1. Introduction
Healthy cells constitutively release nanovesicles which are identified as microvesicles (50–1000 nm diameter), and exosomes (50–200 nm), together referred to as EVs [
1]. Microvesicles (also identified as ectosomes) are shed by the plasma membrane, whereas exosomes are released after inward invagination of endosome membranes, formation of intraluminal vesicles, and release. EVs are an important means of intercellular communication because of their ability to transport DNAs, RNAs, proteins, and lipids from the producer cell to the recipient one [
2].
Nef
mut is a Human Immunodeficiency Virus-1 protein mutated in
G3
C,
V153
L, and
E177
G amino acids. It is a protein mutant defective for all anti-cellular Nef functions, including CD4 and MHC Class I down-regulation, increased HIV-1 infectivity, and p21 activated kinase (PAK)-2 activation [
3]. We previously described the high efficiency of uploading HIV-1 Nef
mut into EVs released by multiple cell types [
4], which remains unchanged when a foreign protein is fused to its C-terminus [
5,
6,
7]. When DNA vectors expressing Nef
mut-based fusion proteins are intramuscularly (i.m.) injected in mice, significant quantities of the fusion proteins are packed into EVs. These in vivo engineered EVs can freely circulate into the body, thereby being internalized by antigen-presenting cells (APCs), which cross-present EV contents to activate antigen-specific CD8
+ T cells. These events result in the induction of potent antigen-specific CD8
+ T cytotoxic lymphocyte (CTL) responses [
6,
7,
8,
9]. Both effectiveness and flexibility of this vaccine platform have been demonstrated with an array of viral products of various origins and sizes, including but not limited to Human Papillomavirus (HPV)16-E6 and -E7 [
8,
10]; Severe acute Respiratory Syndrome Coronavirus (SARS-CoV)-2 S1, S2, M, and N [
11]; HIV-1 Gag p17, Gag p24 and Tat [
12]; Ebola Virus VP24, VP40 and NP [
7]; Hepatitis B Virus Core [
12]; Hepatitis C Virus NS3, West Nile Virus NS3, and Crimean-Congo Hemorrhagic Fever NP [
5,
7].
To translate into the clinic the Nefmut-based vaccine platform, we attempted to maximize its intrinsic safety profile by identifying the minimum part of Nefmut retaining both the EV-anchoring protein and immunogenicity properties. Investigations were carried out by fusing Nefmut-derivatives with antigens from viruses recognizing different pathogenic effects, i.e., HPV16, whose infection can induce tumors, and the acute respiratory disease-inducing SARS-CoV-2.
Pre-clinical studies that we had already carried out provided evidence that the Nef
mut-based CTL vaccine platform can act as an effective therapeutic intervention against HPV16-related and other malignancies [
6,
9]. The benefits expected from a therapeutic cancer vaccine are the possibility to induce a de novo antitumor immunity, as well as widen both potency and breadth of pre-existing immunity [
13,
14]. On the other hand, both experimental and clinical evidence supported the idea that a SARS-CoV-2-specific CD8
+ T cell immunity can be instrumental to mitigate the symptoms related to the viral spread in both upper and lower airways [
15,
16]. Data from experimental infections in rhesus macaques indicated that the virus-specific CD8
+ T cell immunity is critical to protect the animals from virus re-challenge after the rapid decay of neutralizing antibodies [
17]. Consistently, the induction of antiviral CD8
+ T cells was associated with a strongly reduced severity of the disease in humans [
18]. Furthermore, the demonstrated ability of SARS-CoV-2 to spread through cell-to-cell contact [
19] implies the need to induce a robust cellular immunity to contain and clear the virus. In this context, the development of novel preventive strategies focused on the induction of anti-SARS-CoV-2 CD8
+ T cell immunity should be pursued. We recently demonstrated that the Nef
mut-based technology is functional to induce CD8
+ T cell immunity against different SARS-CoV-2 antigens [
11].
To increase the safety profile of the Nefmut-based technology, we found that both EV-incorporation efficiency and immunogenicity of foreign antigens are conserved in the presence of a C-terminal 29 amino acid deletion of Nefmut.
2. Materials and Methods
2.1. DNA Vector Synthesis
The pTargeT (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA)-Nef
mut and -Nef
mutfusion vectors were already described [
6,
12]. The pTargeT-Nef
mut/E6 vector comprised an E6 open reading frame (ORF), which was codon-optimized through an ad hoc algorithm provided by the Codon Optimization On-Line (COOL) service (
https://cool.syncti.org, accessed on 15 January 2018), which introduced 134 base substitutions. In addition, we included the
G130
V amino acid substitution, which generated a loss-of-function of E6 by hindering its interaction with the p53 cell protein partner [
20]. The E6 ORF was inserted in
Apa I/
Sal I sites of the pTargeT-Nef
mutfusion vector. In the pTargeT-Nef
mut/E7 vector, the E7 ORF was codon-optimized through the introduction of 64 base substitutions as described by Cid-Arregui and coll. [
21]. Furthermore, the protein was detoxified through the insertion of three amino acid substitutions, namely three glycines at positions 21, 24, and 26, within the retinoblastoma protein (pRB) binding site. In this way, the E7-specific immortalizing activity was abrogated [
21].
The C-terminal truncated Nef
mut (referred to as Nef
mutPL) was inserted in the pTargeT vector by digesting the pTargeT-Nef
mut vector with
Sma I, which cuts just downstream to the most C-terminal typical Nef
mut mutation (i.e.,
E177
G), as well as at the 3′ end of vector polylinker. The subsequent re-ligation generated a C-terminal 29 amino acid deletion with the generation of a stop codon just downstream the
Sma I restriction site (
Figure 1).
To obtain the pTargeT-Nefmut-PL-based DNA vectors, an intermediate construct referred to as NefmutPLfusion was constructed. In detail, the NefmutPL ORF from pTargeT-NefmutPL was PCR amplified using a forward primer tagged with a Nhe I restriction site, and a reverse primer including an Apa I site together with an overlapping sequence for a GPGP linker. The PCR product was then inserted into the corresponding restriction sites of the pTargeT vector. In this way, the insertion in the unique Apa I restriction site of downstream ORFs resulted in an in-frame sequence. To obtain the pTarget-NefmutPL/E6 and pTarget-NefmutPL/E7 vectors, synthesis and cloning strategy were identical to those described for the DNA vectors expressing full-length Nefmut.
Both Nef
mut/SARS-CoV-2-based fusion proteins were cloned into the pVAX1 plasmid (Thermo Fisher) as already described [
11]. Both pVAX1-Nef
mut and pVAX1-Nef
mutPL vectors were obtained by inserting the respective ORFs in
Nhe I and
Eco RI sites of the vector polylinker. To obtain pVAX1 vectors expressing Nef
mutPL fused with either SARS-CoV-2 S1 or S2 ORFs, an intermediate construct referred to as pVAX1-Nef
mutPLfusion was obtained. To this aim, the Nef
mutPL ORF from the pTargeT-Nef
mutPL vector was PCR amplified using a forward primer tagged with a
Nhe I restriction site, and a reverse primer, including an
Apa I and the overlapping sequence for a GPGP linker. The amplification product was then inserted in the corresponding sites of the pVAX1 vector. In this way, the downstream insertion of S1 or S2 ORFs in
Apa I and
Pme I restriction sites resulted in in-frame sequences, and, upon translation, in Nef
mutPL-based fusion proteins. Eurofins Genomics and Explora Biotech carried out gene synthesis.
2.2. Cell Cultures and Transfection
Human embryonic kidney (HEK)293T cells (ATCC, CRL-11268) were grown in DMEM (Gibco, Thermo Fisher) plus 10% heat-inactivated fetal calf serum (FCS, Gibco, Thermo Fisher). Transfection assays were performed using Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific).
2.3. EV Isolation
Cells transfected with vectors expressing the Nef
mut-based fusion proteins were washed 24 h later and reseeded in a medium supplemented with EV-deprived FCS. The supernatants were harvested from 48 to 72 h after transfection. EVs were recovered through differential centrifugations [
22] by centrifuging supernatants at 500×
g for 10 min, and then at 10,000×
g for 30 min. Supernatants were harvested, filtered with 0.22 µm pore size filters, and ultracentrifuged at 70,000×
g for l h. Pelleted vesicles were resuspended in 1× PBS, and ultracentrifuged again at 70,000×
g for 1 h. Afterward, pellets containing EVs were resuspended in 1:100 of the initial volume.
2.4. Western Blot Analysis
Western blot analyses of both cell lysates and EVs were carried out after resolving samples in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). In brief, the analysis on cell lysates was performed by washing cells twice with 1× PBS (pH 7.4) and lysing them with 1 × SDS-PAGE sample buffer. Samples were resolved by SDS-PAGE and transferred by electroblotting on a 0.45 μM pore size nitrocellulose membrane (GE Healthcare Europe GmbH, Milan, Italy) overnight using a Bio-Rad (Hercules, CA, USA) Trans-Blot. For western blot analysis of EVs, they were lysed and analyzed as described for cell lysates. For immunoassays, membranes were blocked with 5% non-fat dry milk in PBS containing 0.1% Triton X-100 for 1 h at room temperature, then incubated overnight at 4 °C with specific antibodies diluted in PBS containing 0.1% Triton X-100. Filters were revealed using 1:1000-diluted sheep anti-Nef antiserum ARP 444 (MHRC, London, UK), 1:500-diluted anti-β-actin AC-74 mAb from Sigma (St. Louis, MI, USA), and 1:500 diluted anti-Alix H-270 polyclonal Abs from Santa Cruz (Dallas, TX, USA).
2.5. Mice Immunization
Both 6-weeks old C57 Bl/6 and for Nefmut/S2 immunizations (in view of the lack of already characterized H2b immunodominant S2 epitopes), Balb/c female mice were obtained from Charles River (Calco, Italy). They were hosted at the Central Animal Facility of the Istituto Superiore di Sanità (ISS), as approved by the Italian Ministry of Health, authorization n. 565/2020 released on 3 June 2020. Preparations of the DNA vector were diluted in 30 µL of sterile 0.9% saline solution. Both the quality and quantity of the DNA preparations were checked by 260/280 nm absorbance and electrophoresis assays. Each inoculum volume was injected into both quadriceps. Mice were anesthetized with isoflurane as prescribed in the Ministry authorization. Immediately after inoculation, mice underwent electroporation at the site of injection through the Agilpulse BTX (Holliston, MA, USA) device using a 4-needle array 4 mm gap, 5 mm needle length, with the following parameters: 1 pulse of 450 V for 50 µs; 0.2 ms interval; 1 pulse of 450 V for 50 µs; 50 ms interval; 8 pulses of 110 V for 10 ms with 20 ms intervals. The same procedure was repeated for both quadriceps of each mouse. Immunizations were repeated after 14 days. Fourteen days after the second immunization, mice were sacrificed by either cervical dislocation or CO2 inhalation.
2.6. Cell Isolation from Immunized Mice
Spleens were explanted by qualified personnel of the ISS Central Animal Facility and placed into 2 mL Eppendorf tubes filled with 1 mL of RPMI 1640 (Gibco), 50 µM 2-mercaptoethanol (Sigma). Splenocytes were extracted as already detailed [
11]. The procedures for bronchoalveolar lavages were carried out as previously described [
23,
24]. Total lavage volume was approximately 2.5 mL/mouse. Cells were recovered by centrifugation, resuspended in cell culture medium, and counted.
2.7. IFN-γ EliSpot Analysis
A total of 2.5 × 10
5 live cells was seeded in triplicate EliSpot microwells (Millipore, Burlington, MA, USA) pre-coated with the AN18 mAb against mouse IFN-γ (Mabtech, Nacka Strand, Sweden) in RPMI 1640 (Gibco), 10% FCS, 50 µM 2-mercaptoethanol (Sigma) for 16 h in the presence of 5 µg/mL of the following CD8-specific peptides: E6 (H2-K
b): 18–26: KLPQLCTEL [
25]; 50–57: YDFAFRDL [
25]; 109–117: RCINCQKPL [
26]; 127–135: DKKQRFMNI [
25]. HPV-16 E7 (H2-K
b): 49–57 RAHYNIVTF [
25]; 67–75 LCVQSTHVD [
26]. SARS-CoV-2 S1 (H2-K
b): 525–531 VNFNFNGL [
27]; SARS-CoV-2 S2 (H2-K
d): 1079–1089 PAICHDGKAH [
28]. As a negative control, 5 µg/mL of either H2-K
b or H2-K
d-binding peptides were used. More than 70% pure preparations of the peptides were obtained from both UFPeptides, Ferrara, Italy, and JPT, Berlin, Germany. For cell activation control, cultures were treated with 10 ng/mL phorbol 12-myristate 13-acetate (PMA, Sigma) plus 500 ng/mL of ionomycin (Sigma). After 16 h, the cultures were removed, and wells incubated with 100 µL of 1 µg/mL of the R4-6A2 biotinylated anti-IFN-γ (Mabtech) for 2 h at r.t. Wells were then washed and treated for 1 h at r.t. with 1:1000 diluted streptavidine-ALP preparations from Mabtech. After washing, spots were developed by adding 100 µL/well of SigmaFast BCIP/NBT. The spot-forming cells were finally analyzed and counted using an AELVIS EliSpot reader (Hannover, Germany).
2.8. Intracellular Cytokine Staining (ICS)
Splenocytes were seeded at 2 × 106/mL in RPMI medium, 10% FCS, 50 µM 2-mercaptoethanol (Sigma), and 1 µg/mL brefeldin A (BD Biosciences, Franklin Lakes, NJ, USA). Control conditions were carried out either by adding 10 ng/mL PMA (Sigma) and 1 µg/mL ionomycin (Sigma) or with unrelated peptides. After 16 h, cultures were stained with 1 µL of LIVE/DEAD Fixable Aqua Dead Cell reagent (Invitrogen, ThermoFisher) in 1 mL of PBS for 30 min at 4 °C and washed twice with 500 µL of PBS. To minimize nonspecific staining, cells were pre-incubated with 0.5 µg of Fc blocking mAbs (i.e., anti-CD16/CD32 antibodies, Invitrogen/eBioscience, Thermo Fisher, Waltham, MA) in 100 µL of PBS with 2% FCS for 15 min at 4 °C. For the detection of cell surface markers, cells were stained with 2 µL of the following Abs: FITC conjugated anti-mouse CD3, APC-Cy7 conjugated anti-mouse CD8a, and PerCP conjugated anti-mouse CD4 (BD Biosciences) and incubated for 1 h at 4 °C. After washing, cells were permeabilized and fixed through the Cytofix/Cytoperm kit (BD Biosciences) as per the manufacturer’s recommendations and stained for 1 h at 4 °C with 2 µL of the following Abs: PE-Cy7 conjugated anti-mouse IFN-γ (BD Biosciences), PE-conjugated anti-mouse IL-2 (Invitrogen eBioscience), and BV421 anti-mouse TNF-α (BD Biosciences) in a total of 100 µL of 1× Perm/Wash Buffer (BD Biosciences). After two washes, cells were fixed in 200 µL of 1× PBS/formaldehyde (2% v/v). Samples were then assessed by a Gallios flow cytometer and analyzed using Kaluza software (Beckman Coulter, Brea, CA, USA).
Gating strategy was as follows: live cells as detected by Aqua LIVE/DEAD Dye vs. FSC-A, singlet cells from FSC-A vs. FSC-H (singlet 1) and SSC-A vs SSC-W (singlet 2), CD3 positive cells from CD3 (FITC) vs. SSC-A, CD8 or CD4 positive cells from CD8 (APC-Cy7) vs. CD4 (PerCP). The CD8+ cell population was gated against APC-Cy7, PE, and BV421 to observe changes in IFN-γ, IL-2, and TNF-α production, respectively. Boolean gates were created to determine any cytokine co-expression pattern.
2.9. Statistical Analysis
When appropriate, data are presented as mean + standard deviation (SD). In some instances, the Mann-Whitney U test was used. p < 0.05 was considered significant.
3. Results
3.1. Design and EV-Uploading of a C-Terminal Truncated Nefmut
Removing unnecessary sequences from the Nefmut EV-anchoring protein can increase the safety profile of the Nefmut CTL vaccine platform. We tried to identify the shortest Nefmut amino acid sequence retaining the ability to incorporate into EVs at high levels.
HIV-1 Nef is a 206 to 210 amino acid long protein recognizing an extended structured N-moiety, and an unstructured C-terminal flexible region [
29]. To leave untouched the Nef
mut secondary structure, which is assumed to be critical for the EV-uploading activity, isoforms deleted of part of the unstructured C-terminus were considered. Another relevant constraint was represented by the most C-terminal unique amino acid substitution of Nef
mut (i.e.,
E177
G) whose presence is mandatory to preserve the high efficiency of EV uploading [
4]. On this basis, a Nef
mut protein truncated at the amino acid 178 (hereinafter referred to as Nef
mutPL), hence deprived of 29 C-terminal amino acids, was tested (
Figure 1 and
Supplementary Figure S1).
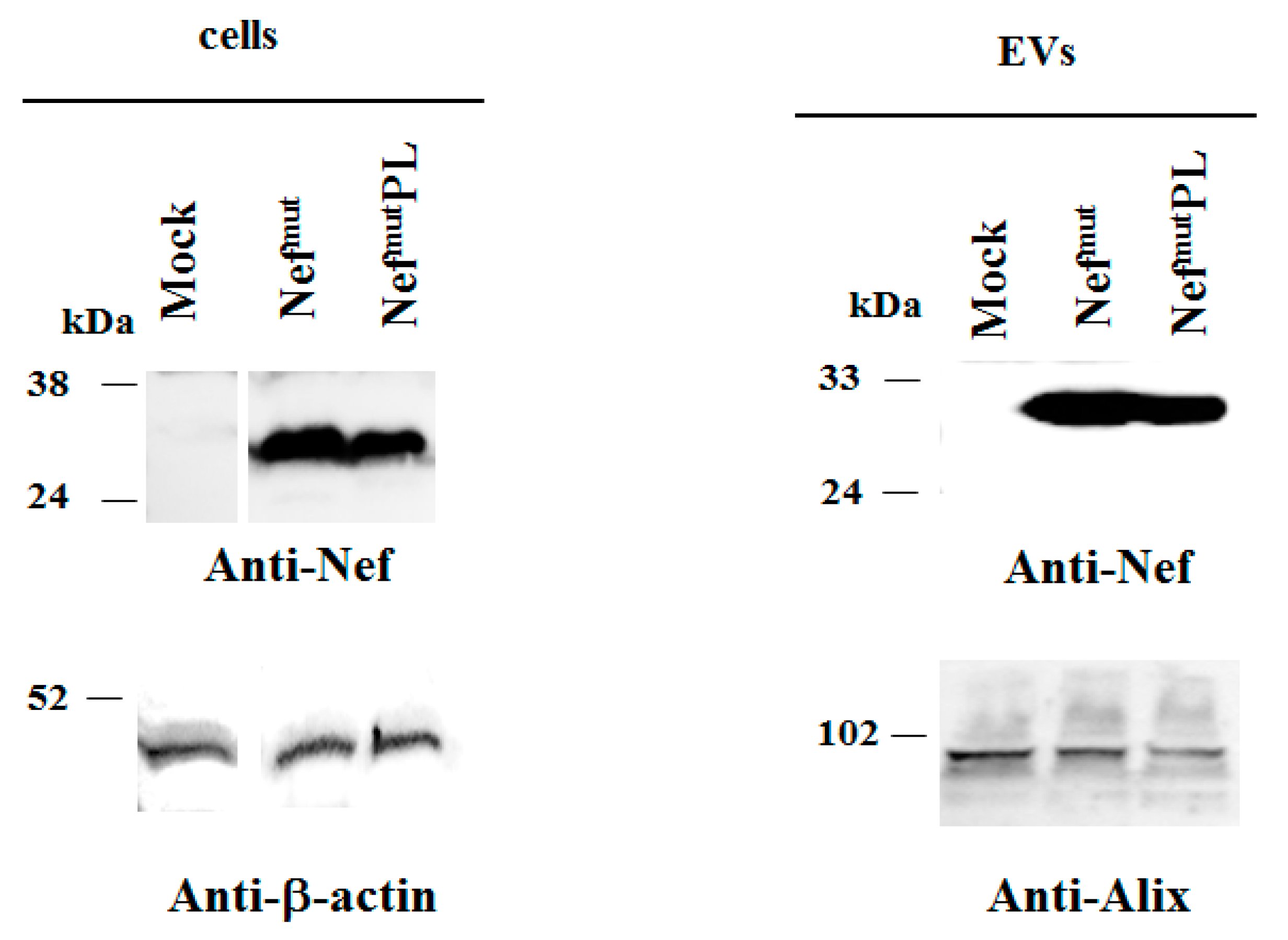
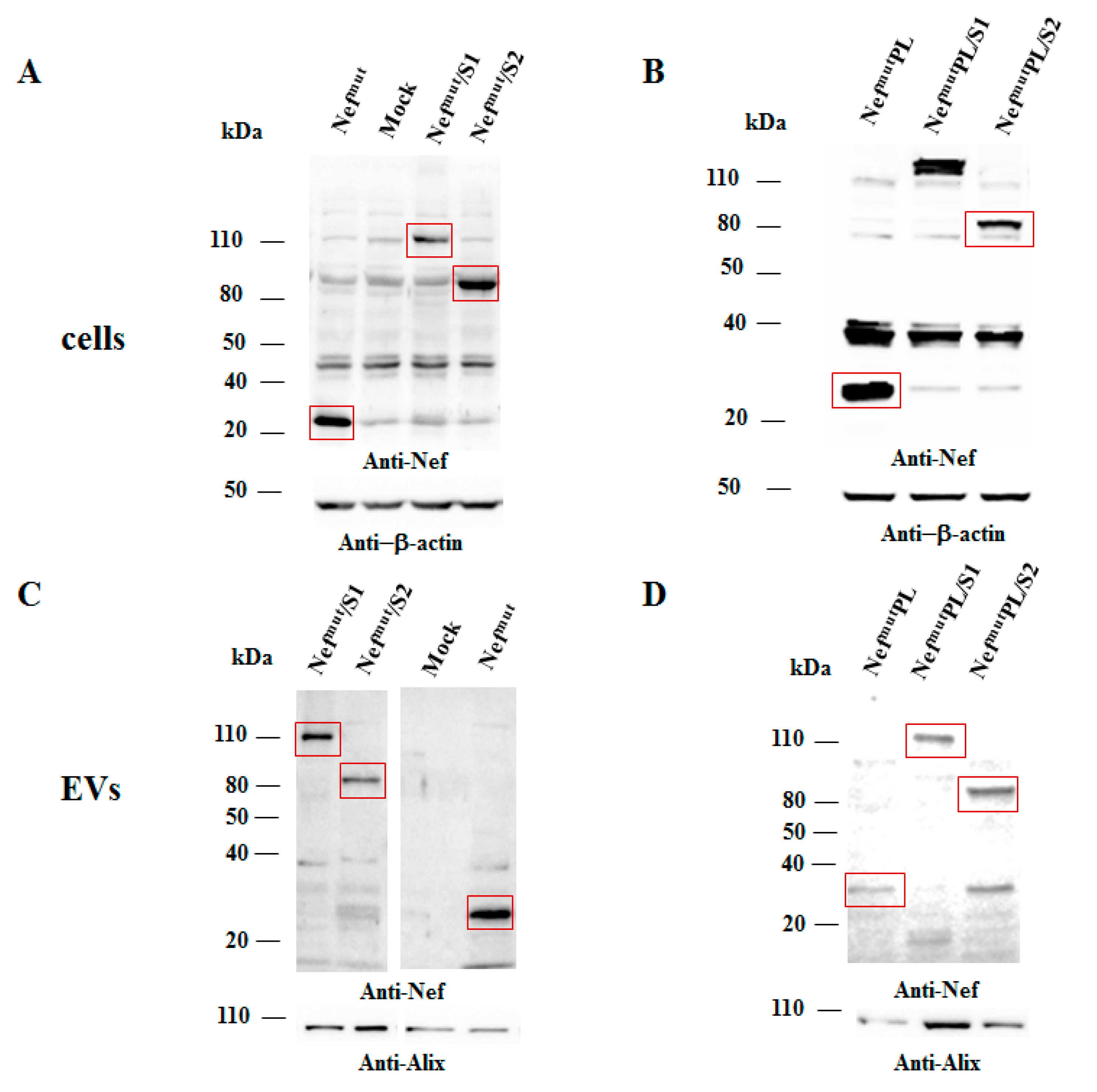
Both cell accumulation and EV association of Nef
mutPL compared to the full-length isoform were analyzed. To this aim, western blot analysis on lysates of both transiently transfected HEK293T cells and EVs isolated from respective supernatants were carried out. Representative results are shown in
Figure 2 and
Supplementary Figure S2 indicated that Nef
mut and Nef
mutPL accumulated into transfected cells at comparable extents, suggesting that the C-terminal truncation did not affect the protein stability. Most important, Nef
mutPL associated with EVs at levels like those of full-length Nef
mut, indicating that the presence of the 29 C-terminal amino acids does not influence the uploading efficiency of Nef
mut into EVs.
3.2. Intracellular Expression and EV-Uploading of HPV16-E6 and -E7 Fused with NefmutPL
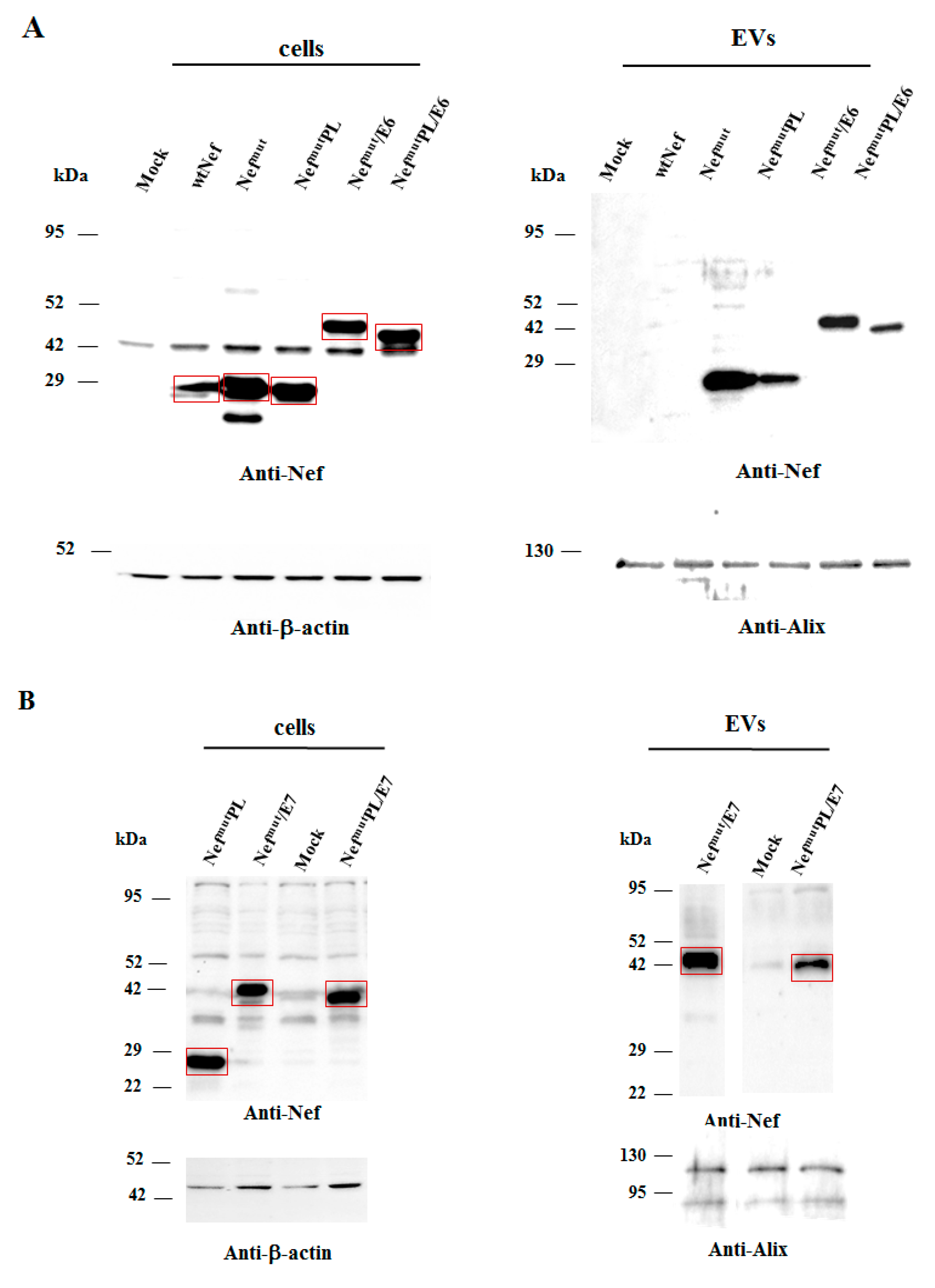
Next, we tested the efficiency of Nef
mutPL to vehiculate foreign proteins fused to it into EVs compared to parental Nef
mut. We considered both HPV16-E6 and -E7 as foreign antigens since they are uploaded in EVs very efficiently upon fusion with full-length Nef
mut [
6,
10]. The Nef
mut-related sequences were fused with either HPV16-E6 or -E7 ORFs, which were optimized for translation in eukaryotic cells, and whose protein domains involved in the respective pathogenic effects were inactivated as hereabove described. Steady-state levels in both transfected cells and EVs isolated from the respective supernatants were evaluated by western blot analysis. The E6-based fusion products accumulated in HEK293T transfected cells, as well as in EVs at similar levels whatever the anchoring protein. (
Figure 3A and
Supplementary Figure S3). Similarly, E7 fused with either Nef
mut or Nef
mutPL was expressed and uploaded in EVs at similar extents (
Figure 3B and
Supplementary Figure S4).
We concluded that the presence of the C-terminal 29 amino acids of Nefmu is not essential for the efficient EV-uploading of products fused to it.
In both panels, polyclonal anti-Nef Abs served to detect Nefmut-based products, while β-actin and Alix were markers for cell lysates and EVs, respectively. Where relevant, specific signals are highlighted. Molecular markers are given in kDa.
3.3. Induction of Antigen-Specific Polyfunctional CD8+ T Lymphocytes in Mice Immunized with DNA Vectors Expressing NefmutPL/E6 and /E7
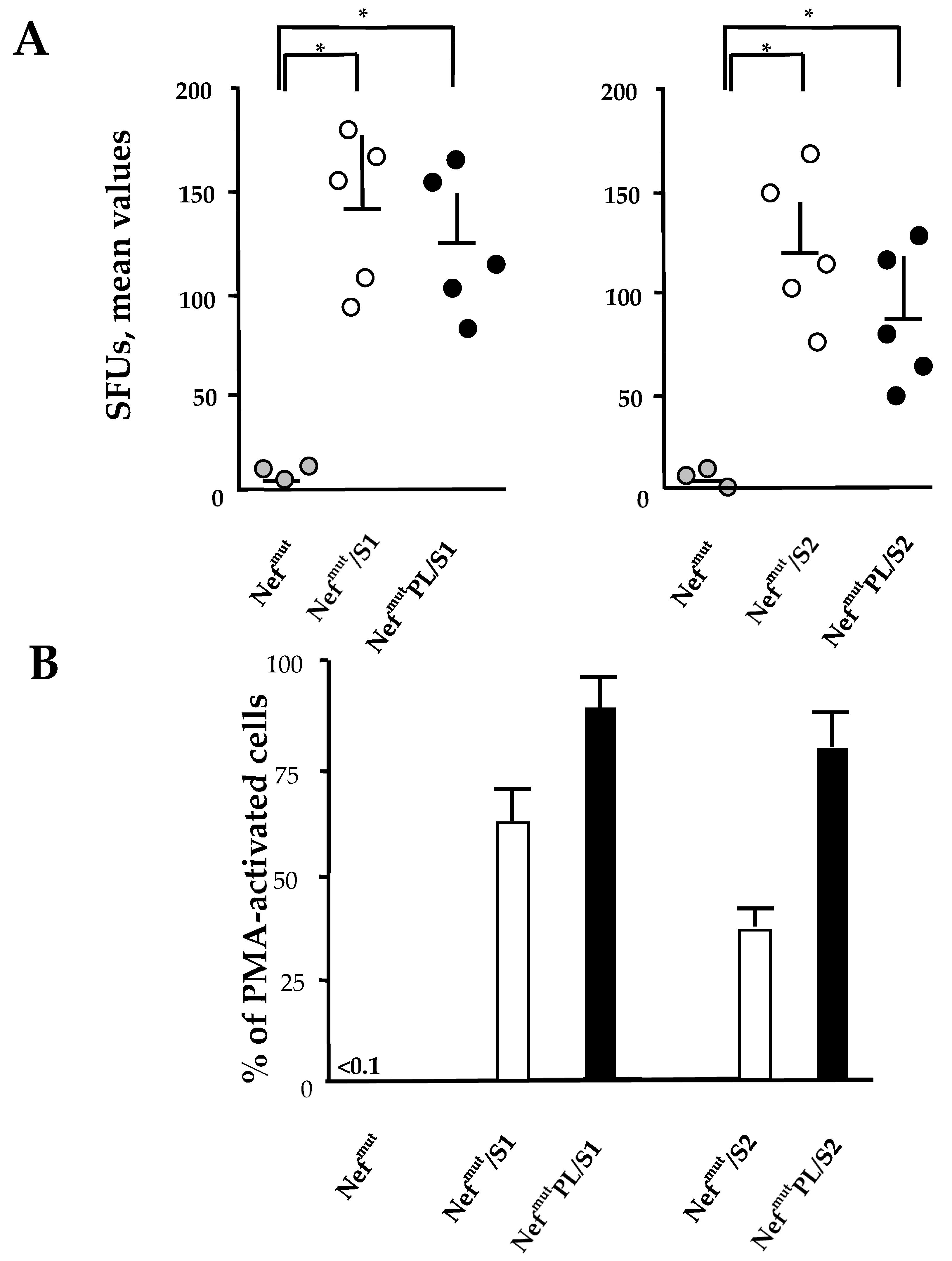
The induction of a strong CTL immunity implies the generation of antigen-specific CD8+ T lymphocytes co-expressing inflammatory Th-1 cytokines including IFN-γ, IL-2, and TNF-α (i.e., polyfunctional CD8+ T lymphocytes). We compared the effectiveness of Nefmut and Nefmut/PL in eliciting the antigen-specific CD8+ T cell immune response in terms of induction of polyfunctional CD8+ T lymphocytes.
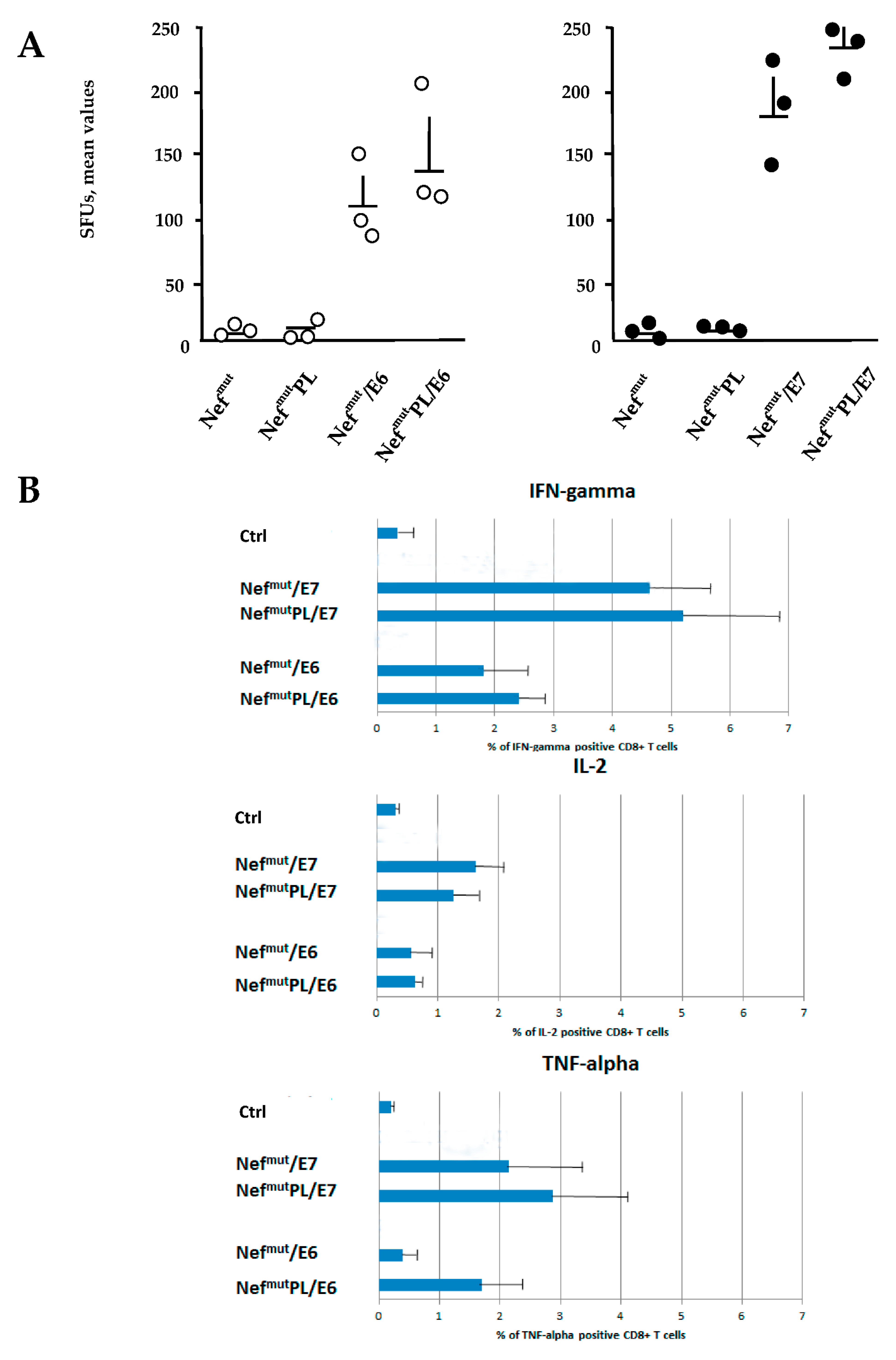
DNA vectors expressing HPV16-E6 and-E7 fused with either Nef
mut or Nef
mutPL were injected in mice. Fourteen days after the second immunization, IFN-γ EliSpot assays showed that all injected mice developed a well detectable antigen-specific CD8
+ T cell immunity (
Figure 4A), with DNA vectors expressing E7-derivatives inducing stronger immune responses compared to E6-derivatives. We noticed that the levels of CD8
+ T cell immune response in mice immunized with Nef
mutPL-based vectors appeared similar to those detected in mice injected with DNA vectors expressing full-length Nef
mut (
Figure 4A). The immune response in mice injected with the vectors expressing either Nef
mut or Nef
mutPL remained at the background levels.
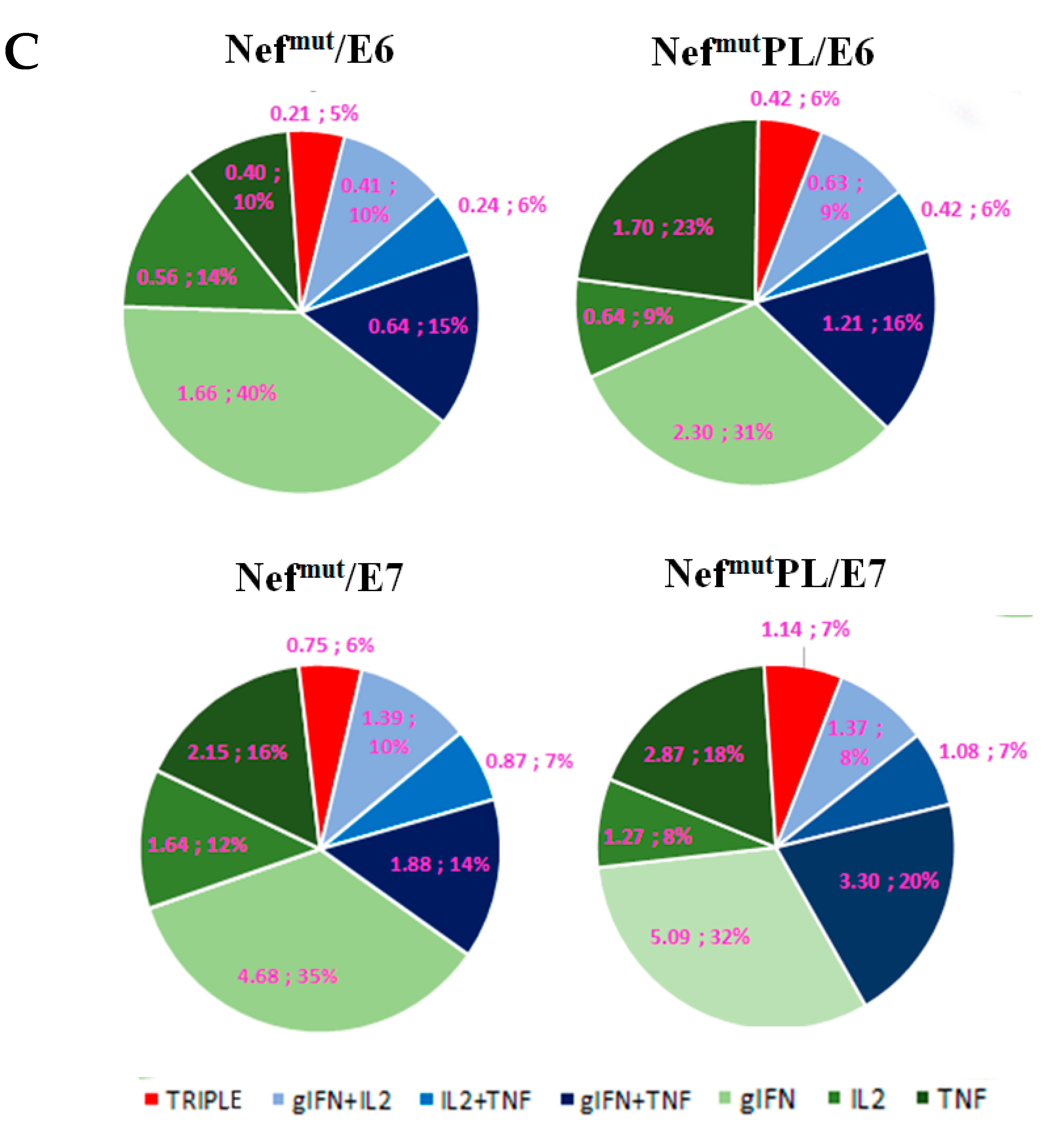
Single-cytokine ICS analysis showed that the percentages of E6/E7-specific CD8
+ T lymphocytes expressing either IFN-γ, IL-2, or TNF-α in no cases decreased when the antigen was fused to Nef
mutPL (
Figure 4B). Most importantly, the C-terminal truncation of Nef
mut did not affect the generation of antigen-specific, triple positive, polyfunctional CD8
+ T lymphocytes (
Figure 4C).
We concluded that the anti-HPV16-E6 and -E7 immunogenicity remained largely unchanged when the anchoring protein was deleted of 29 C-terminal amino acids.
3.4. Intracellular Expression and EV-Uploading of SARS-CoV-2 Antigens Fused with NefmutPL
Very recently, we demonstrated that the Nef
mut-based vaccine platform could be instrumental to induce strong CD8
+ T cell immunity against multiple SARS-CoV-2 antigens as detected in both spleens and lung airways [
11]. To establish whether these immunogenic properties are preserved also in the presence of the Nef
mut C-terminal truncation, we first evaluated the EV-uploading efficiency of the products of fusion of either Nef
mut or Nef
mutPL with S1 and S2 SARS-CoV-2 antigens.
Intracellular expression of the fusion products was evaluated by transient transfection in HEK293T cells, and the relative levels of uploading into EVs were scored upon their isolation from the supernatants of transfected cells. Results from western blot assays indicated that S1- and S2-based fusion proteins accumulated in cells at comparable extents whatever the EV-anchoring protein considered (
Figure 5 and
Supplementary Figure S5). Most important, no evident differences in the levels of EV-uploading of the fusion products were assessed in the presence of a slight decrease in EV uploading of both Nef
mut/S1 and /S2 compared to Nef
mut alone (
Figure 5 and
Supplementary Figure S5).
We concluded that, as already observed with HPV16 products, the efficiency of EV uploading of SARS-CoV-2 antigens was not influenced by the Nefmut C-terminal truncation.
3.5. CD8+ T Cell Immunity Induced in Both Spleens and Lungs of Mice Injected with Vectors Expressing NefmutPL-S1 and -S2
Both SARS-CoV-2 S1 and S2 were previously shown to induce a strong CD8
+ T cell immunity in spleens and lung airways when expressed by a DNA vector as products of fusion with Nef
mut and injected in mice [
11]. On this basis, we compared the CD8
+ T cell responses following the injection of DNA vectors expressing the two SARS-CoV-2 antigens fused with either Nef
mut or Nef
mut-PL. Fourteen days after the second immunization, splenocytes were isolated from injected mice and cultured overnight in IFN-γ EliSpot microwells. Comparable antigen-specific CD8
+ T cell activations were observed in splenocytes from mice inoculated with vectors expressing the diverse fusion products, with a slight increase in the case of vectors expressing full-length Nef
mut (
Figure 6A). On the other hand, the levels of immune responses measured in cells isolated from bronchoalveolar lavage fluids (BALFs) of injected mice appeared similar, with a small increase in cells from mice injected with Nef
mutPL-based vectors (
Figure 6B).
These data strongly supported the idea that the Nefmut C-terminus is dispensable for the induction of SARS-CoV-2 specific immunity in spleens and lung airways, i.e., the district primarily involved in SARS-CoV-2-related pathogenesis. Hence, NefmutPL can be considered an effective and safe alternative to full-length Nefmut for anti-SARS-CoV-2 CTL vaccines based on the technology of endogenously engineered EVs.
4. Discussion
The perspective to translate into the clinic the Nef
mut-based CTL vaccine platform requires the optimization of its safety profile. To this aim, the elimination of unnecessary sequences in the Nef
mut-based vectors would be of utility. Concerning the DNA vector, we already proved that Nef
mut-based fusion antigens retain their immunogenic properties when expressed by the pVAX1 vector [
11], i.e., a vector where prokaryotic sequences are reduced compared to the more complex pTargeT vector we previously utilized. More advanced, extremely small DNA vectors lacking antibiotic resistance (e.g., minicircle DNA vectors) [
30] would represent a feasible alternative for translation into the clinic of the Nef
mut-based vaccine platform. Regarding the possibility to reduce the sequences required for efficient EV-uploading, we recently demonstrated that the presence of the N-terminal domain with associated post-translational modifications is required [
31]. On the other hand, it should be considered that the Nef
mut defectiveness relies on the co-existence of two amino acid substitutions at positions 153 and 177. Unpredictable, rare, however, theoretically possible events of back-mutation might occur, especially when the DNA vectors undergo large-scale production and delivery. In the case of unwanted back-mutations, possible anti-cellular effects are expected to be strongly mitigated by the deletion of a significant part of the protein. The 29 amino acid C-terminal region deleted in Nef
mutPL includes domains involved in both signaling and trafficking functions typical of wt Nef [
32]. In this way, possibly back-mutated Nef
mutPL molecules are expected to lose the most detrimental anti-cellular effects.
The truncation of a C-terminal region of 29 amino acids had no impact on both efficiencies of EV uploading and immunogenicity of diverse fusion products. In some instances, these results can be considered unexpected since the Nef C-terminal region comprises the cholesterol recognizing motif (CRM) (
Figure 1) already characterized as an important domain for the association of Nef with the viral envelope [
33]. Considering the convergence in the biogenesis of HIV-1 and EVs [
34], this domain has the potential to play a role in the interaction of Nef with the nanovesicles released by Nef-expressing cells. The CRM amino acid sequence, i.e., LHPEYYK in SF2-like Nef alleles and LHPEYFK in HXB2-like ones, is well conserved among HIV-1 types B strains, including that from which Nef
mut originates, i.e., F12/HIV-1 [
35]. We hypothesized that the effects of both N-terminal myristoylation and palmitoylation present in Nef
mut, together with the stretch of basic amino acids located in alpha helix 1, are prevalent over those induced by CRM in terms of stable interaction with lipid rafts of both plasma and endosomal membranes.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






