.; Helin, K.; Kunkel, S.L.; Lukacs, N.W. RSV-Induced H3K4 Demethylase KDM5B Leads to Regulation of Dendritic Cell-Derived Innate Cytokines and Exacerbates Pathogenesis In Vivo. PLoS Pathog. 2015, 11, e1004978. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zou, J.; Wang, M.; Ding, X.; Chepelev, I.; Zhou, X.; Zhao, W.; Wei, G.; Cui, J.; Zhao, K.; et al. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nat. Commun. 2014, 5, 5780. [Google Scholar] [CrossRef] [Green Version]
- De Araujo-Souza, P.S.; Hanschke, S.C.; Viola, J.P. Epigenetic control of interferon-gamma expression in CD8 T cells. J. Immunol. Res. 2015, 2015, 849573. [Google Scholar] [CrossRef] [Green Version]
- Deschoolmeester, V.; Baay, M.; Lardon, F.; Pauwels, P.; Peeters, M. Immune Cells in Colorectal Cancer: Prognostic Relevance and Role of MSI. Cancer Microenviron. 2011, 4, 377–392. [Google Scholar] [CrossRef] [Green Version]
- Ling, A.; Lundberg, I.V.; Eklof, V.; Wikberg, M.L.; Oberg, A.; Edin, S.; Palmqvist, R. The infiltration, and prognostic importance, of Th1 lymphocytes vary in molecular subgroups of colorectal cancer. J. Pathol. Clin. Res. 2016, 2, 21–31. [Google Scholar] [CrossRef]
- Amicarella, F.; Muraro, M.G.; Hirt, C.; Cremonesi, E.; Padovan, E.; Mele, V.; Governa, V.; Han, J.; Huber, X.; Droeser, R.A.; et al. Dual role of tumour-infiltrating T helper 17 cells in human colorectal cancer. Gut 2017, 66, 692–704. [Google Scholar] [CrossRef] [Green Version]
- De Simone, V.; Pallone, F.; Monteleone, G.; Stolfi, C. Role of TH17 cytokines in the control of colorectal cancer. Oncoimmunology 2013, 2, e26617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells. Immunol. Cell Biol. 2018, 96, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Parekh, S.; Ziegenhain, C.; Vieth, B.; Enard, W.; Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci. Rep. 2016, 6, 25533. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRC Patients | |
|---|---|
| Number | 18 |
| Age (median) | 62 (23–78) |
| Gender (Male:Female) | 10:8 |
| TNM stage | |
| I | 5 |
| II | 5 |
| III | 3 |
| IV | 5 |
| Tumor histological grade | |
| G2 (Moderately differentiated) | 17 |
| G3 (Poorly differentiated) | 1 |
| Lymphovascular invasion | 2 |
| MSI-H/BRAF mutation | 1 * |
| Tumor location | |
| Cecum | 3 |
| Ascending colon | 2 |
| Decending colon | 3 |
| Sigmoid | 5 |
| Rectosigmoid | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toor, S.M.; Sasidharan Nair, V.; Saleh, R.; Taha, R.Z.; Murshed, K.; Al-Dhaheri, M.; Khawar, M.; Ahmed, A.A.; Kurer, M.A.; Abu Nada, M.; et al. Transcriptome of Tumor-Infiltrating T Cells in Colorectal Cancer Patients Uncovered a Unique Gene Signature in CD4+ T Cells Associated with Poor Disease-Specific Survival. Vaccines 2021, 9, 334. https://doi.org/10.3390/vaccines9040334
Toor SM, Sasidharan Nair V, Saleh R, Taha RZ, Murshed K, Al-Dhaheri M, Khawar M, Ahmed AA, Kurer MA, Abu Nada M, et al. Transcriptome of Tumor-Infiltrating T Cells in Colorectal Cancer Patients Uncovered a Unique Gene Signature in CD4+ T Cells Associated with Poor Disease-Specific Survival. Vaccines. 2021; 9(4):334. https://doi.org/10.3390/vaccines9040334
Chicago/Turabian StyleToor, Salman M., Varun Sasidharan Nair, Reem Saleh, Rowaida Z. Taha, Khaled Murshed, Mahmood Al-Dhaheri, Mahwish Khawar, Ayman A. Ahmed, Mohamed A. Kurer, Mohamed Abu Nada, and et al. 2021. "Transcriptome of Tumor-Infiltrating T Cells in Colorectal Cancer Patients Uncovered a Unique Gene Signature in CD4+ T Cells Associated with Poor Disease-Specific Survival" Vaccines 9, no. 4: 334. https://doi.org/10.3390/vaccines9040334
APA StyleToor, S. M., Sasidharan Nair, V., Saleh, R., Taha, R. Z., Murshed, K., Al-Dhaheri, M., Khawar, M., Ahmed, A. A., Kurer, M. A., Abu Nada, M., & Elkord, E. (2021). Transcriptome of Tumor-Infiltrating T Cells in Colorectal Cancer Patients Uncovered a Unique Gene Signature in CD4+ T Cells Associated with Poor Disease-Specific Survival. Vaccines, 9(4), 334. https://doi.org/10.3390/vaccines9040334