3.1. Open Circuit Voltage (OCV) for Membranes with 1, 2, 4-Triazole Additive
The melt blown membrane with 1, 2, 4-triazole additive was fabricated and assembled without any further post-treatment into a single cell to evaluate its fuel cell performance. Conditioning curve for this sample was not able to be collected since the cell performance was too low to draw any load. Alternatively, OCV hold testing was performed.
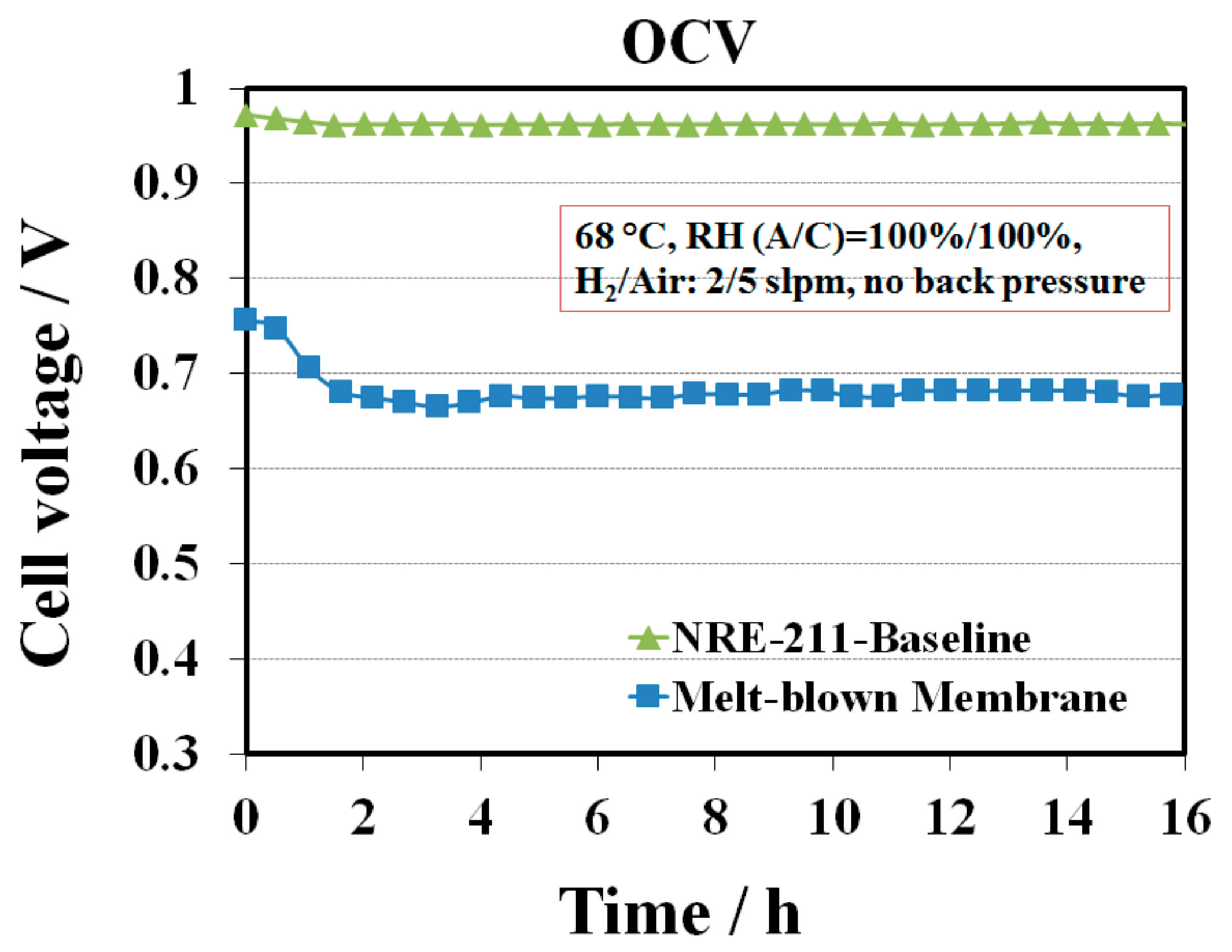
Figure 1 shows a comparison of a single cell OCV profile between the melt blown membrane and the baseline NRE-211 at 68 °C and 100% RH. The OCV curve for the baseline sample was flat with only 1% variation in cell voltage compared to the initial voltage. In contrast, the OCV for the melt blown sample decreased during the first few hours, then slightly increased, and next kept constant over time. The lowest OCV points for the melt blown sample was at ~ 3.5 h with OCV of 665 mV. Compared to the OCV of the baseline in the same range of time (~970 mV), the OCV of this sample was ~300 mV lower, implying that the 1, 2, 4-triazole additive has a considerable impact on the cell performance. Generally, the OCV depends on the hydrogen permeation rate across the membrane as well as the mixed potential of the electrochemical reactions of Pt surface oxidation and the oxygen reduction reaction (ORR). Lower OCV value indicates either high reactant crossover and/or electronic short through the membrane, or poisoning of the catalyst or electrolyte [
15]. In our case, the lower OCV phenomena could be explained by the contamination of the catalyst layers by the 1, 2, 4-triazole additive, as it likely migrated from the membrane into the catalyst layers, leading to a decrease in OCV in the first few hours. While the accumulated additive in the catalyst layers achieved its maximum value, the OCV started to slightly increase probably due to partial contaminant removal from the cell via convection. However, the real mechanism of additive migration is not clear. It is hypothesized that at the beginning of the MEA assembly, 1, 2, 4-triazole additive might migrate from the membrane into the catalyst layer and be retained at the Pt surface by forming covalent bonds between the nitrogen (N) in 1, 2, 4-triazole and the Pt atom causing the initial relatively low OCV [
16]. MEA humidification during OCV test exacerbated the catalyst layers contamination afterwards because the additives could keep migrating along with the water from humidified gas streams. Meanwhile, a portion of retained 1, 2, 4-triazole could be washed out from the catalyst layer into the GDLs or released from the MEA by water. The amount of triazole remaining in the catalyst layer relies on the dynamic balance between the amount of additive migrated from the membrane and the amount of additive moved away from the catalyst layer.
The hypothesis can be supported by UV-vis spectra of the tested membranes and GDEs. 1, 2, 4-triazole is one type of heterocyclic compounds with the molecular formula C
2H
3N
3, which has a five-membered ring containing two carbon and three nitrogen atoms and can easily be detected by UV spectra.
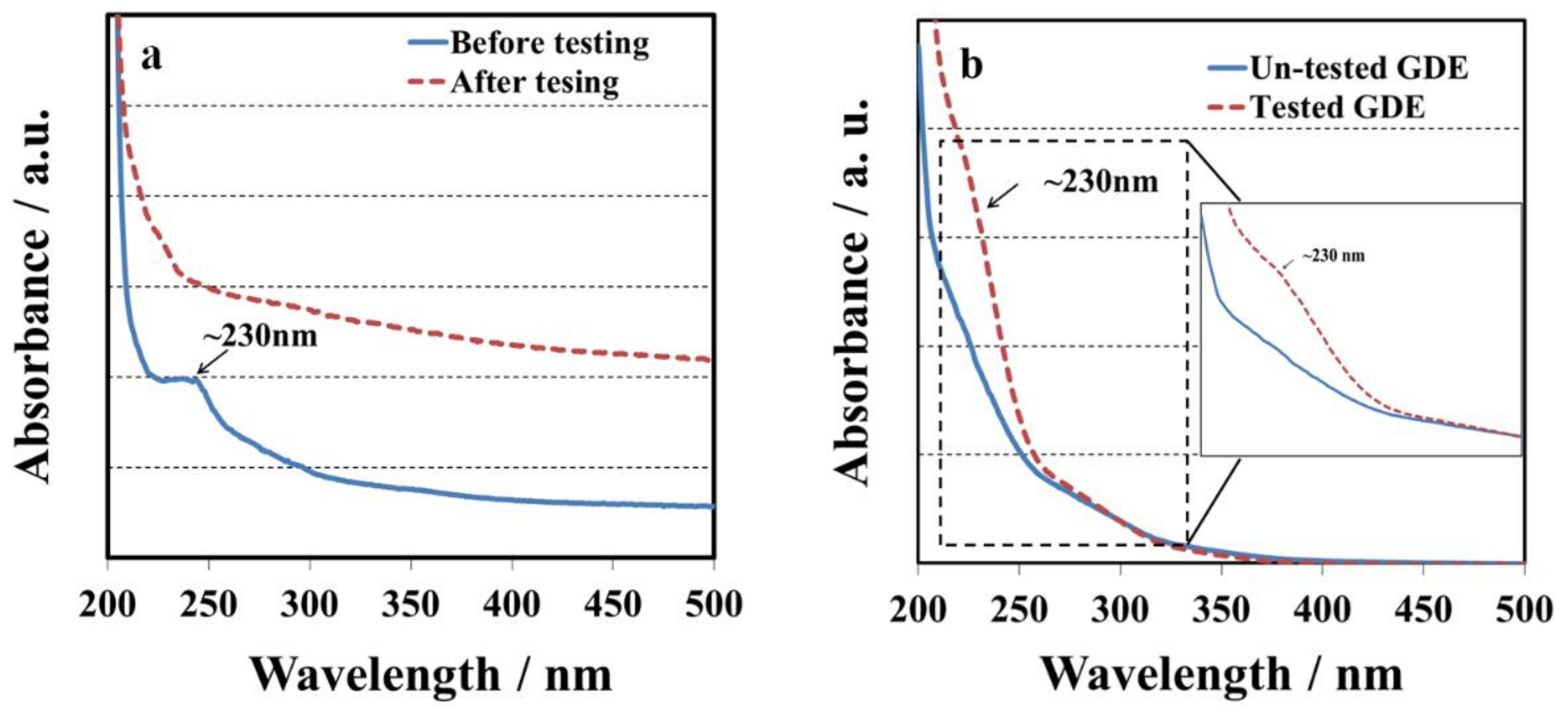
Figure 2a shows the UV spectra of the membrane with 1, 2, 4-triazole before and after OCV hold test. There was one absorption peak at ~230 nm associated with 1, 2, 4-triazole before OCV hold test. However, the absorption peak at ~230 nm completely disappeared after 16 hours’ OCV hold test. The changes in UV spectra could be explained by the fact that 1, 2, 4-triazole moved away from the membrane during fuel cell testing. Although the absorption peak at ~230 nm disappeared after fuel cell testing, there might be some leftover species which were undetectable by the spectrometer. In order to further prove our hypothesis, the tested GDE on the cathode side and a standard GDE before testing were immersed in ethanol/water solvents (7: 3) for 12 h, and then their solution samples were examined by UV-vis.
Figure 2b displays one weak absorption peaks at ~230 nm associated with 1, 2, 4-triazole diffusion to the soaking solution from the tested GDE, confirming that additive migration from the membrane into the catalyst layer occurs via convection of either humidified gas or water. For the soaking solutions with the un-tested GDE, the peak at ~230 nm could not be detected indicating the amount of triazole in catalyst layer is insignificant. However, trace amount of triazole could cause a significant impact on the OCV.
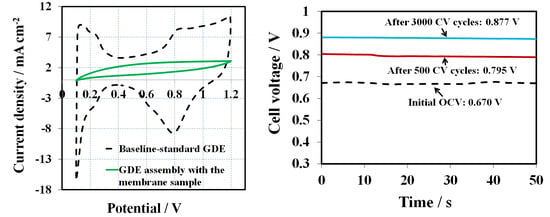
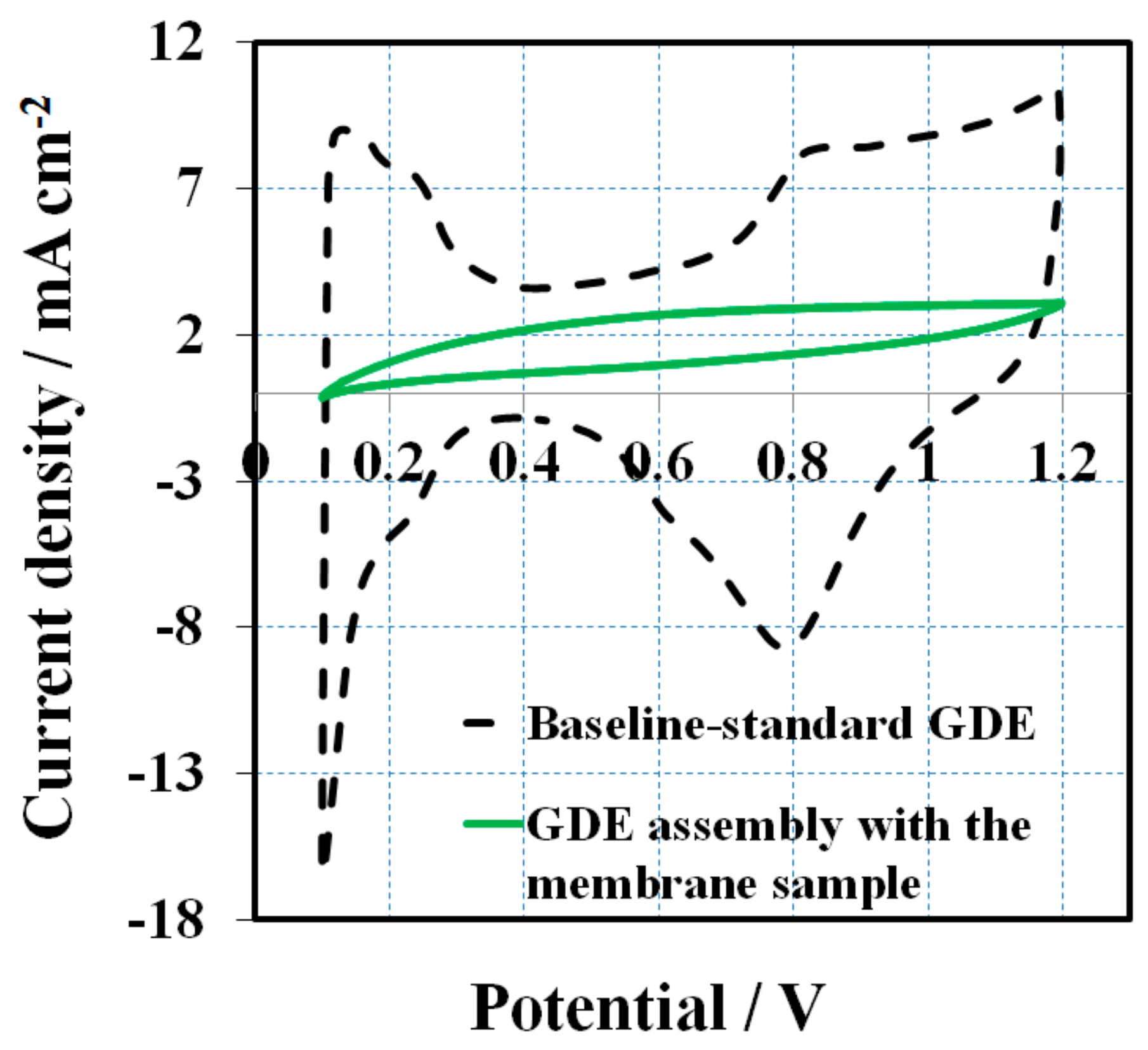
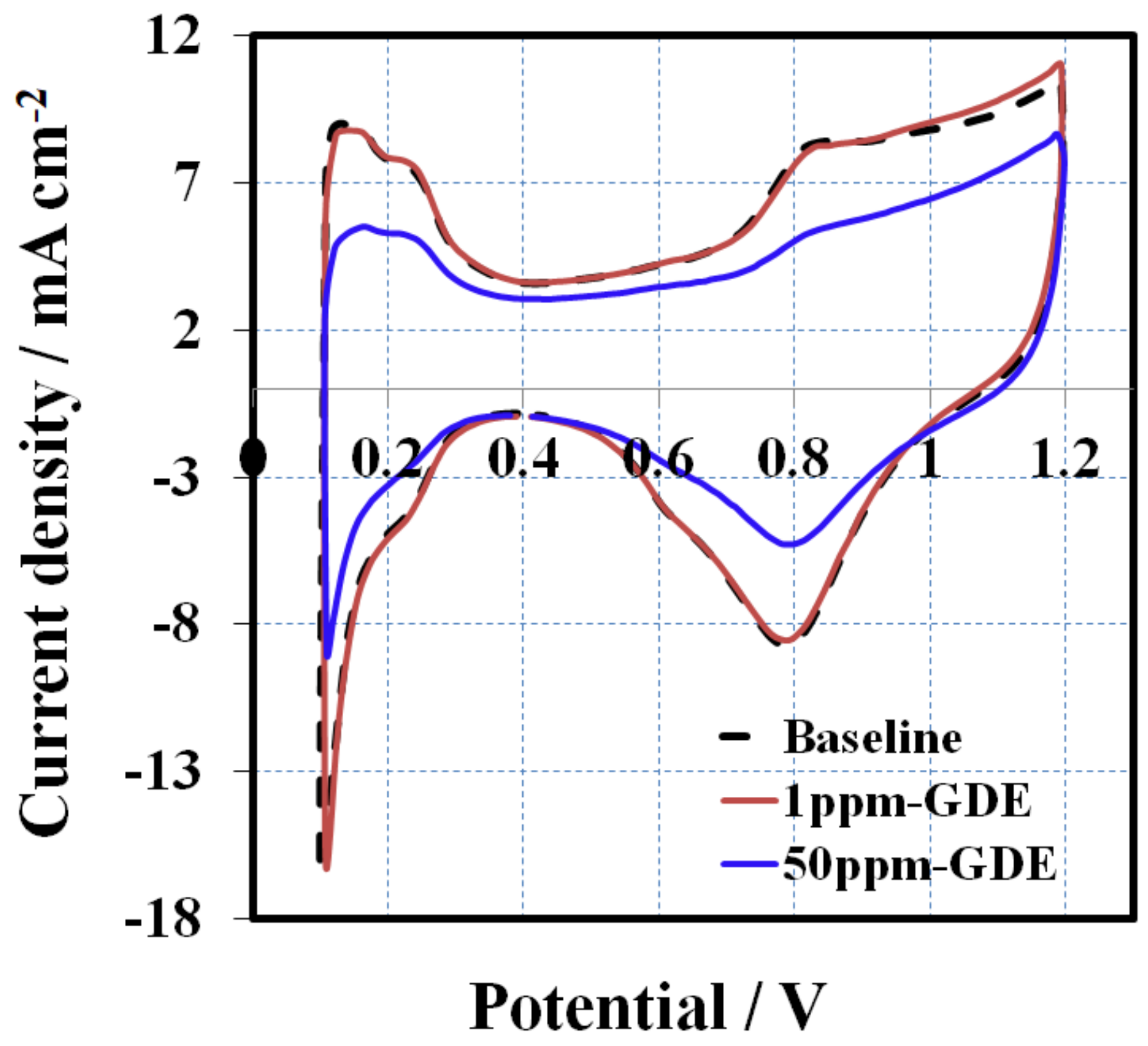
After a 16 h OCV hold test, CV scans were performed to compare the cathode side GDE against the baseline to determine if the additive had contaminated the cathodic catalyst layer. All of the GDEs before MEA assembly is assumed to have an identical CV (commercial standard GDEs from the same batch).
Figure 3 presents the CVs obtained at a scan rate of 50 mV s
−1 after the OCV hold test. The baseline CV (black dashed line) shows characteristic CV peaks for hydrogen adsorption/desorption of Pt electrode. The initial baseline ECSA (H-des) was about 26 m
2 g
−1, calculated from hydrogen desorption peak. In contrast, there are no obvious features of hydrogen adsorption/desorption for the cathode electrode of the melt blown membrane, assuming the 1, 2, 4-triazole additive migrated from the membrane to the surface of the catalyst where it was absorbed. This proves that the catalyst layer was contaminated, leading to ECSA losses and inferior fuel cell performance.
It is worthwhile to mention that the contaminated catalyst layers can be partially cleaned by CV cycling to remove the additives from the surface of catalyst. The clean-up efficacy depends on the amount of 1, 2, 4-triazole initially present in the membrane. As reported, some organic additive contaminants could be effectively removed by oxidation under higher potentials [
17]. However, the CV cycling process may also cause the formation of Pt oxide, Pt particle sintering, Pt dissolution/re-deposition, and carbon support corrosion [
18]. In this work, 3000 CV cycles under a wide potential window (0.1 to 1.2 V) were used as one of the approaches for triazole additive removal.
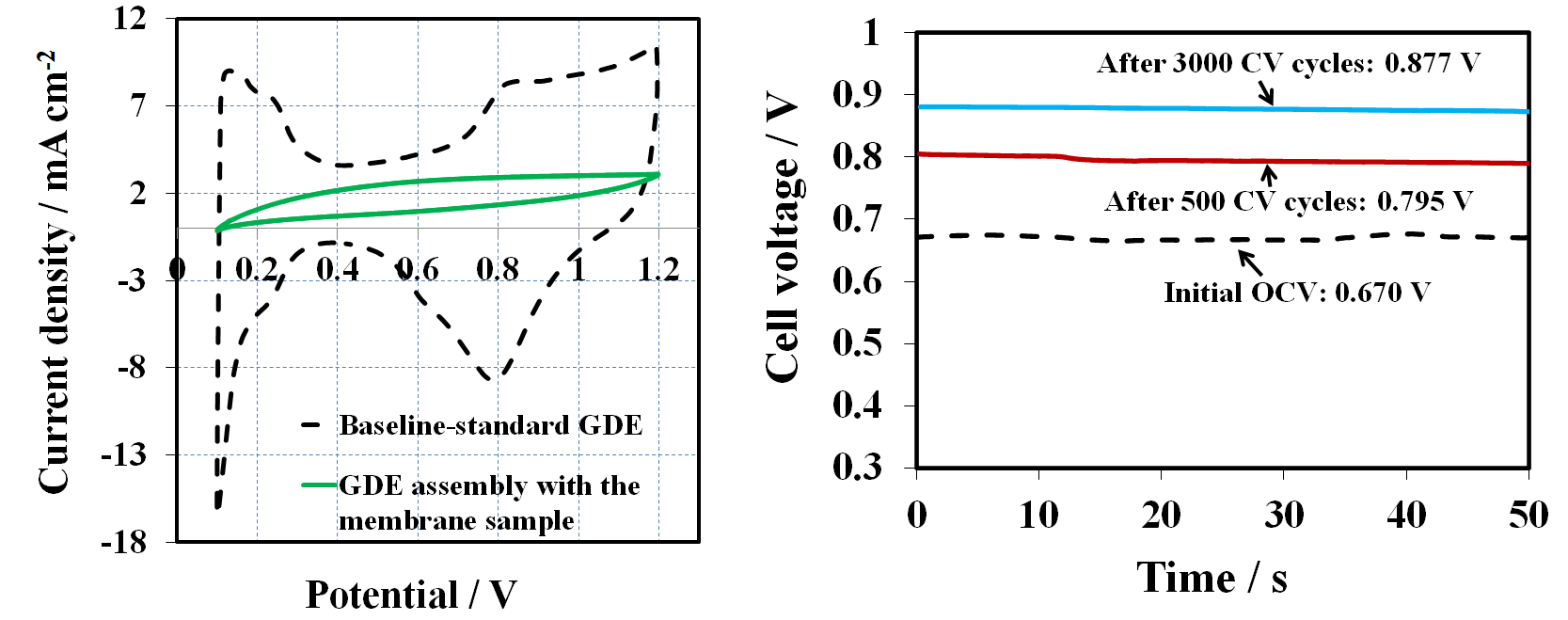
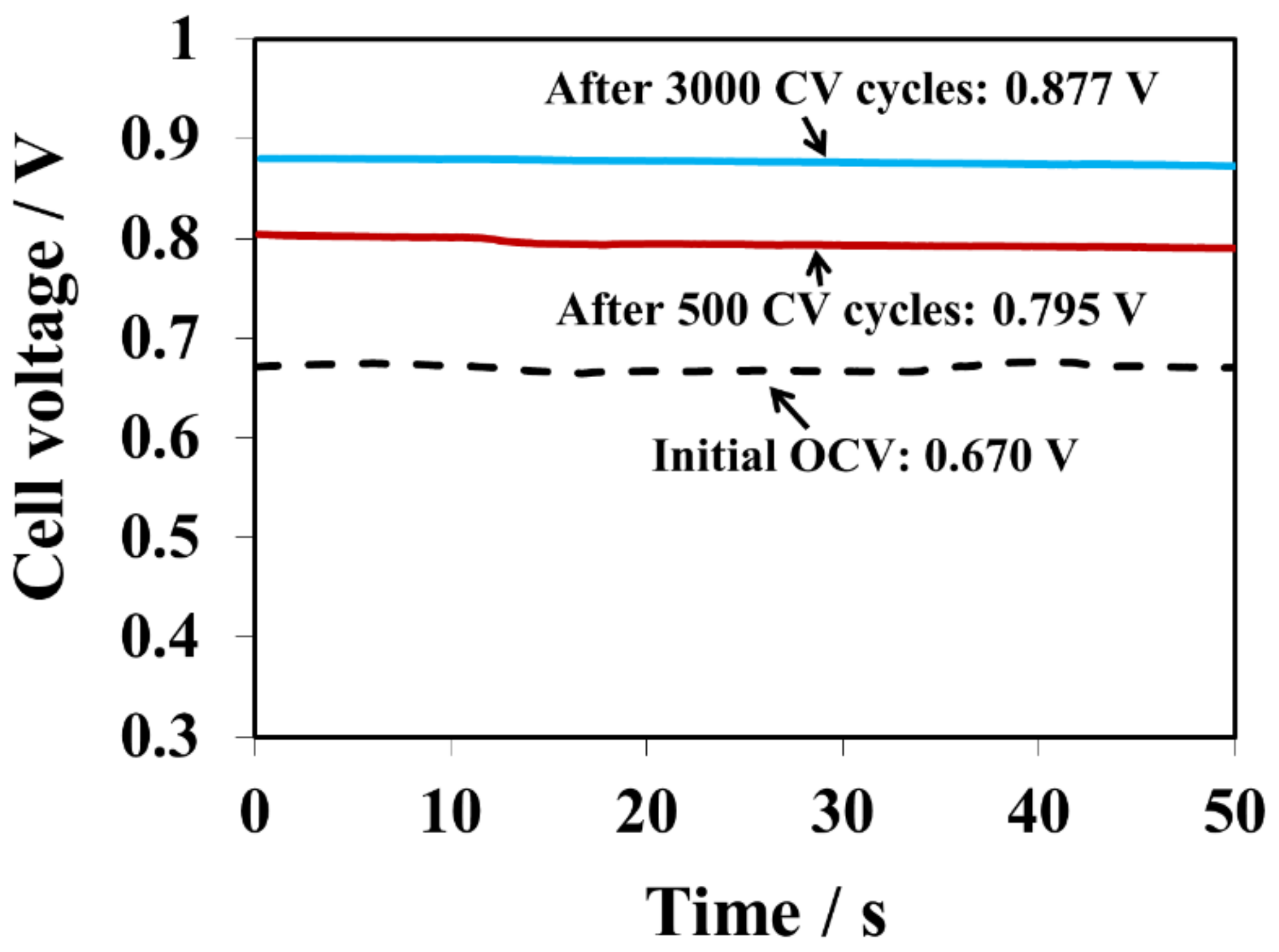
Figure 4 and
Table 1 show the OCV changes after 500, 1000, 1500, 2500, and 3000 CV cycles for the 1, 2, 4-triazole contaminated GDE. The OCV were compared with the baseline and the OCV ratio between the sample and the baseline was calculated. Clearly, the OCV gradually increased after every 500 CV cycles. For example, the OCV were 0.795V and 0.877V, which reached 82% and 90% of the baseline OCV value, after 500 and 3000 CV cycles, respectively. Therefore, CV cycling is a practical approach for removing the triazole additive from the catalyst layer. However, the OCV baseline value was not recoverable and no distinguishable features of hydrogen adsorption/desorption for the cathode electrode were observed, implying that the amount of 1, 2, 4-triazole additive in the membrane and GDEs was too high to be fully cleansed solely by CV cycling. It further demonstrates that a trace amount of triazole in the catalyst layer could have a huge impact on fuel cell performance.
The migration of triazole additive from membrane into the cathode catalyst layer was also confirmed by XPS. The survey spectra of untested and tested cathodic GDE assembly with melt blown membrane sample were collected in
Figures S1 and S2, in which Pt 4f, C1s, O1s, N1s, F1s, and S2p orbitals can be observed and each individual element content was presented. As N is major element of the 1, 2, 4-triazole, it was considered as a GDE contamination indicator. The atomic ratio of N 1s: Pt 4f of GDE before and after fuel cell testing increased from 0.16 to 0.87 (see
Table 2), indicating a non-negligible amount of additive moved out of the membrane into the GDE and remained in the cathode catalyst layer. The XPS trend is consistent with the CV results shown in
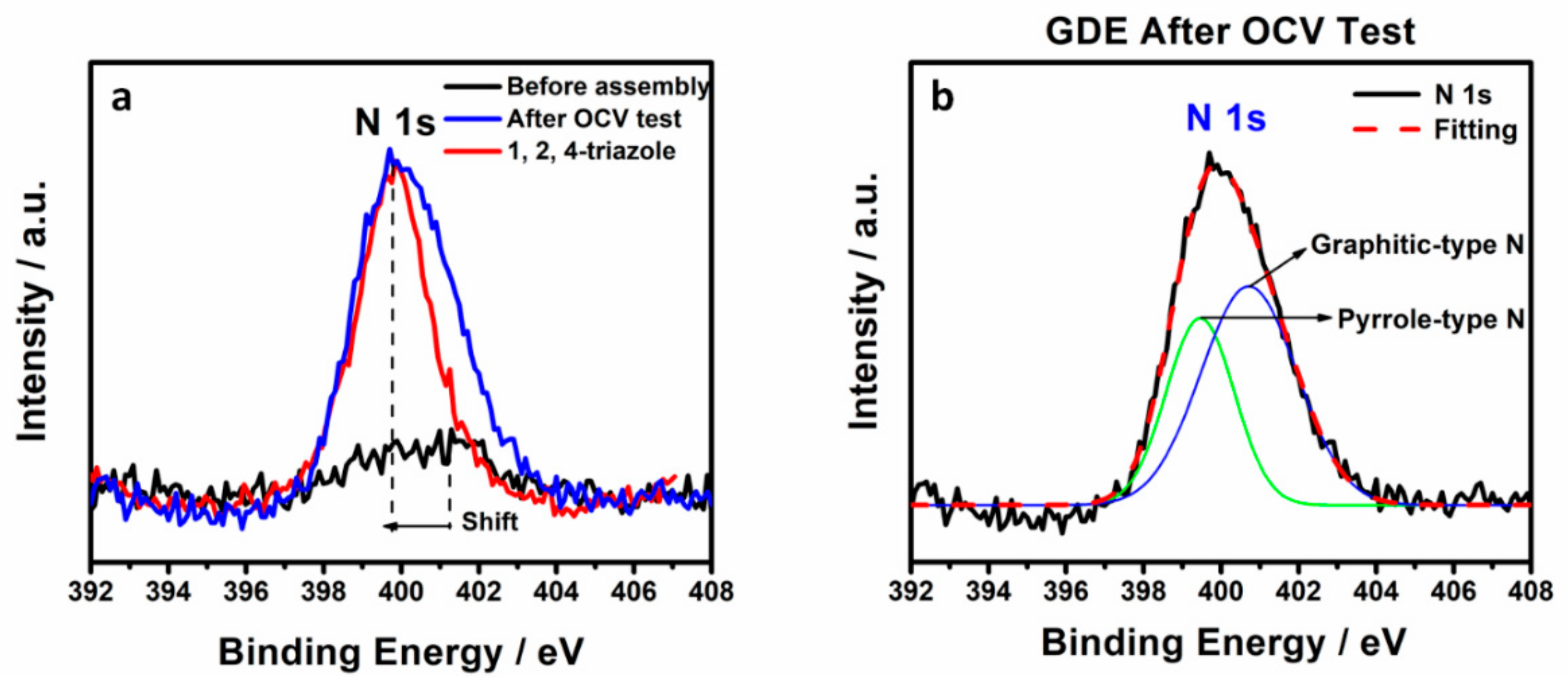
Figure 3. The core-level XPS spectra of N 1s for untested, tested cathodic GDE and 1, 2, 4-triazole as a comparison are shown in
Figure 5a. The tested cathodic GDE shows a negative shift of the N 1s binding energy and a relatively high peak intensity of N 1s, which has a similar N 1s binding energy as 1, 2, 4-triazole, compared to untested cathodic GDE, suggesting a considerable amount of N associated with triazole observed in the tested cathodic GDE. The N1s XPS spectra for tested cathodic GDE could be reasonably deconvoluted into two kinds of nitrogen functional groups: pyrrole-type N (400.2 eV) and graphitic-type N (401.1 eV) [
19,
20]. The deconvoluted N1s XPS spectra and calculated relative area of pyrrole-type N and graphitic-type N were shown in
Figure 5b and
Table 3, respectively, which identified the nature of triazole. Meanwhile, the core-level XPS spectra of Pt 4f is shown in
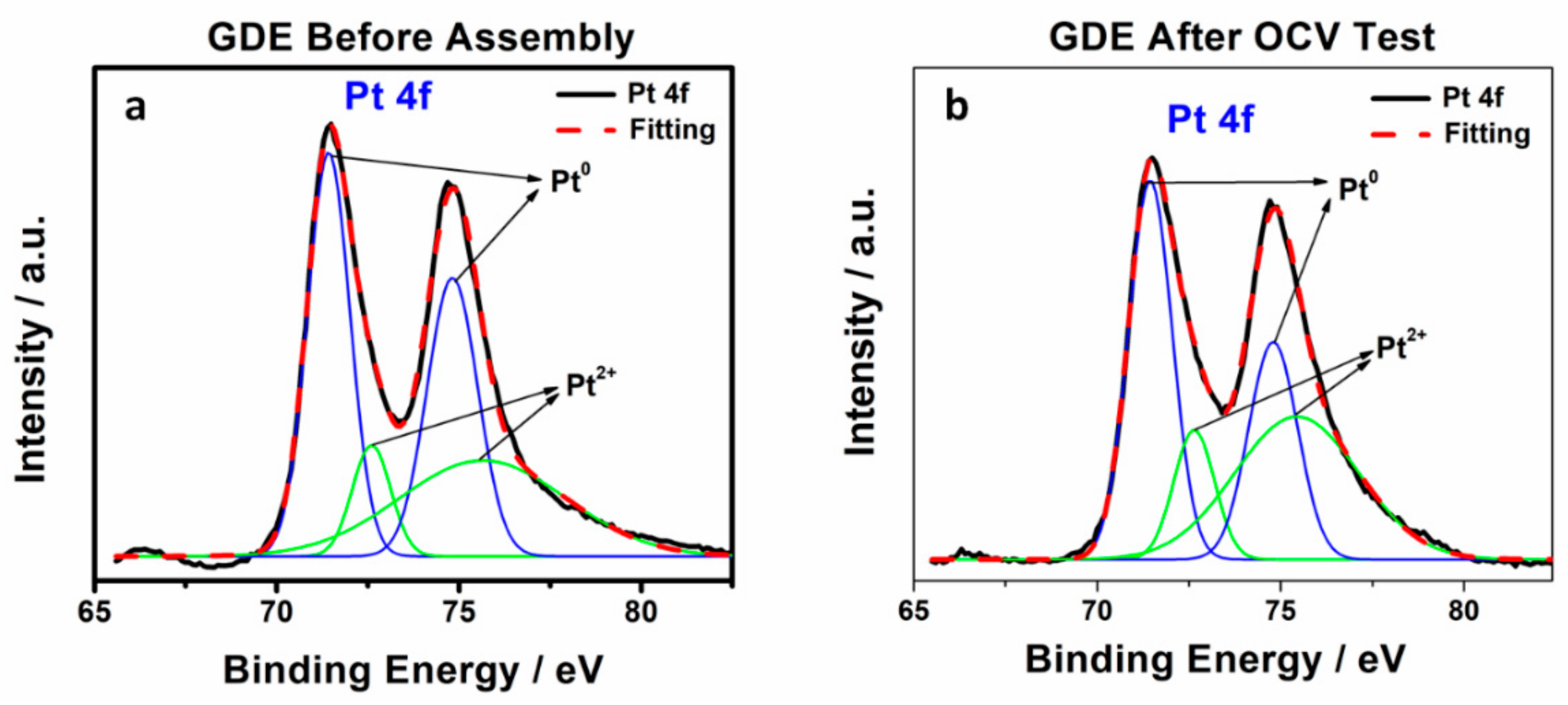
Figure 6 and the principle peaks were attributed to Pt
0 at 71.4 eV (4f
7/2) and 74.7 eV (4f
5/2), while 72.8 eV and 76.1 eV were assigned to Pt in 2+ state [
21]. The results of different Pt species were calculated based on the deconvolution of these two kinds of Pt states and listed in
Table 3. The relative area of Pt
0 and Pt
2+ was calculated to be 61% and 39%, respectively, for standard untested cathodic GDE. After fuel cell testing, the proportion of Pt
0 on the surface decreased to 50%, while Pt
2+ increased to 50%, indicating that the additional nitrogen containing coordination sites from 1, 2, 4-triazole possibly bonded to Pt. The XPS results proved that triazole additives migrated from the membrane into the catalyst and then attached to the Pt surface during OCV testing.
3.2. Conditioning Behaviour of 1, 2, 4-Triazole Additive Contaminated GDE
In order to further investigate the effects of 1, 2, 4-triazole additive on fuel cell performance, the triazole was intentionally introduced into GDEs by immerging the GDEs in either 1 ppm or 50 ppm 1, 2, 4-triazole additive solution for 48 h, which were named 1 ppm-GDE (GDE contaminated in 1 ppm of 1, 2, 4-triazole solution) and 50 ppm-GDE (GDE contaminated in 50 ppm of 1, 2, 4-triazole solution), respectively. The baseline was a standard GDE without any 1, 2, 4-triazole pre-treatment.
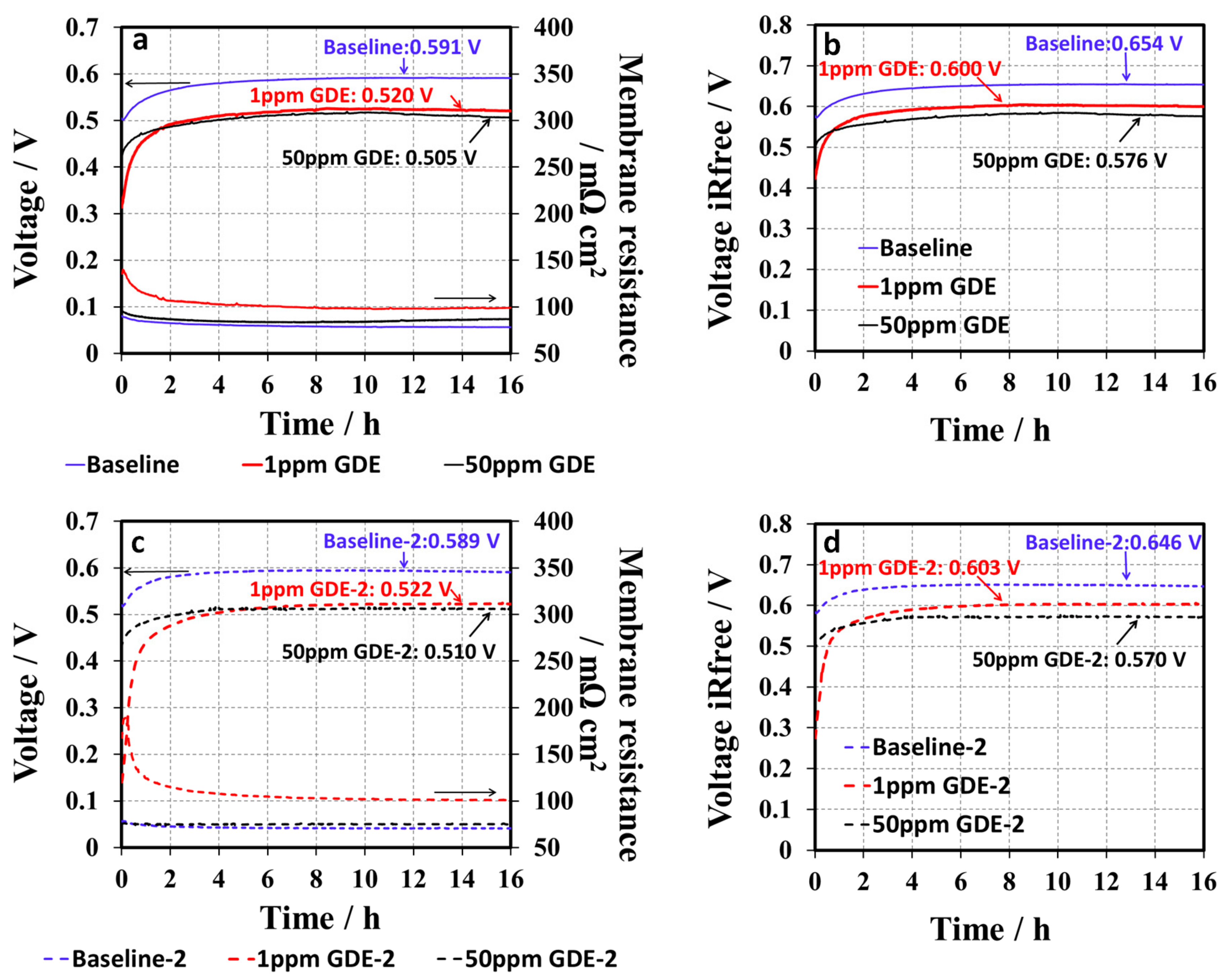
Figure 7a shows the H
2/air conditioning curves for baseline, 1 ppm, and 50 ppm GDE samples obtained at 68 °C and 100% RH at a current density of 0.8 A cm
−2. Each GDE sample was tested with two duplicated samples for examining the experimental reproducibility, which was labelled as baseline-2, 1 ppm-GDE-2, and 50 ppm-GDE-2, respectively. The results are shown in
Figure 7c. The repeatability of the test was acceptable because the difference in cell voltage between the two measurements (
Figure 7a,c) was within 10 mV after conditioning. The cell voltage in the conditioning curves of the baseline GDEs gradually increased from 505 mV to 587 mV in the first 6 h and then reached a plateau, suggesting that the MEA be “well-conditioned.” Moreover, the high frequency resistance (HFR) collected by the current interrupt technique is mostly contributed from the membrane’s ionic resistance (see
Figure 7a,c), which slightly decreased by ~15 mΩ cm
2 during the conditioning, which is mainly attributed to the membrane hydration under 100% RH. Like the baseline, the conditioning curves of the 1 ppm-GDE showed a similar trend: the cell voltage gradually increased, and then reached its steady state (see
Figure 7a). However, the 1 ppm-GDE sample showed inferior cell performance. The cell voltage for the 1 ppm-GDE sample was 190 mV and 70 mV lower than that of baseline at initial and after conditioning, respectively. For the 50 ppm-GDE, the performance drop was more obvious than the 1 ppm-GDE, since the cell performance was too low to draw a load at 20A. Alternatively, the 50 ppm-GDE sample was conditioned at a relatively low current of 0.4 A cm
−2 for 16 h. After that, the conditioning curve was collected at an increased load, 0.8 A cm
−2, as shown in
Figure 7a. After conditioning, the cell voltages of the MEA samples at a current of 0.8 A cm
−2 were in the order of 590 mV for the baseline, >520 mV for the 1 ppm-GDE, and >505 mV for the 50 ppm-GDE. The 50 ppm-GDE sample exhibited the lowest cell performance compared to the baseline and the 1 ppm-GDE. The absorption of 1, 2, 4-triazole in the catalyst layers led to a decrease in cell voltage, thus explaining the initial low fuel cell performance, while the removal of the contaminant from the cell by water generated from the electrochemical reaction or humidified gas streams resulted in a gradual improvement in cell performance. Additionally, the final cell performance for the contaminated GDE was still inferior to the baseline, implying that a certain amount of additive remained in the catalyst layer and the 50 ppm-GDE retained the most additive. IR-compensated conditioning curves were obtained by membrane resistance correction [
22], as shown in
Figure 7b,d. Similar to the non-corrected conditioning curves, IR-compensated conditioning curves showed the same trend: the cell voltages gradually went up, and then reached a plateau without further increases. It also demonstrated that HFR cannot be the main factor resulting in the differences of conditioning behavior between the MEAs. To further confirm the inferior cell performance of triazole doped GDEs, other in-situ characterizations were conducted and discussed in the following sections.
3.4. 2, 4-Triazole Additive Influence on ORR Kinetics
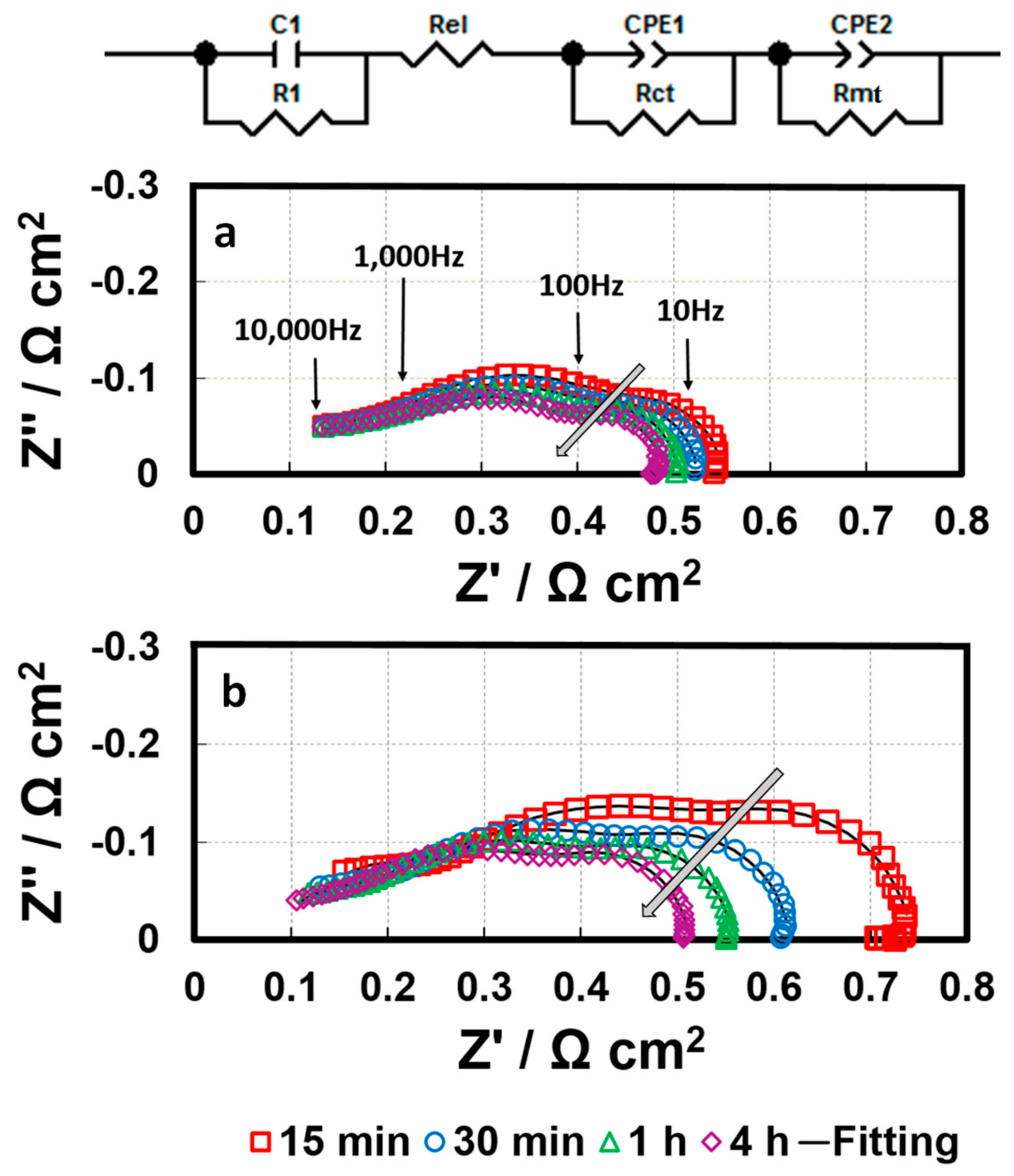
During conditioning, in-situ EIS was conducted to improve understanding of the mechanism behind the impacts of triazole additives on fuel cell performance. Generally, when the cell is operated at high current densities, a double semicircle in the Nyquist plot could be observed, which is attributed to the sum of the charge transfer resistance (Rct) and mass transport resistance (Rmt).
Figure 9 shows the spectrum (Nyquist plots) of the baseline and the 1 ppm-GDE, including one high frequency (HF) capacitive loop and one medium frequency (MF) capacitive loop, as well as one low frequency (LF) capacitive loop and one LF inductive loop [
23]. The small HF capacitive loop can be fitted by a contact resistance R1 in parallel with a contact capacitance C1, associated with either the electronic contact impedance or the ionic ohmic drop inside the active layer [
24]. The MF impedance arc mainly corresponds to the charge transfer resistance associated with the ORR, and the LF domain is predominantly attributed to the mass transfer resistance, representing the resistance to mass transfer in the gas phase within the gas diffusion layer and the catalyst layer. Based on the essential feature of fuel cell reactions and the observed electrochemical phenomena, the MF and LF loops can be fitted using the equivalent circuit shown in
Figure 9, consisting of a charge transfer resistance Rct in parallel with a constant phase element (CPE1) and a mass transfer resistance Rmt in parallel with a constant phase element (CPE2), respectively [
25]. With increasing conditioning time, the MF capacitive loop changes significantly. In contrast, there was negligible variation in the LF inductive loops. Therefore, the fitting for LF inductive loops was not conducted in this work.
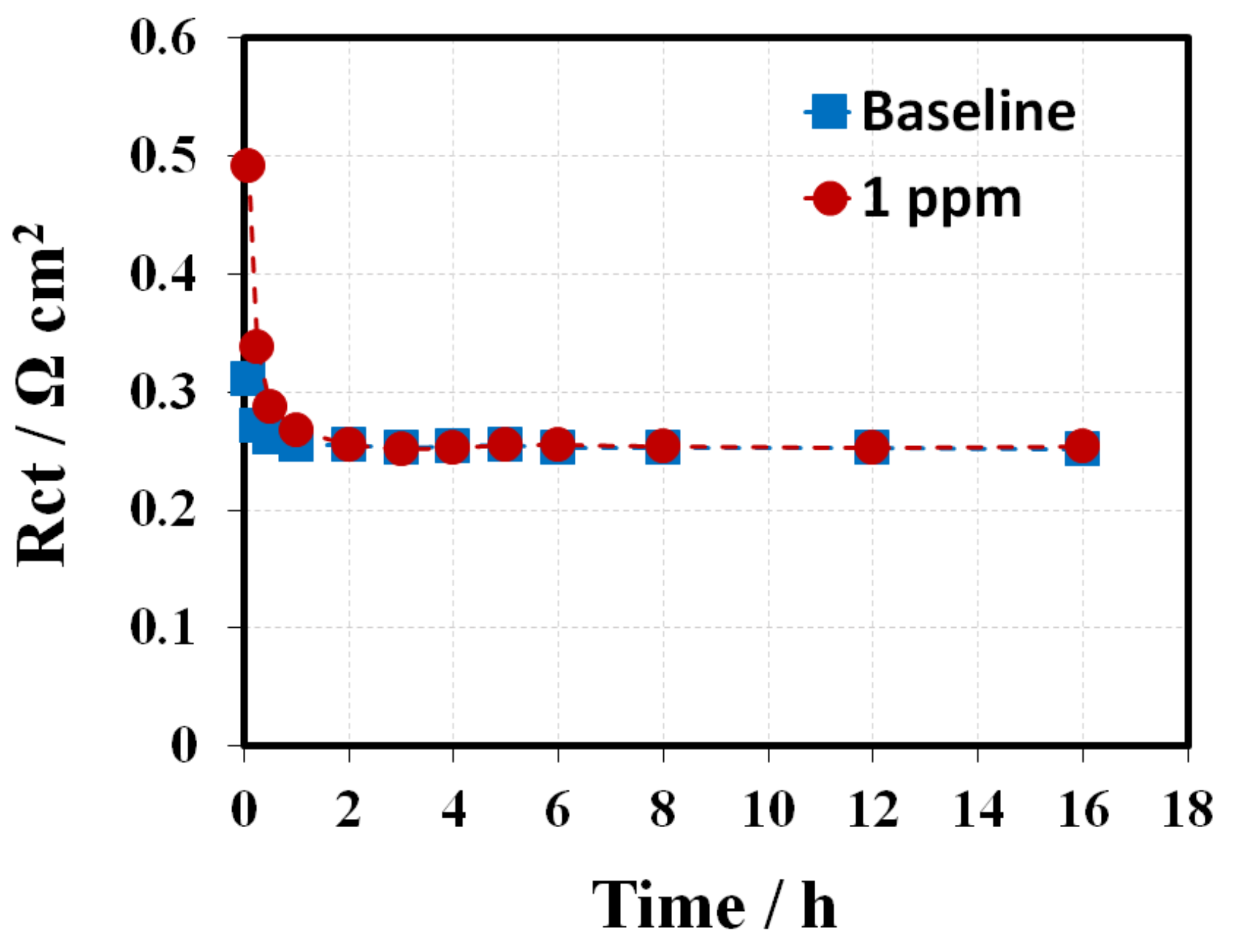
Figure 9 and
Figure 10 present the fitting curves (solid line) and fitting results, respectively. The Rct for the baseline have a ~15% decrease compared to their initial values during the entire conditioning period. In comparison, the initial Rct for the 1 ppm-GDE was 180 mΩ cm
2 (~50%) higher than that of the baseline. The results implied the triazole additive bonding/poisoning with/of the Pt catalyst, which led to a significant increment in Rct at the beginning. Meanwhile, the Rct for 1 ppm-GDE exhibited a quick decrease from ~490 mΩ cm
2 to ~250 mΩ cm
2 in the first 4 h, accounting for more than 50%. The results indicate that the triazole additive bonded on Pt catalyst surface could be washed away either by humidified gas streams or the water generated from the electrochemical reaction [
25,
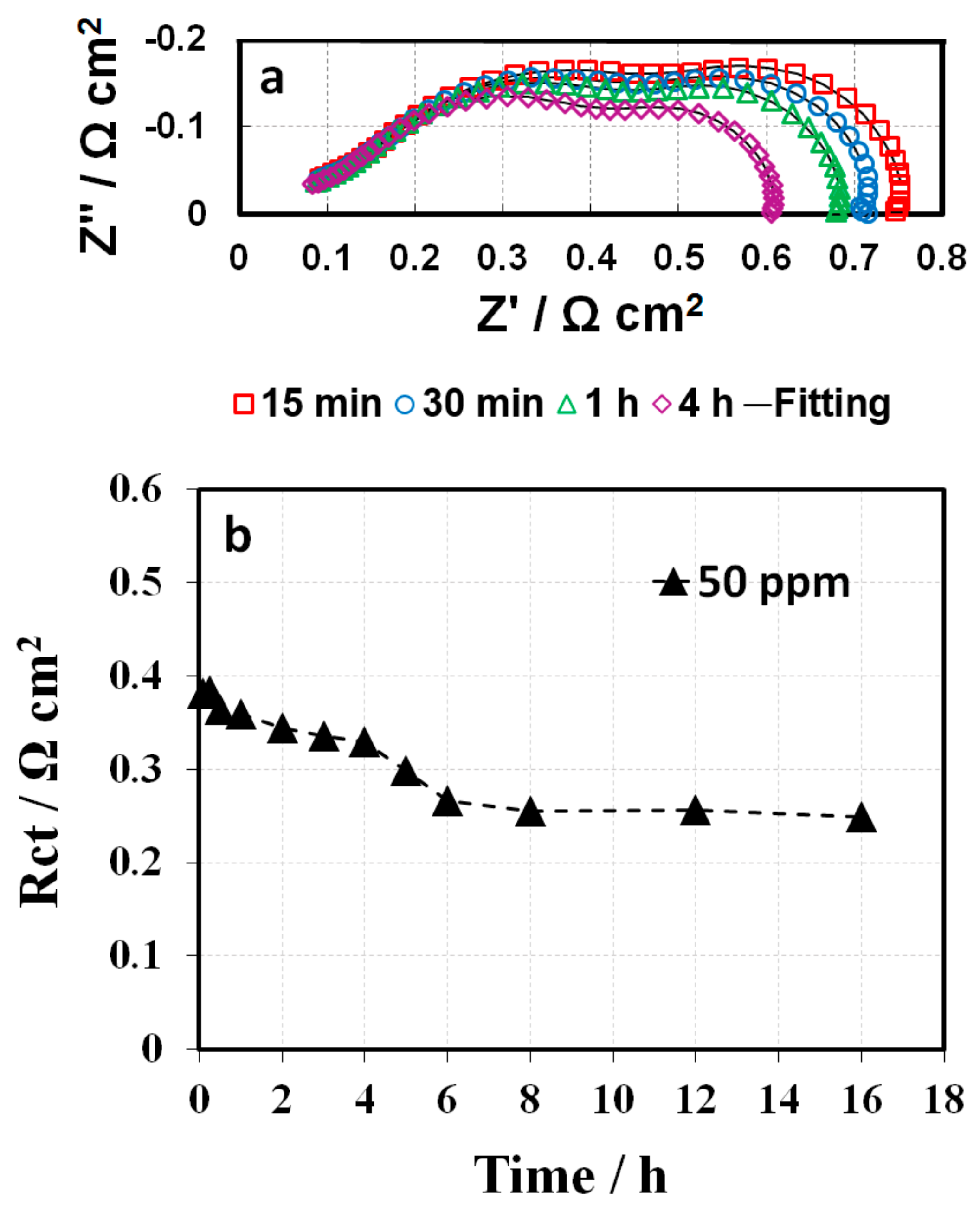
26]. The Rct gradually reduced and then nearly approached the baseline after a certain amount of additive was rinsed off from the catalyst surface by water. Alternatively, the 50 ppm-GDE sample needed to be conditioned in two consecutive steps, at a current density of 0.4 A cm
−2 for 16 h and then at a current density of 0.8 A cm
−2 for another 16 h, due to the triazole additive severe poisoning of the catalyst layer. As a result, Rct decreased gradually and reached the same value as the baseline (
Figure 11), which suggests that contaminant removal requires additional energy or extended conditioning time. Moreover, the inferior voltage compared to the baseline after conditioning suggests that the anode might be contaminated, or the additive remains in GDLs. Further study is ongoing to determine the hypothesis.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}