
Hierarchically Assembled Type I Collagen Fibres as Biomimetic Building Blocks of Biomedical Membranes



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Extraction of Type I Collagen
2.3. Sequential Functionalisation of Rat Tail Collagen
2.4. Chemical Characterisation of Photoactive Collagen Precursors
2.5. Fabrication of UV-Cured Hydrogels and Wet Spun Fibres
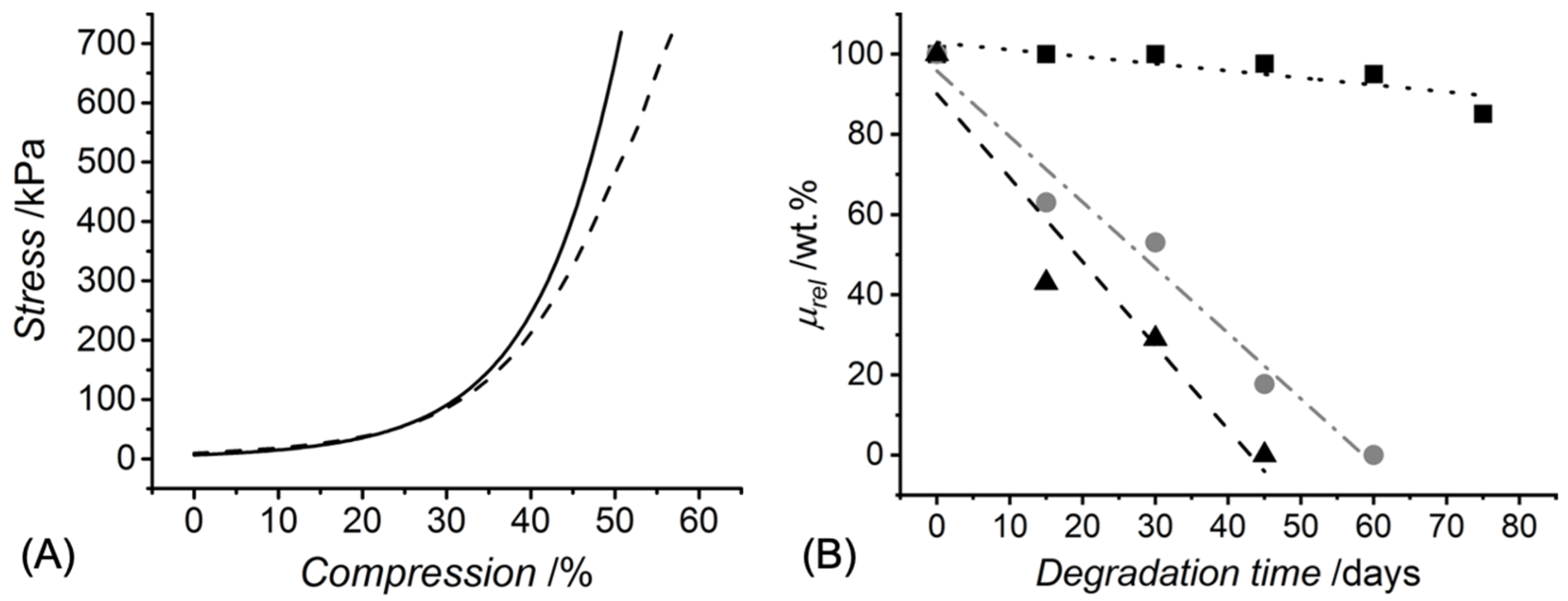
2.6. Hydrolytic Degradation Tests
2.7. Fibre Conditioning and Transmission Electron Microscopy (TEM)
2.8. Gel Content and Swelling Ratio Tests
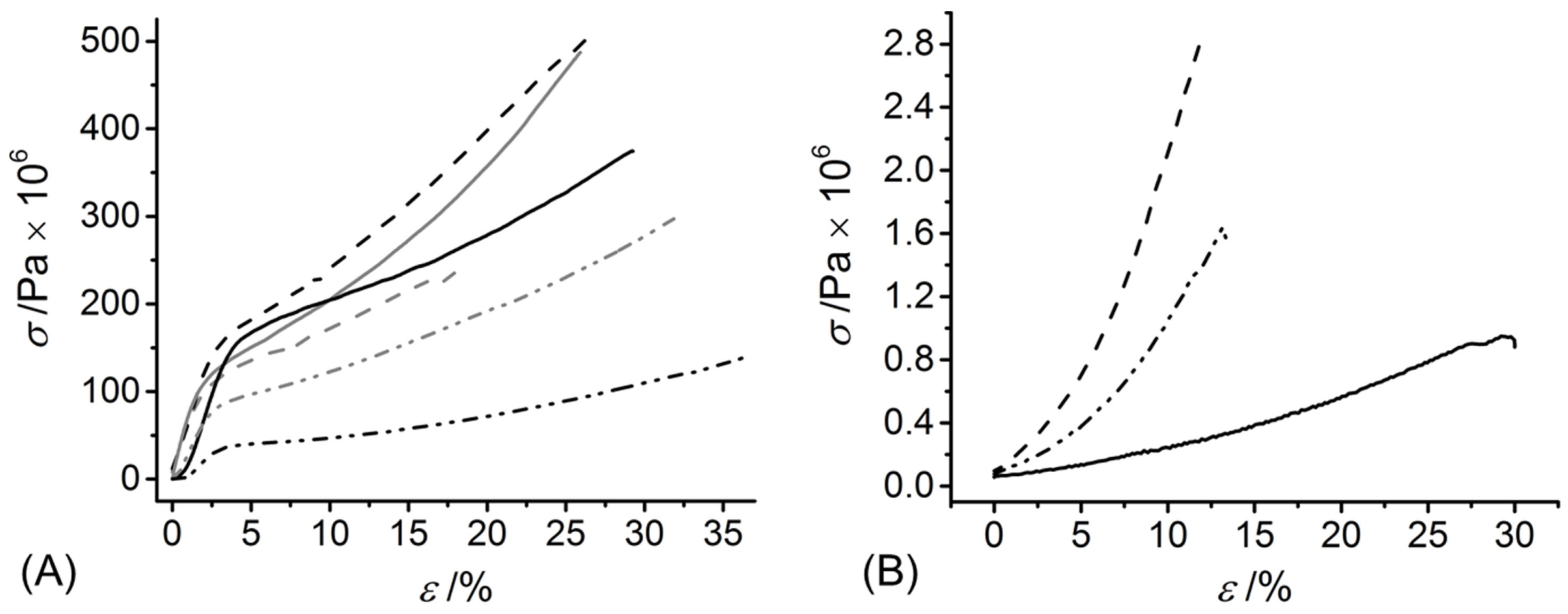
2.9. Mechanical Property Characterisation
2.10. Chemical, Thermal and Structural Analysis
2.11. Scanning Electron Microscopy
2.12. Extract Cytotoxicity Tests
3. Results and Discussion
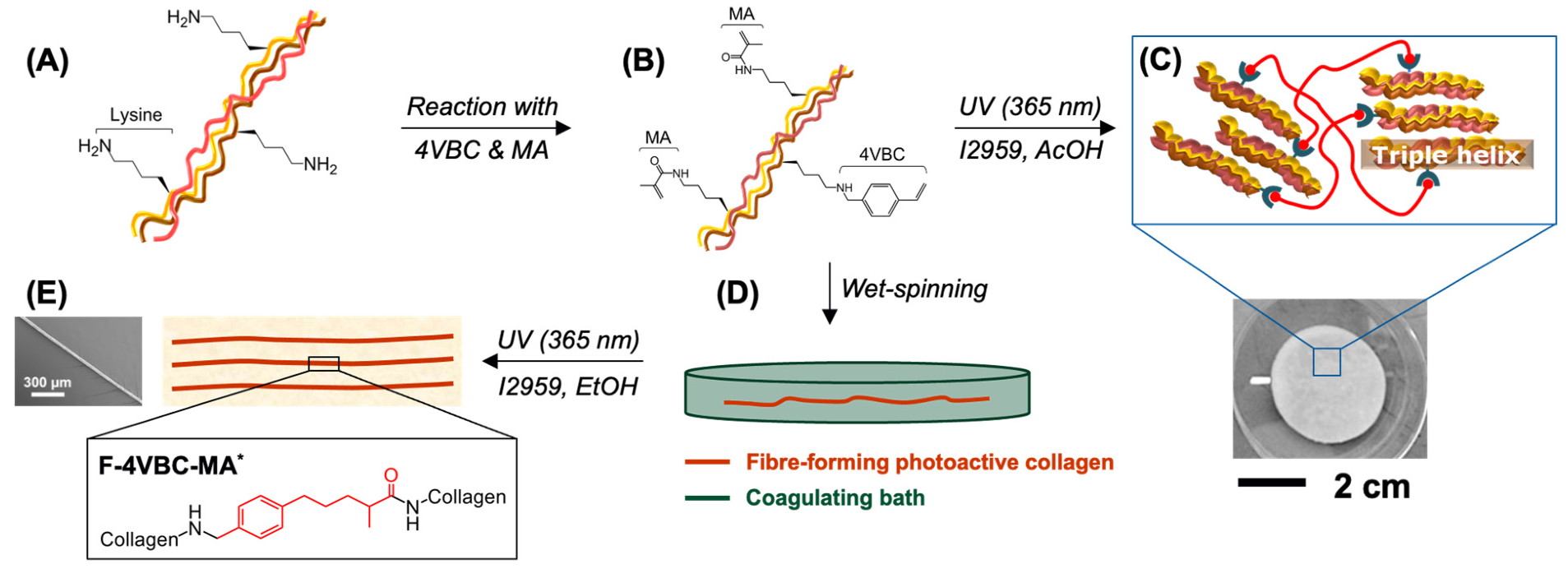
3.1. Synthesis of Photoactive Collagen Precursors
3.2. Macroscopic Characterisation of UV-Cured Hydrogels
3.3. Thermal Properties and Structural Analysis of UV-Cured Hydrogels
3.4. Fabrication of Wet Spun Fibres of Photoactive Collagen and UV-Cured Networks
3.5. Fibrillogenesis Study in Wet Spun Fibre
3.6. Cell Tolerability of Fibre Extracts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Naik, G.; Harding, K.G. Assessment of acceptability and ease of use of gelling fiber dressings in the management of heavily exuding wounds. Chronic Wound Care Manag. Res. 2019, 6, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Contreras, A.; Raxworthy, M.J.; Wood, S.; Schiffman, J.D.; Tronci, G. Photodynamically Active Electrospun Fibers for Antibiotic-Free Infection Control. ACS Appl. BioMater. 2019, 2, 4258–4270. [Google Scholar] [CrossRef]
- No, Y.J.; Castilho, M.; Ramaswamy, Y.; Zreiqat, H. Role of Biomaterials and Controlled Architecture on Tendon/Ligament Repair and Regeneration. Adv. Mater. 2019, 32, e1904511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Du, L.; Zhao, J.; Ding, J.; Zhang, P.; Wang, L.; Xu, B. Biomimetic angle-ply multi-lamellar scaffold for annulus fibrosus tissue engineering. J. Mater. Sci. Mater. Med. 2020, 31, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Tamayol, A.; Bagherifard, S.; Serex, L.; Mostafalu, P.; Faramarzi, N.; Mohammadi, M.H.; Khademhosseini, A. Textile Technologies and Tissue Engineering: A Path Toward Organ Weaving. Adv. Health Mater. 2016, 5, 751–766. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Li, F.; Zhao, X.; Ma, Y.; Li, Y.; Lin, M.; Jin, G.; Lu, T.J.; Genin, G.M.; Xu, F. Functional and Biomimetic Materials for Engineering of the Three-Dimensional Cell Microenvironment. Chem. Rev. 2017, 117, 12764–12850. [Google Scholar] [CrossRef] [PubMed]
- Tronci, G.; Kanuparti, R.S.; Arafat, M.T.; Yin, J.; Wood, D.J.; Russell, S.J. Wet-spinnability and crosslinked fibre properties of two collagen polypeptides with varied molecular weight. Int. J. Biol. Macromol. 2015, 81, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynl, C.; Hofmann, E.; Pawar, K.; Förster, S.; Scheibel, T. Microfluidics-Produced Collagen Fibers Show Extraordinary Mechanical Properties. Nano Lett. 2016, 16, 5917–5922. [Google Scholar] [CrossRef] [PubMed]
- Malladi, S.; Miranda-Nieves, D.; Leng, L.; Grainger, S.J.; Tarabanis, C.; Nesmith, A.P.; Kosaraju, R.; Haller, C.A.; Parker, K.K.; Chaikof, E.L.; et al. Continuous Formation of Ultrathin, Strong Collagen Sheets with Tunable Anisotropy and Compaction. ACS Biomater. Sci. Eng. 2020, 6, 4236–4246. [Google Scholar] [CrossRef]
- Bazbouz, M.B.; Liang, H.; Tronci, G. A UV-cured nanofibrous membrane of vinylbenzylated gelatin-poly(ε-caprolactone) dimethacrylate co-network by scalable free surface electrospinning. Mater. Sci. Eng. C 2018, 91, 541–555. [Google Scholar] [CrossRef]
- Wang, X.; Ronsin, O.; Gravez, B.; Farman, N.; Baumberger, T.; Jaisser, F.; Coradin, T.; Hélary, C. Nanostructured Dense Collagen-Polyester Composite Hydrogels as Amphiphilic Platforms for Drug Delivery. Adv. Sci. 2021, 8, 2004213. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.; Kirk, S.; Tronci, G.; Yang, X.; Wood, D. Influence of telopeptides on the structural and physical properties of polymeric and monomeric acid-soluble type I collagen. Mater. Sci. Eng. C 2017, 77, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Bürck, J.; Aras, O.; Bertinetti, L.; Ilhan, C.A.; Ermeydan, M.A.; Schneider, R.; Ulrich, A.S.; Kazanci, M. Observation of triple helix motif on electrospun collagen nanofibersand its effect on the physical and structural properties. J. Mol. Struct. 2018, 1151, 73–80. [Google Scholar] [CrossRef]
- Sorushanova, A.; Delgado, L.M.; Wu, Z.; Shologu, N.; Kshirsagar, A.; Raghunath, R.; Mullen, A.M.; Bayon, Y.; Pandit, A.; Raghunath, M.; et al. The Collagen Suprafamily: From Biosynthesis to Advanced Biomaterial Development. Adv. Mater. 2018, 31, e1801651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djabourov, M.; Leblond, J.; Papon, P. Gelation of aqueous gelatin solutions. I. Structural investigation. J. Phys. 1988, 49, 319–332. [Google Scholar] [CrossRef]
- Tronci, G. The application of collagen in advanced wound dressings. In Advanced Textiles for Wound Care, 2nd ed.; Rajendran, S., Ed.; Elsevier Ltd.: London, UK, 2019; pp. 363–389. [Google Scholar]
- Green, E.C.; Zhang, Y.; Li, H.; Minus, M.L. Gel-spinning of mimetic collagen and collagen/nano-carbon fibers: Understanding multi-scale influences on molecular ordering and fibril alignment. J. Mech. Behav. Biomed. Mater. 2017, 65, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.M.; AbuSamra, D.B.; Chivu, A.; Argüeso, P.; Dohlman, C.H.; Patra, H.K.; Chodosh, J.; González-Andrades, M. Optimization of Collagen Chemical Crosslinking to Restore Biocompatibility of Tissue-Engineered Scaffolds. Pharmaceutics 2021, 13, 832. [Google Scholar] [CrossRef]
- Bazrafshan, Z.; Stylios, G.K. Spinnability of collagen as a biomimetic material: A review. Int. J. Biol. Macromol. 2019, 129, 693–705. [Google Scholar] [CrossRef]
- Tronci, G.; Doyle, A.; Russell, S.J.; Wood, D.J. Triple-helical collagen hydrogels via covalent aromatic functionalisation with 1,3-phenylenediacetic acid. J. Mater. Chem. B 2013, 1, 5478–5488. [Google Scholar] [CrossRef]
- Arafat, M.T.; Tronci, G.; Yin, J.; Wood, D.J.; Russell, S.J. Biomimetic wet-stable fibres via wet spinning and diacid-based crosslinking of collagen triple helices. Polymer 2015, 77, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Tonndorf, R.; Aibibu, D.; Cherif, C. Collagen multifilament spinning. Mater. Sci. Eng. C 2019, 106, 110105. [Google Scholar] [CrossRef]
- Tonndorf, R.; Gossla, E.; Aibibu, D.; Lindner, M.; Gelinsky, M.; Cherif, C. Corrigendum: Wet spinning and riboflavin crosslinking of collagen type I/III filaments (2019 Biomed. Mater. 14 015007). Biomed. Mater. 2019, 14, 039501. [Google Scholar] [CrossRef]
- Caves, J.M.; Kumar, V.A.; Wen, J.; Cui, W.; Martinez, A.; Apkarian, R.; Coats, J.E.; Berland, K.; Chaikof, E.L. Fibrillogenesis in continuously spun synthetic collagen fiber. J. Biomed. Mater. Res. Part B Appl. Biomater. 2009, 93, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pins, G.; Christiansen, D.; Patel, R.; Silver, F. Self-assembly of collagen fibers. Influence of fibrillar alignment and decorin on mechanical properties. Biophys. J. 1997, 73, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- Pins, G.D.; Huang, E.K.; Christiansen, D.L.; Silver, F.H. Effects of static axial strain on the tensile properties and failure mechanisms of self-assembled collagen fibers. J. Appl. Polym. Sci. 1997, 63, 1429–1440. [Google Scholar] [CrossRef]
- Tronci, G.; Russell, S.J.; Wood, D.J. Photo-active collagen systems with controlled triple helix architecture. J. Mater. Chem. B 2013, 1, 3705–3715. [Google Scholar] [CrossRef] [Green Version]
- Tronci, G.; Grant, C.; Thomson, N.; Russell, S.J.; Wood, D.J. Multi-scale mechanical characterization of highly swollen photo-activated collagen hydrogels. J. R. Soc. Interface 2015, 12, 20141079. [Google Scholar] [CrossRef]
- Liang, H.; Russell, S.J.; Wood, D.J.; Tronci, G. Monomer-Induced Customization of UV-Cured Atelocollagen Hydrogel Networks. Front. Chem. 2018, 6, 626. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Russell, S.J.; Wood, D.J.; Tronci, G. A hydroxamic acid-methacrylated collagen conjugate for the modulation of inflammation-related MMP upregulation. J. Mater. Chem. B 2018, 6, 3703–3715. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Yang, X.; Jones, E.; Bingham, P.A.; Scrimshire, A.; Thornton, P.D.; Tronci, G. An injectable, self-healing and MMP-inhibiting hyaluronic acid gel via iron coordination. Int. J. Biol. Macromol. 2020, 165, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Tronci, G.; Yin, J.; Holmes, R.A.; Liang, H.; Russell, S.J.; Wood, D.J. Protease-sensitive atelocollagen hydrogels promote healing in a diabetic wound model. J. Mater. Chem. B 2016, 4, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Van Bochove, B.; Grijpma, D.W. Photo-crosslinked synthetic biodegradable polymer networks for biomedical applications. J. Biomater. Sci. Polym. Ed. 2019, 30, 77–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neffe, A.T.; Tronci, G.; Alteheld, A.; Lendlein, A. Controlled Change of Mechanical Properties during Hydrolytic Degradation of Polyester Urethane Networks. Macromol. Chem. Phys. 2010, 211, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Bazrafshan, Z.; Sylios, G.K. A novel approach to enhance the spinnability of collagen fibers by graft polymeryzation. Mater. Sci. Eng. C 2019, 94, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Du, J.; Cao, Y.; Yue, C.; Cheng, B. Effects of the aspect ratio of multi-walled carbon nanotubes on the structure and properties of regenerated collagen fibers. Int. J. Biol. Macromol. 2018, 126, 595–602. [Google Scholar] [CrossRef]
- Salim, N.V.; Jin, X.; Mateti, S.; Lin, H.; Glattauer, V.; Fox, B.; Ramshaw, J.A.M. Porous carbon fibers made from collagen derived fron an animal by-product. Mater. Today Adv. 2019, 1, 100005. [Google Scholar] [CrossRef]
- Lu, T.; Hu, H.; Li, Y.; Jiang, Q.; Su, J.; Lin, H.; Xiao, Y.; Zhu, X.; Zhang, X. Bioactive scaffolds based on collagen filaments with tunable physico-chemical and biological features. Soft Matter 2020, 16, 4540–4548. [Google Scholar] [CrossRef]
- Yaari, A.; Schilt, Y.; Tamburu, C.; Raviv, U.; Shoseyov, O. Wet Spinning and Drawing of Human Recombinant Collagen. ACS Biomater. Sci. Eng. 2016, 2, 349–360. [Google Scholar] [CrossRef]
- Zeugolis, D.I.; Paul, R.G.; Attenburrow, G. Extruded Collagen-Polyethylene Glycol Fibers for Tissue Engineering Applications. J. Biomed. Mater. Res. Part B Appl. Biomater. 2008, 85, 343–352. [Google Scholar] [CrossRef]
- Calejo, I.; Costa-Almeida, R.; Reis, R.L.; Gomes, M.E. A Textile platform using continuous aligned and textured composite microfibres to engineer tendon-to-bone interface gradient scaffolds. Adv. Health Mater. 2019, 8, 1900200. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Christiansen, D.L.; Hahn, R.A.; Shieh, S.-J.; Goldstein, J.D.; Silver, F.H. Mechanical properties of collagen fibres: A comparison of reconstituted and rat tail tendon fibres. Biomaterials 1989, 10, 38–42. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | F/mol.-% | c/wt.% | G/wt.-% | SR/wt.-% | Ec/kPa | σb/kPa | εb/% |
|---|---|---|---|---|---|---|---|
| 4VBC-MA* | 93 ± 1 | 0.8 | 90 | 900 ± 180 | 153 ± 85 | 112 ± 43 | 56 ± 4 |
| 0.4 | 67 | 1560 ± 100 | 292 ± 205 | 190 ± 75 | 62 ± 7 | ||
| 4VBC* | 45 | 0.8 | 100 | 1965 | 81 | n.a. | n.a. |
| GMA* | 65 | 0.8 | 100 | 1262 | 39 | n.a. | n.a. |
| Sample ID | Ø/μm | SR/wt.-% | σb/MPa | εb/% | E/MPa |
|---|---|---|---|---|---|
| F-C | 80 ± 3 | n.a. | 72 ± 3 | 24 ± 3 | 2189 ± 220 |
| F-GMA | 39 ± 10 | n.a. | 252 ± 40 | 26 ± 5 | 2010 ± 600 |
| F-GMA* | 40 ± 10 | 3130 ± 300 | 191 ± 50 | 22 ± 6 | 3210 ± 900 |
| 150 ± 20 (a) | 0.4 ± 0.1 (a) | 19 ± 5 (a) | 5 ± 1 (a) | ||
| F-4VBC | 54 ± 5 | n.a. | 252 ± 40 | 26 ± 5 | 3130 ± 600 |
| F-4VBC* | 51 ± 10 | 3350 ± 500 | 338 ± 50 | 26 ± 5 | 4340 ± 500 |
| 140 ± 30 (a) | 1.6 ± 0.5 (a) | 13 ± 3 (a) | 9 ± 1 (a) | ||
| F-4VBC-MA | 51 ± 5 | n.a. | 504 ± 30 | 29 ± 4 | 4190 ± 500 |
| F-4VBC-MA* | 49 ± 10 | 1880 ± 200 | 542 ± 80 | 30 ± 6 | 5160 ± 400 |
| 130 ± 30 (a) | 2.6 ± 0.4 (a) | 15 ± 4 (a) | 11 ± 4 (a) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, J.; Wood, D.J.; Russell, S.J.; Tronci, G. Hierarchically Assembled Type I Collagen Fibres as Biomimetic Building Blocks of Biomedical Membranes. Membranes 2021, 11, 620. https://doi.org/10.3390/membranes11080620
Yin J, Wood DJ, Russell SJ, Tronci G. Hierarchically Assembled Type I Collagen Fibres as Biomimetic Building Blocks of Biomedical Membranes. Membranes. 2021; 11(8):620. https://doi.org/10.3390/membranes11080620
Chicago/Turabian StyleYin, Jie, David J. Wood, Stephen J. Russell, and Giuseppe Tronci. 2021. "Hierarchically Assembled Type I Collagen Fibres as Biomimetic Building Blocks of Biomedical Membranes" Membranes 11, no. 8: 620. https://doi.org/10.3390/membranes11080620
APA StyleYin, J., Wood, D. J., Russell, S. J., & Tronci, G. (2021). Hierarchically Assembled Type I Collagen Fibres as Biomimetic Building Blocks of Biomedical Membranes. Membranes, 11(8), 620. https://doi.org/10.3390/membranes11080620