

Polystyrene Sulfonate Particles as Building Blocks for Nanofiltration Membranes



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
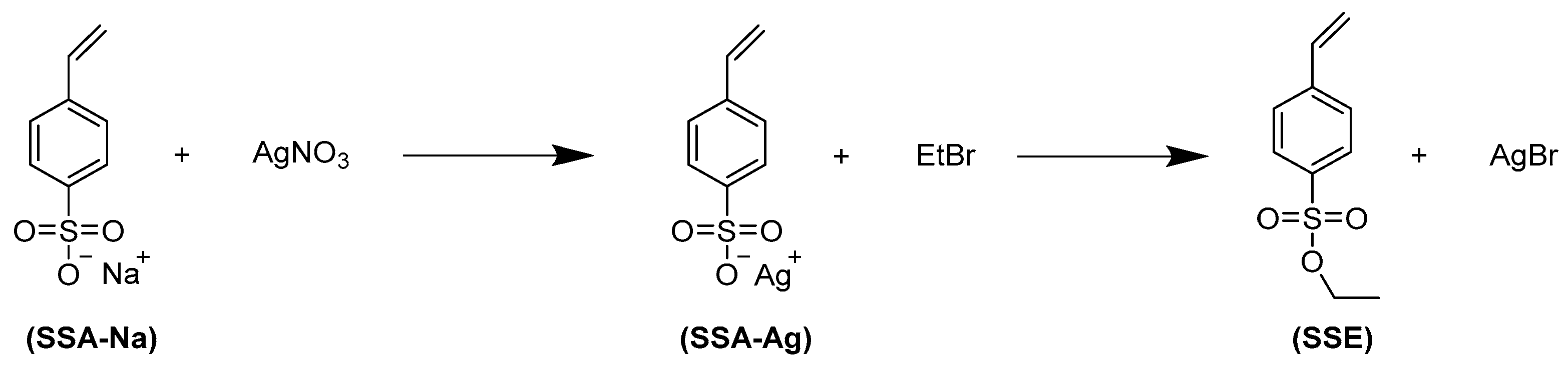
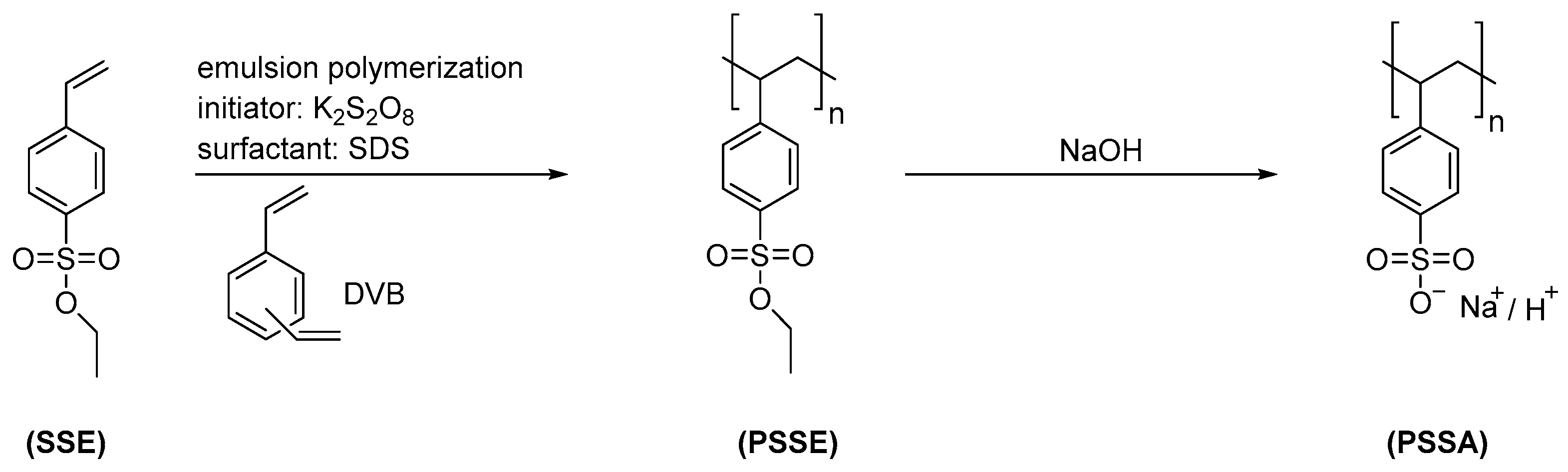
2.2. Preparation of Negatively Charged Polyelectrolyte Particles (Polystyrene Sulfonate)
2.3. Particle Characterization
2.3.1. Calculation of Charged Group Density
2.3.2. Zeta Potential and Particle Size
2.3.3. Scanning Electron Microscopy (SEM)
2.4. Membrane Fabrication
2.5. Membrane Testing and Characterization
2.5.1. Water Permeance and Single Salt Rejection
2.5.2. Zeta Potential
2.5.3. ATR-IR Spectroscopy
3. Results and Discussion
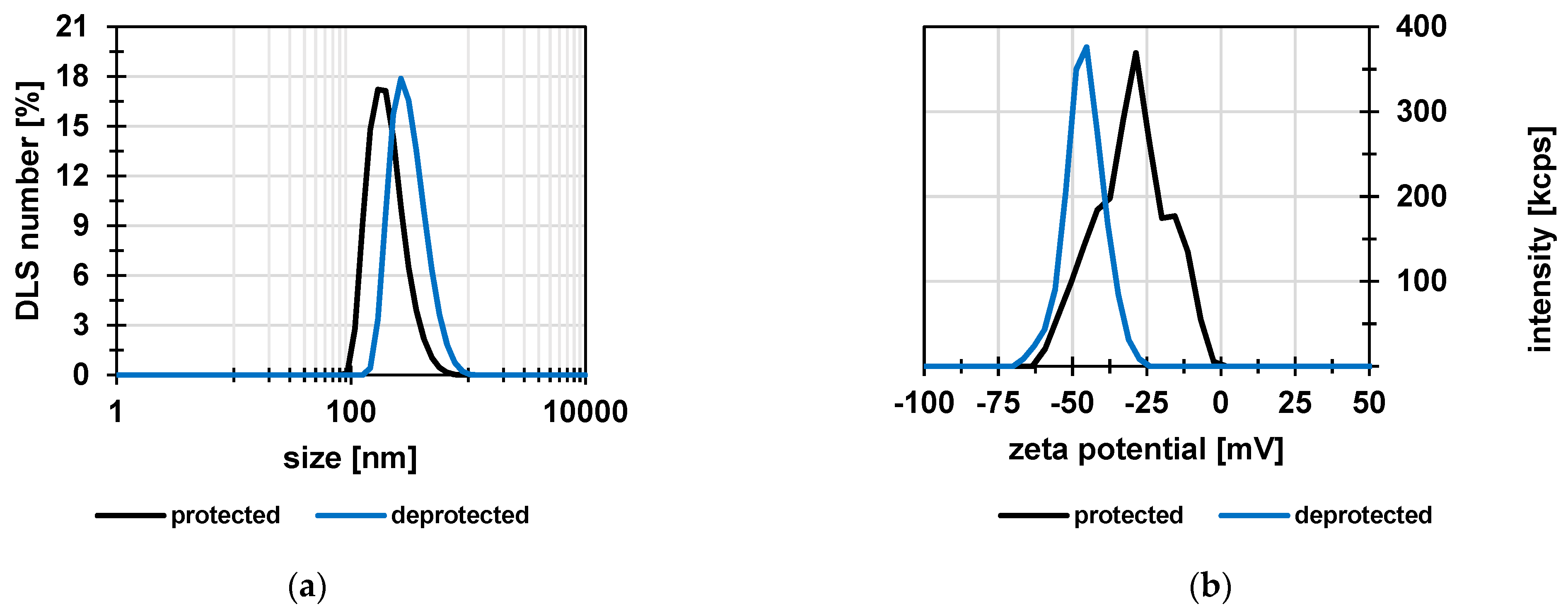
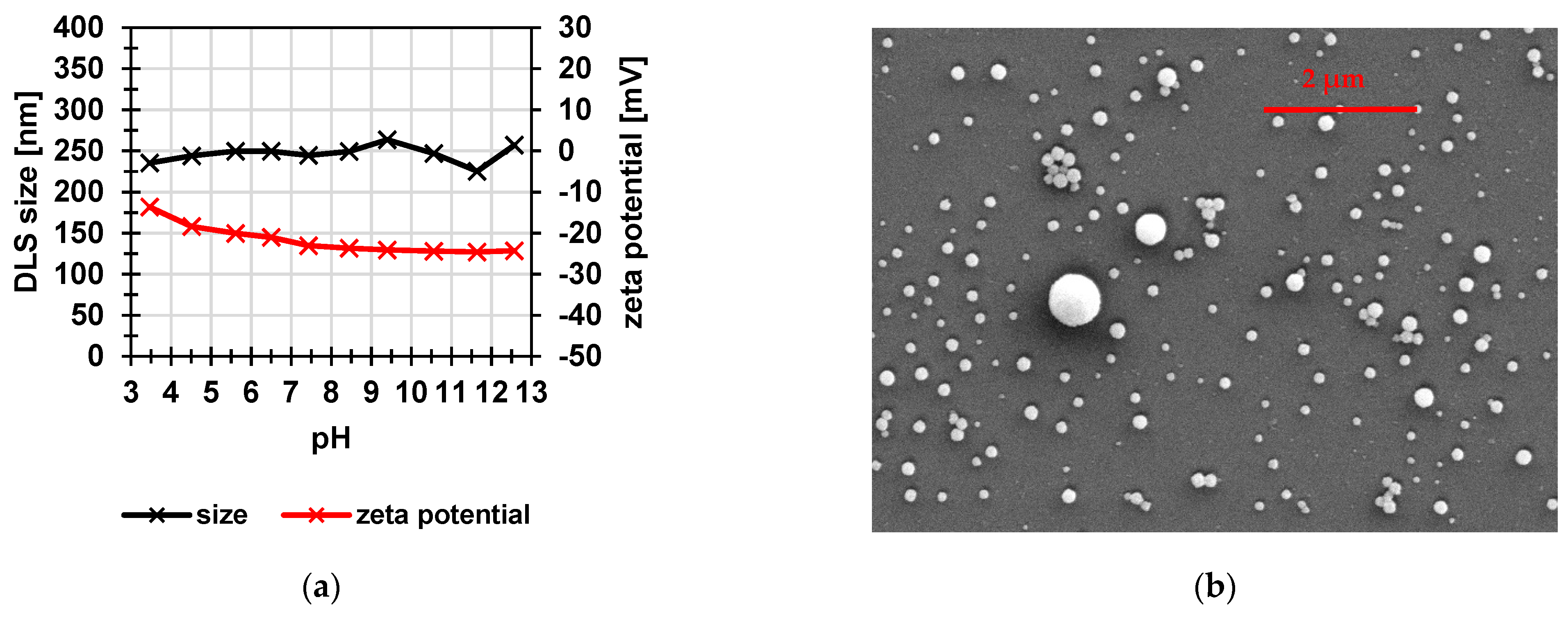
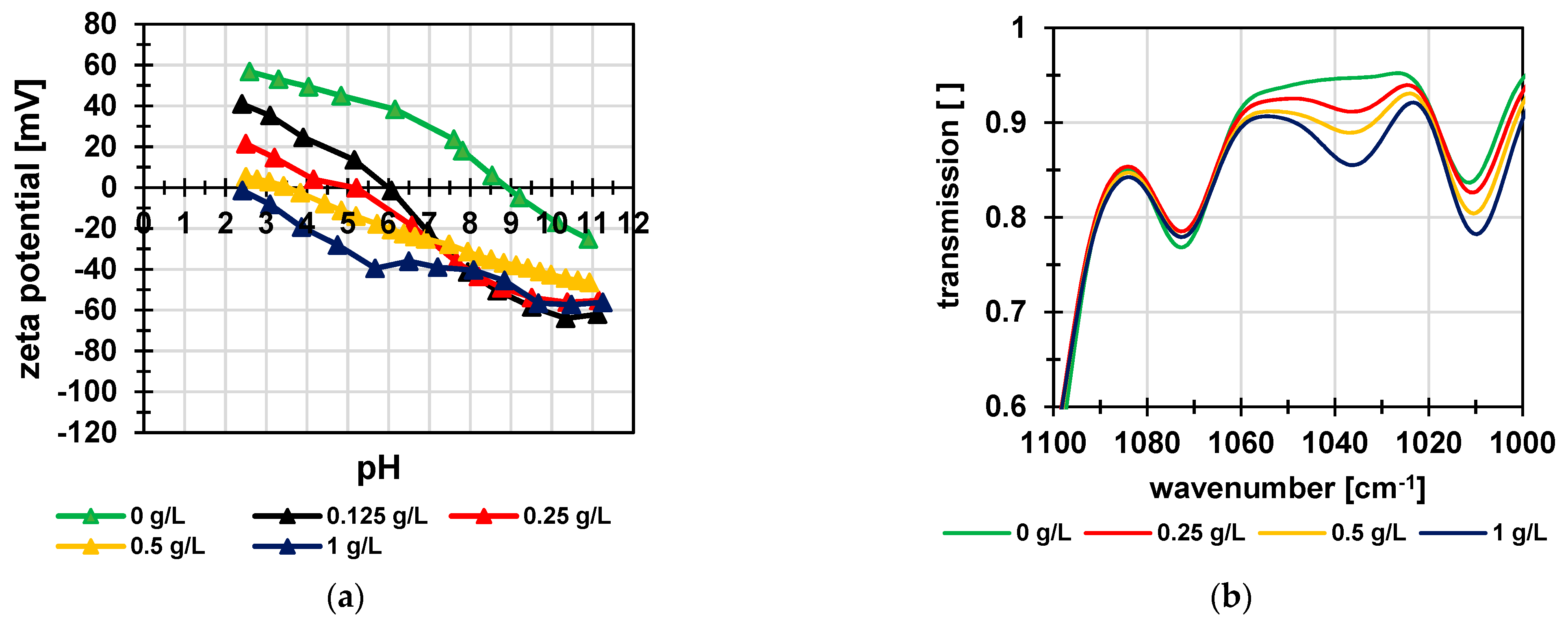
3.1. Poly(Styrene Sulfonic Acid) Particles
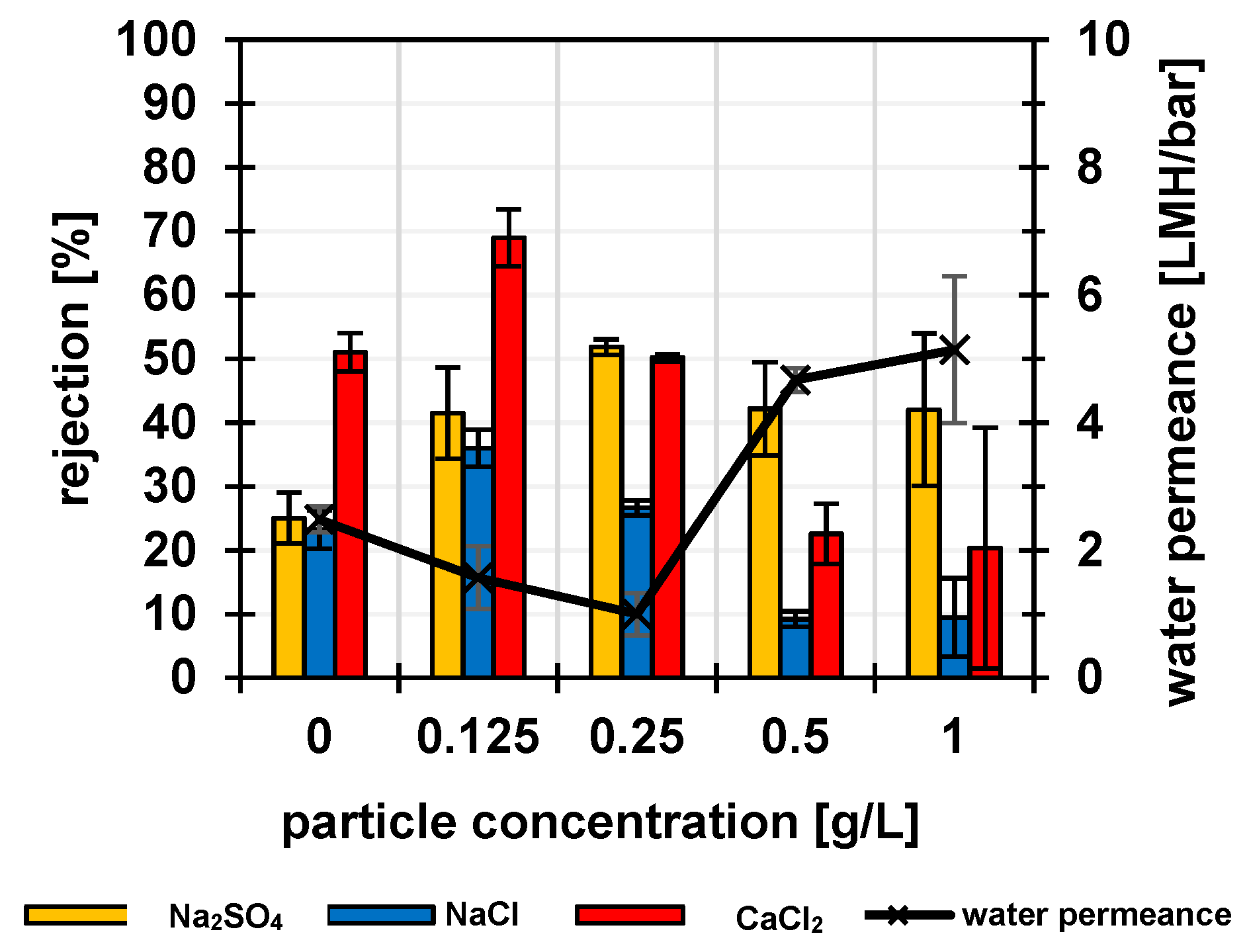
3.2. Membrane Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, H.; He, Q.; Luo, J.; Wan, Y.; Darling, S.B. Sharpening Nanofiltration: Strategies for Enhanced Membrane Selectivity. ACS Appl. Mater. Interfaces 2020, 12, 39948–39966. [Google Scholar] [CrossRef]
- Freger, V.; Ramon, G.Z. Polyamide desalination membranes: Formation, structure, and properties. Prog. Polym. Sci. 2021, 122, 101451. [Google Scholar] [CrossRef]
- Ji, Y.-L.; Gu, B.-X.; An, Q.-F.; Gao, C.-J. Recent Advances in the Fabrication of Membranes Containing “Ion Pairs” for Nanofiltration Processes. Polymers 2017, 9, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durmaz, E.N.; Sahin, S.; Virga, E.; de Beer, S.; de Smet, L.C.P.M.; de Vos, W.M. Polyelectrolytes as Building Blocks for Next-Generation Membranes with Advanced Functionalities. ACS Appl. Polym. Mater. 2021, 3, 4347–4374. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.; Ahmadiannamini, P.; Hoogenboom, R.; Vankelecom, I.F.J. Layer-by-layer preparation of polyelectrolyte multilayer membranes for separation. Polym. Chem. 2014, 5, 1817–1831. [Google Scholar] [CrossRef]
- Lee, D.; Rubner, M.F.; Cohen, R.E. All-Nanoparticle Thin-Film Coatings. Nano Lett. 2006, 6, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Ghostine, R.A.; Markarian, M.Z.; Schlenoff, J.B. Asymmetric Growth in Polyelectrolyte Multilayers. J. Am. Chem. Soc. 2013, 135, 7636–7646. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.; Ahmadiannamini, P.; Jishna, P.S.; Volodin, A.; Vankelecom, I.F. ‘Up-scaling’ potential for polyelectrolyte multilayer membranes. J. Membr. Sci. 2015, 492, 271–280. [Google Scholar] [CrossRef]
- Meyer, K.H.; Sievers, J.-F. La perméabilité des membranes I. Théorie de la perméabilité ionique. Helv. Chim. Acta 1936, 19, 649–664. [Google Scholar] [CrossRef]
- Meyer, K.H.; Sievers, J.-F. La perméabilité des membranes. II. Essais avec des membranes sélectives artificielles. Helv. Chim. Acta 1936, 19, 665–677. [Google Scholar] [CrossRef]
- Fievet, P. SEDE (Steric, Electric, and Dielectric Exclusion) Model: Approximated Versions. In Encyclopedia of Membranes; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1755–1758. [Google Scholar]
- Levchenko, S.; Freger, V. Breaking the Symmetry: Mitigating Scaling in Tertiary Treatment of Waste Effluents Using a Positively Charged Nanofiltration Membrane. Environ. Sci. Technol. Lett. 2016, 3, 339–343. [Google Scholar] [CrossRef]
- Bernstein, R.; Antón, E.; Ulbricht, M. UV-Photo Graft Functionalization of Polyethersulfone Membrane with Strong Polyelectrolyte Hydrogel and Its Application for Nanofiltration. ACS Appl. Mater. Interfaces 2012, 4, 3438–3446. [Google Scholar] [CrossRef] [PubMed]
- Kaganovich, M.; Zhang, W.; Freger, V.; Bernstein, R. Effect of the membrane exclusion mechanism on phosphate scaling during synthetic effluent desalination. Water Res. 2019, 161, 381–391. [Google Scholar] [CrossRef]
- Sollner, K. Über Mosaikmembranen. Biochem. Z. 1932, 244, 370. [Google Scholar]
- Linder, C.; Kedem, O. Asymmetric ion exchange mosaic membranes with unique selectivity. J. Membr. Sci. 2001, 181, 39–56. [Google Scholar] [CrossRef]
- Zelner, M.; Jahn, P.; Ulbricht, M.; Freger, V. A mixed-charge polyelectrolyte complex nanofiltration membrane: Preparation, performance and stability. J. Membr. Sci. 2021, 636, 119579. [Google Scholar] [CrossRef]
- Nunes, S.P.; Culfaz-Emecen, P.Z.; Ramon, G.Z.; Visser, T.; Koops, G.H.; Jin, W.; Ulbricht, M. Thinking the future of membranes: Perspectives for advanced and new membrane materials and manufacturing processes. J. Membr. Sci. 2020, 598, 117761. [Google Scholar] [CrossRef]
- Bassyouni, M.; Abdel-Aziz, M.H.; Zoromba, M.S.; Abdel-Hamid, S.M.S.; Drioli, E. A review of polymeric nanocomposite membranes for water purification. J. Ind. Eng. Chem. 2019, 73, 19–46. [Google Scholar] [CrossRef]
- Ibrahim, G.P.S.; Isloor, A.M.; Bavarian, M.; Nejati, S. Integration of Zwitterionic Polymer Nanoparticles in Interfacial Polymerization for Ion Separation. ACS Appl. Polym. Mater. 2020, 2, 1508–1517. [Google Scholar] [CrossRef]
- Tiwari, R.; Walther, A. Strong anionic polyelectrolyte microgels. Polym. Chem. 2015, 6, 5550–5554. [Google Scholar] [CrossRef]
- Woeste, G.; Meyer, W.H.; Wegner, G. Copolymers of ethyl p-vinylbenzenesulfonate for the preparation of polyelectrolytes of reproducible ion content. Die Makromol. Chem. 1993, 194, 1237–1248. [Google Scholar] [CrossRef]
- Emmons, W.D.; Ferris, A.F. Metathetical Reactions of Silver Salts in Solution. II. The Synthesis of Alkyl Sulfonates1. J. Am. Chem. Soc. 1953, 75, 2257. [Google Scholar] [CrossRef]
- Chern, C. Emulsion polymerization mechanisms and kinetics. Prog. Polym. Sci. 2006, 31, 443–486. [Google Scholar] [CrossRef]
- Priest, W.J. Partice Growth in the Aqueous Polymerization of Vinyl Acetate. J. Phys. Chem. 1952, 56, 1077–1082. [Google Scholar] [CrossRef]
- Ghiasi, S.; Behboudi, A.; Mohammadi, T.; Ulbricht, M. High-performance positively charged hollow fiber nanofiltration membranes fabricated via green approach towards polyethyleneimine layer assembly. Sep. Purif. Technol. 2020, 251, 117313. [Google Scholar] [CrossRef]
- Schlenoff, J.B.; Yang, M.; Digby, Z.A.; Wang, Q. Ion Content of Polyelectrolyte Complex Coacervates and the Donnan Equilibrium. Macromolecules 2019, 52, 9149–9159. [Google Scholar] [CrossRef]
- Yang, M.; Shi, J.; Schlenoff, J.B. Control of Dynamics in Polyelectrolyte Complexes by Temperature and Salt. Macromolecules 2019, 52, 1930–1941. [Google Scholar] [CrossRef]
- Zelner, M.; Stolov, M.; Tendler, T.; Jahn, P.; Ulbricht, M.; Freger, V. Elucidating ion transport mechanism in polyelectrolyte-complex membranes. J. Membr. Sci. 2022, 658, 120757. [Google Scholar] [CrossRef]
- Fan, H.; Huang, Y.; Billinge, I.H.; Bannon, S.M.; Geise, G.M.; Yip, N.Y. Counterion Mobility in Ion-Exchange Membranes: Spatial Effect and Valency-Dependent Electrostatic Interaction. ACS ES&T Eng. 2022, 2, 1274–1286. [Google Scholar] [CrossRef]
- Freger, V. Ion partitioning and permeation in charged low-T* membranes. Adv. Colloid Interface Sci. 2020, 277, 102107. [Google Scholar] [CrossRef]
- van der Poel, S. Parting Ways—Removal of Salts and Organic Micropollutants by Direct Nanofiltration: Pretreatment of Surface Water for the Production of Dune Infiltration Water. 2020. Available online: https://repository.tudelft.nl/islandora/object/uuid%3A6774b91c-6850-4c82-b3c0-a3110f0c40b9 (accessed on 4 August 2022).
- Han, Y.; Jiang, Y.; Gao, C. High-Flux Graphene Oxide Nanofiltration Membrane Intercalated by Carbon Nanotubes. ACS Appl. Mater. Interfaces 2015, 7, 8147–8155. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shi, L.; Wang, R. Crosslinked layer-by-layer polyelectrolyte nanofiltration hollow fiber membrane for low-pressure water softening with the presence of SO42− in feed water. J. Membr. Sci. 2015, 486, 169–176. [Google Scholar] [CrossRef]
- de Grooth, J.; Oborný, R.; Potreck, J.; Nijmeijer, K.; de Vos, W.M. The role of ionic strength and odd–even effects on the properties of polyelectrolyte multilayer nanofiltration membranes. J. Membr. Sci. 2015, 475, 311–319. [Google Scholar] [CrossRef]
- Reurink, D.M.; Willott, J.D.; Roesink, H.D.W.; de Vos, W.M. Role of Polycation and Cross-Linking in Polyelectrolyte Multilayer Membranes. ACS Appl. Polym. Mater. 2020, 2, 5278–5289. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, G.Q.; Yang, X.; Deng, H. Preparation of Layer-by-Layer Nanofiltration Membranes by Dynamic Deposition and Crosslinking. Membranes 2019, 9, 20. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Size (nm) | PDI | S (wt%) | Functional/Charged Group Density, Experimental (mmol/g) |
|---|---|---|---|---|
| Protected | 207 ± 12 | 0.14 ± 0.01 | 14.9 ± 0.1 | 4.7 |
| Deprotected | 344 ± 40 | 0.14 ± 0.06 | 13.6 ± 0.1 | 4.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahn, P.; Zelner, M.; Freger, V.; Ulbricht, M. Polystyrene Sulfonate Particles as Building Blocks for Nanofiltration Membranes. Membranes 2022, 12, 1138. https://doi.org/10.3390/membranes12111138
Jahn P, Zelner M, Freger V, Ulbricht M. Polystyrene Sulfonate Particles as Building Blocks for Nanofiltration Membranes. Membranes. 2022; 12(11):1138. https://doi.org/10.3390/membranes12111138
Chicago/Turabian StyleJahn, Philipp, Michael Zelner, Viatcheslav Freger, and Mathias Ulbricht. 2022. "Polystyrene Sulfonate Particles as Building Blocks for Nanofiltration Membranes" Membranes 12, no. 11: 1138. https://doi.org/10.3390/membranes12111138
APA StyleJahn, P., Zelner, M., Freger, V., & Ulbricht, M. (2022). Polystyrene Sulfonate Particles as Building Blocks for Nanofiltration Membranes. Membranes, 12(11), 1138. https://doi.org/10.3390/membranes12111138