

Influenza A Virus M1 Protein Non-Specifically Deforms Charged Lipid Membranes and Specifically Interacts with the Raft Boundary

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of the M1 Protein
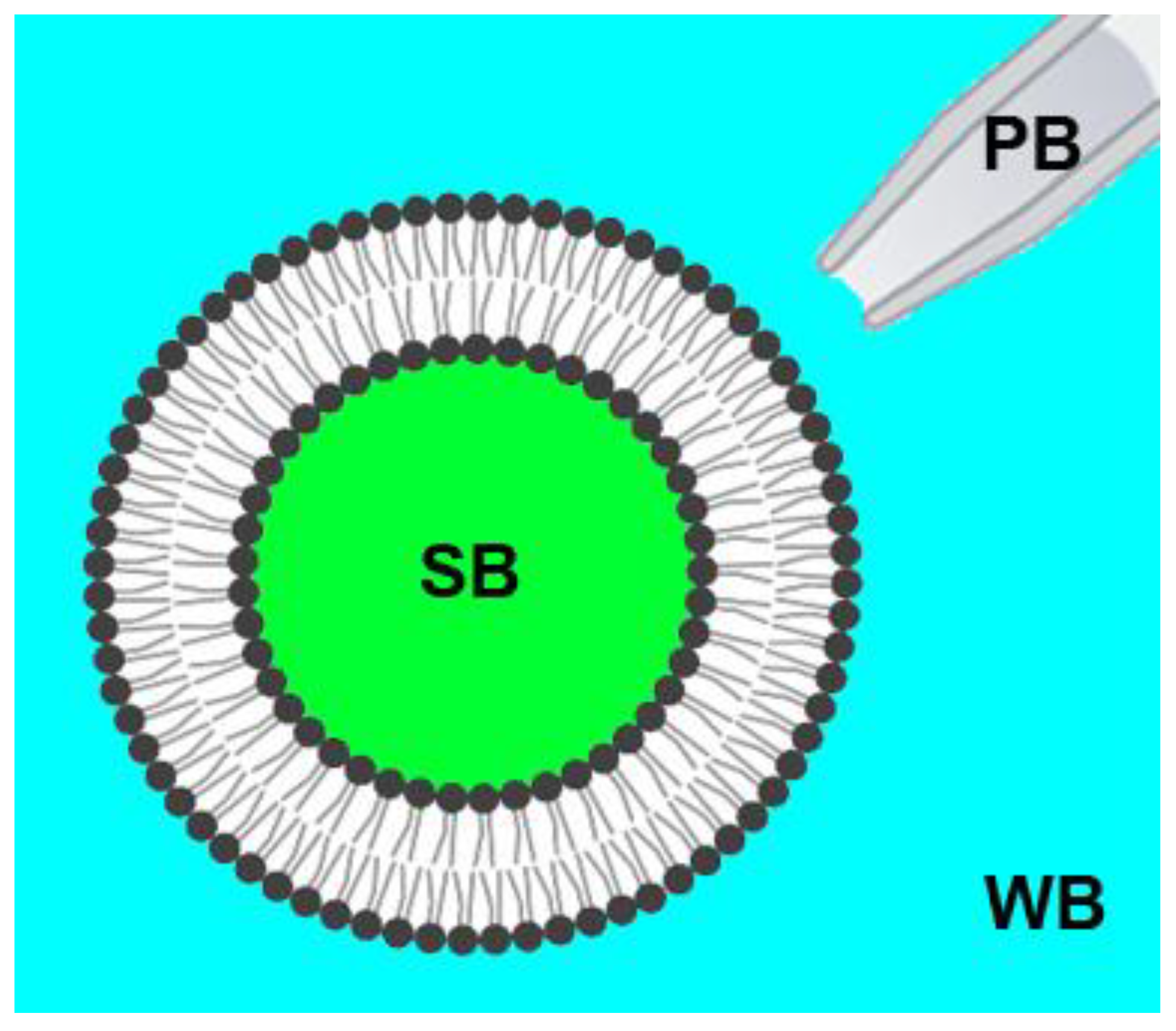
2.2. Preparation of Giant Unilamellar Vesicles (GUVs)
2.3. Confocal Fluorescence Microscopy
3. Results
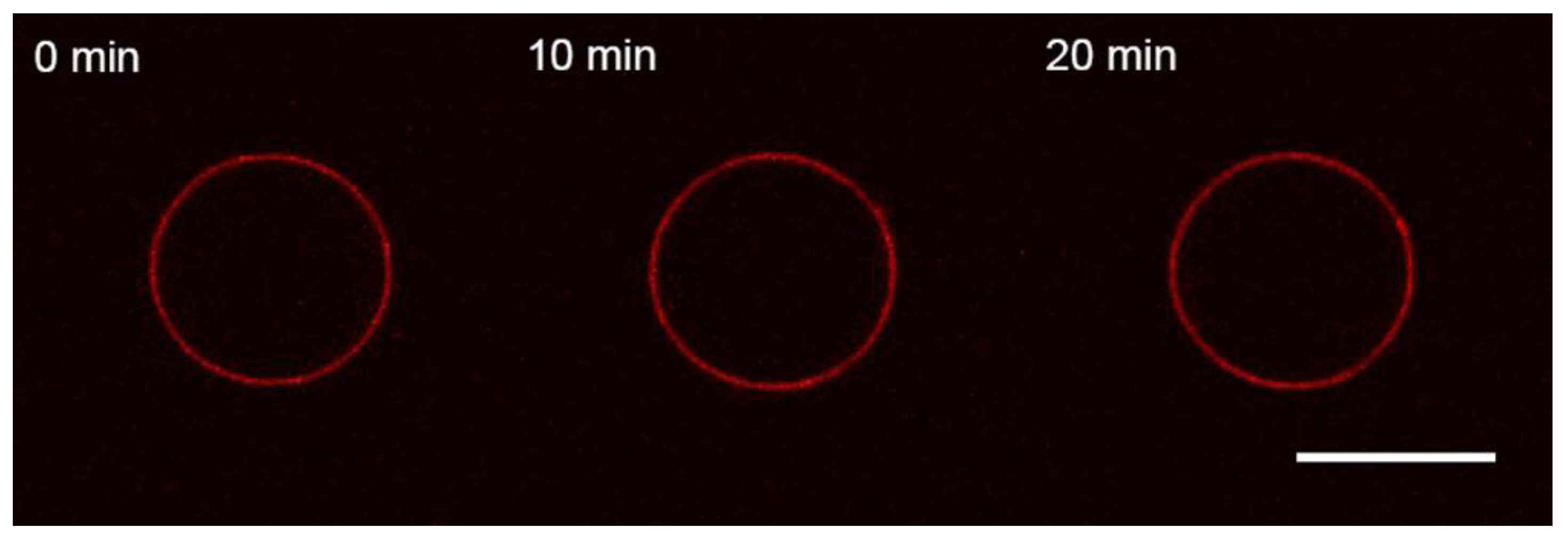
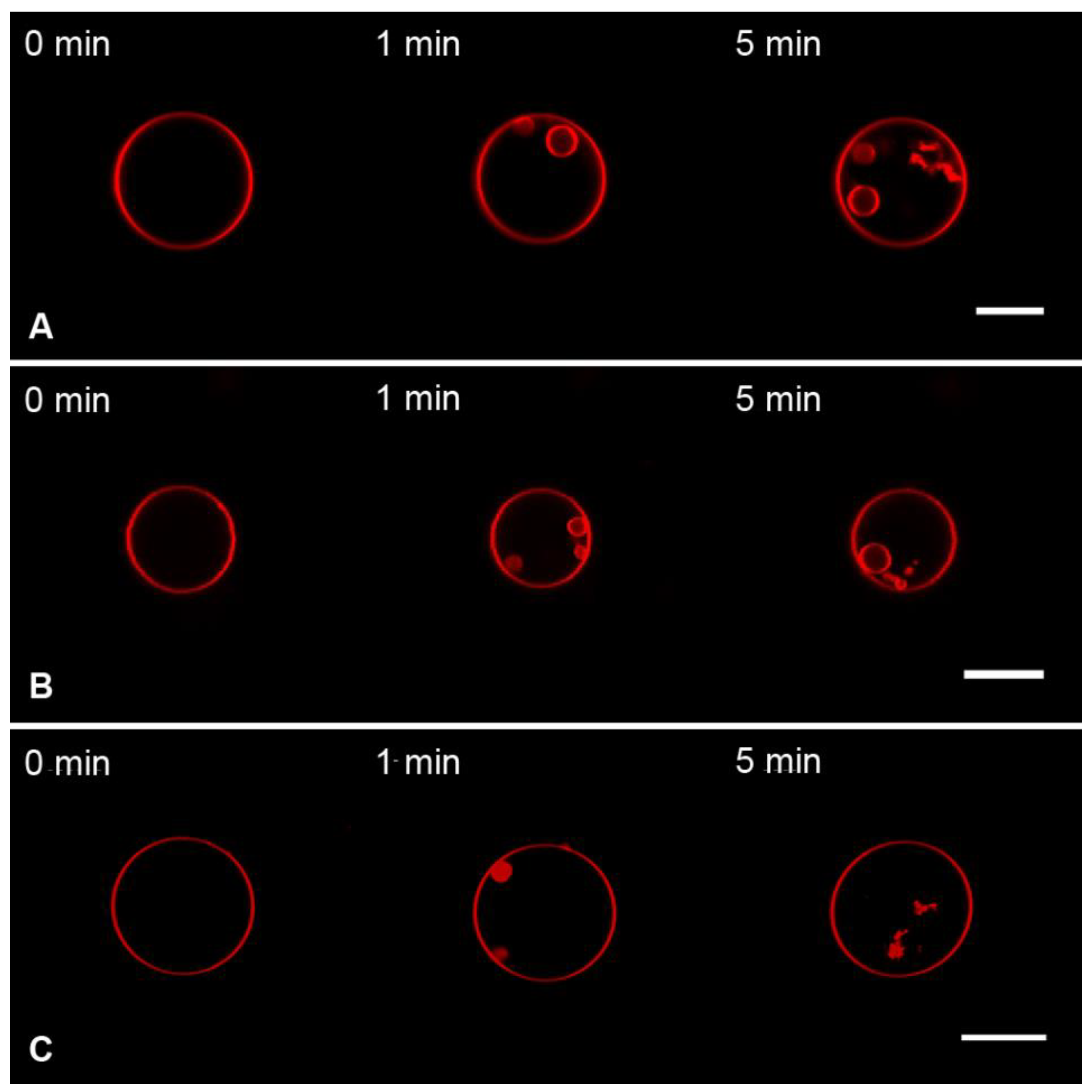
3.1. Interaction of the M1 Protein with GUVs in Isosmotic Conditions
- (1)
- DOPC:DOPE:Chol:Rho-PE = 59.99 mol%:30 mol%:10 mol%:0.01 mol% (herein referred to as Mixture 1);
- (2)
- DOPC:DOPE:Chol:DOPS:Rho-PE = 50 mol%:24 mol%:10 mol%:15.99 mol%:0.01 mol% (herein referred to as Mixture 2).
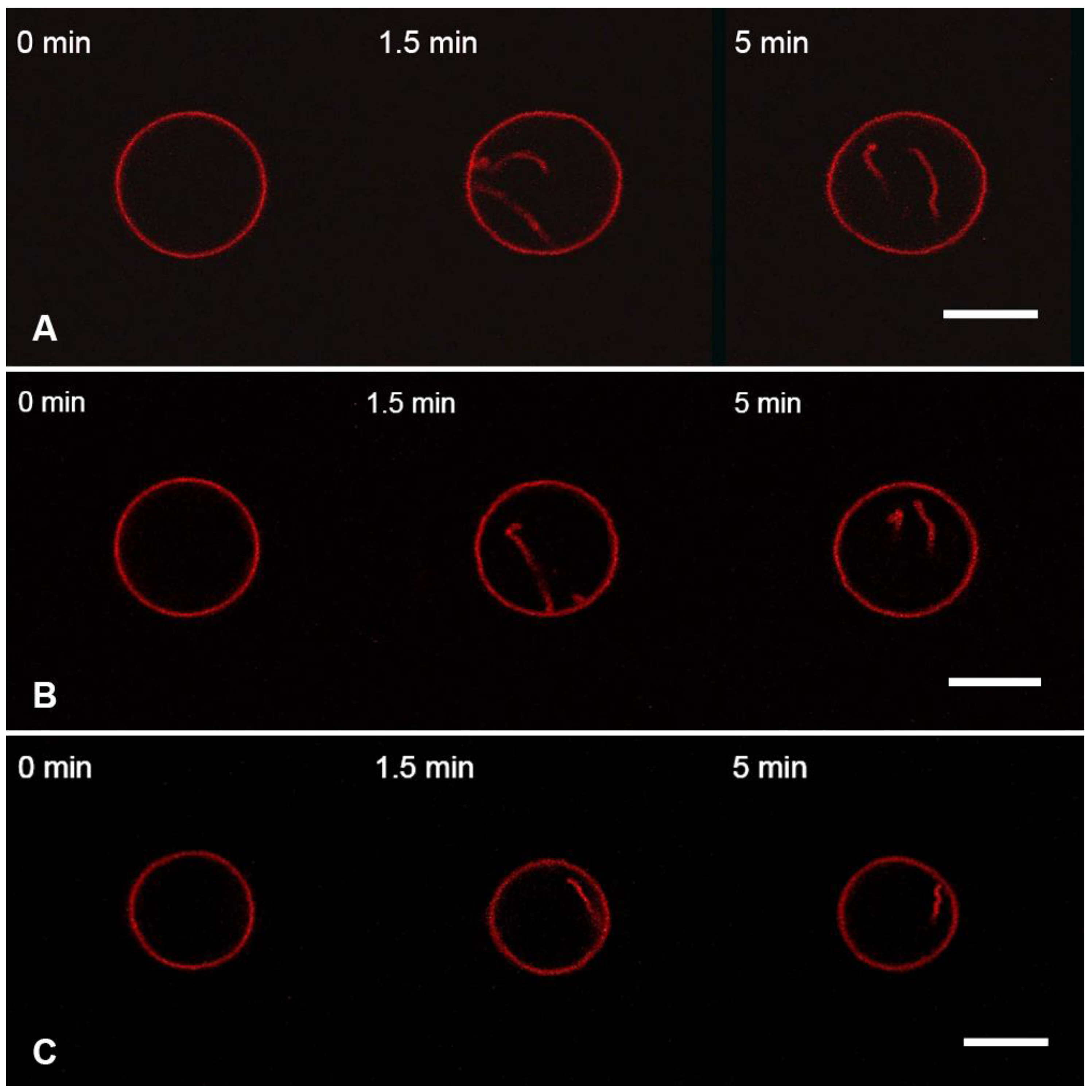
3.2. Interaction of the M1 Protein with GUVs in Hyperosmotic Conditions
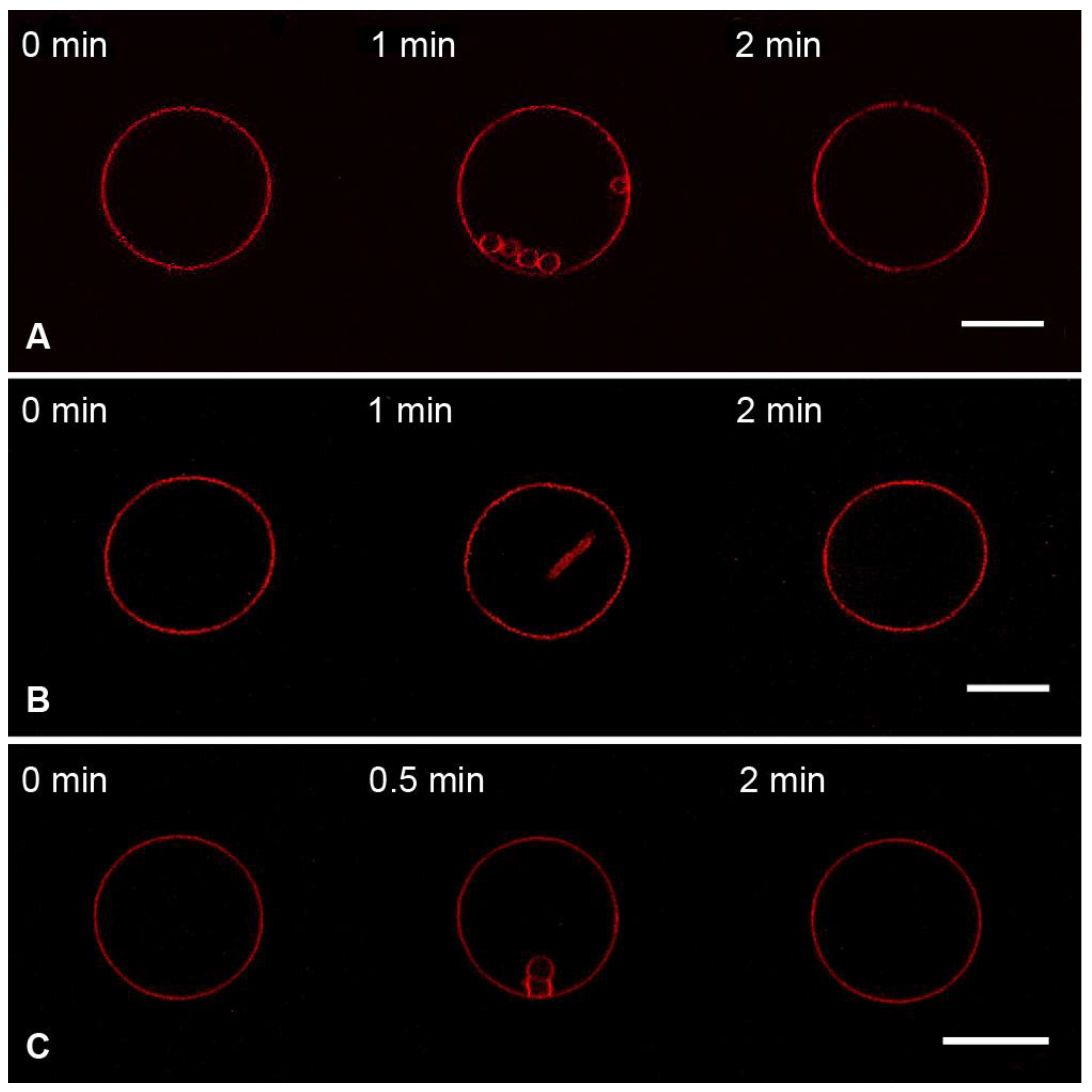
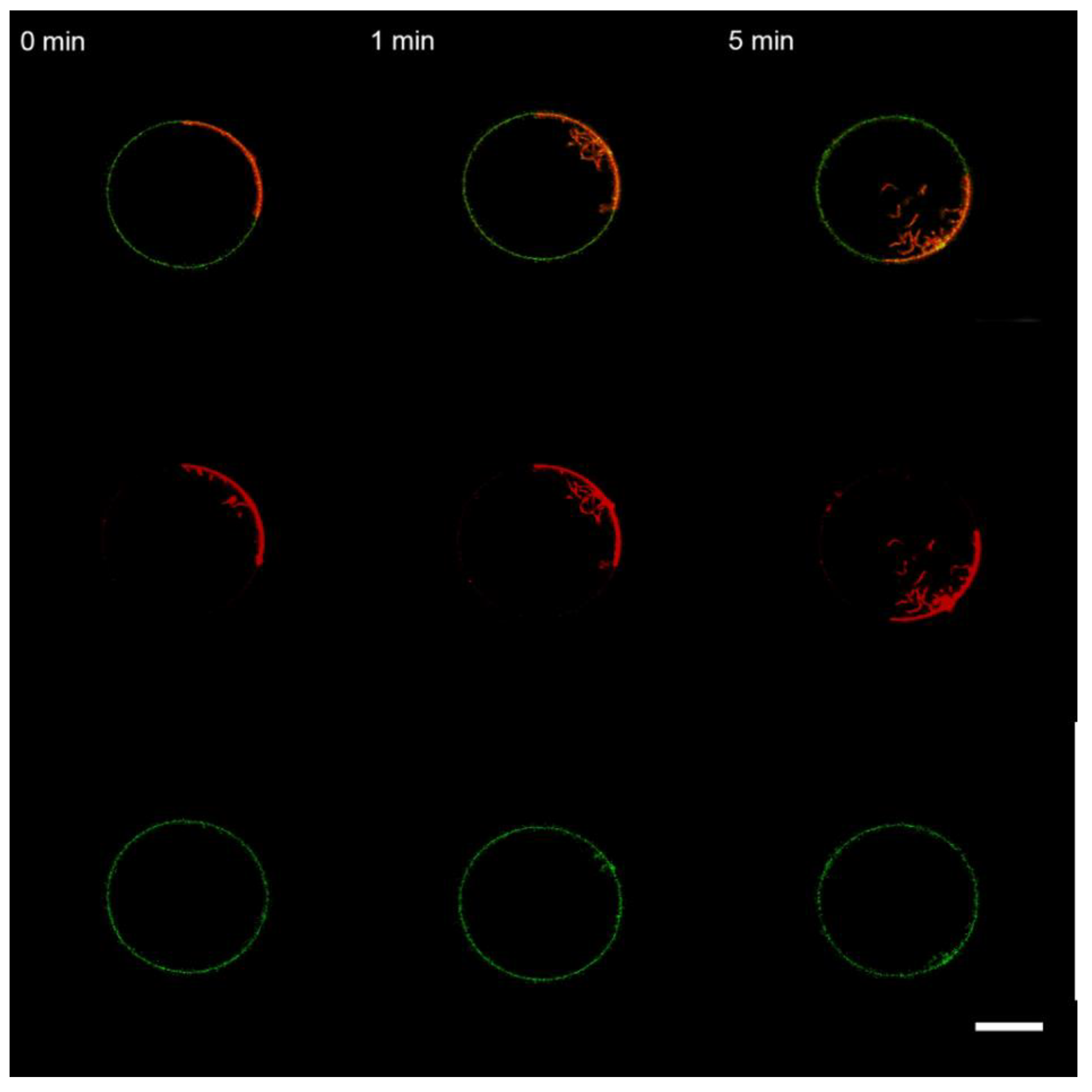
3.3. Interaction of the M1 Protein with the “Raft” GUVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rey, F.A.; Lok, S.-M. Common Features of Enveloped Viruses and Implications for Immunogen Design for Next-Generation Vaccines. Cell 2018, 172, 1319–1334. [Google Scholar] [CrossRef] [PubMed]
- Garoff, H.; Hewson, R.; Opstelten, D.-J.E. Virus Maturation by Budding. Microbiol. Mol. Biol. Rev. 1998, 62, 1171–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremser, M.; Nickel, W.; Schweikert, M.; Ravazzola, M.; Amherdt, M.; Hughes, C.A.; Söllner, T.H.; Rothman, J.E.; Wieland, F.T. Coupling of Coat Assembly and Vesicle Budding to Packaging of Putative Cargo Receptors. Cell 1999, 96, 495–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerberg, J.; Kozlov, M.M. How Proteins Produce Cellular Membrane Curvature. Nat. Rev. Mol. Cell Biol. 2006, 7, 9–19. [Google Scholar] [CrossRef]
- Schekman, R.; Orci, L. Coat Proteins and Vesicle Budding. Science 1996, 271, 1526–1533. [Google Scholar] [CrossRef]
- McMahon, H.T.; Kozlov, M.M.; Martens, S. Membrane Curvature in Synaptic Vesicle Fusion and Beyond. Cell 2010, 140, 601–605. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Jardetzky, T.S.; Lamb, R.A. Timing Is Everything: Fine-Tuned Molecular Machines Orchestrate Paramyxovirus Entry. Virology 2015, 479–480, 518–531. [Google Scholar] [CrossRef] [Green Version]
- White, J.M.; Whittaker, G.R. Fusion of Enveloped Viruses in Endosomes: Virus Fusion in Endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [Green Version]
- Jasenosky, L.D.; Neumann, G.; Lukashevich, I.; Kawaoka, Y. Ebola Virus VP40-Induced Particle Formation and Association with the Lipid Bilayer. J. Virol. 2001, 75, 5205–5214. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Puertas, P.; Albo, C.; Pérez-Pastrana, E.; Vivo, A.; Portela, A. Influenza Virus Matrix Protein Is the Major Driving Force in Virus Budding. J. Virol. 2000, 74, 11538–11547. [Google Scholar] [CrossRef]
- Justice, P.A.; Sun, W.; Li, Y.; Ye, Z.; Grigera, P.R.; Wagner, R.R. Membrane Vesiculation Function and Exocytosis of Wild-Type and Mutant Matrix Proteins of Vesicular Stomatitis Virus. J. Virol. 1995, 69, 3156–3160. [Google Scholar] [CrossRef] [Green Version]
- Saletti, D.; Radzimanowski, J.; Effantin, G.; Midtvedt, D.; Mangenot, S.; Weissenhorn, W.; Bassereau, P.; Bally, M. The Matrix Protein M1 from Influenza C Virus Induces Tubular Membrane Invaginations in an in Vitro Cell Membrane Model. Sci. Rep. 2017, 7, 40801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCauley, J.W.; Mahy, B.W.J. Structure and Function of the Influenza Virus Genome. Biochem. J. 1983, 211, 281–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hom, N.; Gentles, L.; Bloom, J.D.; Lee, K.K. Deep Mutational Scan of the Highly Conserved Influenza A Virus M1 Matrix Protein Reveals Substantial Intrinsic Mutational Tolerance. J. Virol. 2019, 93, e00161-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peukes, J.; Xiong, X.; Erlendsson, S.; Qu, K.; Wan, W.; Calder, L.J.; Schraidt, O.; Kummer, S.; Freund, S.M.V.; Kräusslich, H.-G.; et al. The Native Structure of the Assembled Matrix Protein 1 of Influenza A Virus. Nature 2020, 587, 495–498. [Google Scholar] [CrossRef]
- Ge, P.; Tsao, J.; Schein, S.; Green, T.J.; Luo, M.; Zhou, Z.H. Cryo-EM Model of the Bullet-Shaped Vesicular Stomatitis Virus. Science 2010, 327, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Liljeroos, L.; Huiskonen, J.T.; Ora, A.; Susi, P.; Butcher, S.J. Electron Cryotomography of Measles Virus Reveals How Matrix Protein Coats the Ribonucleocapsid within Intact Virions. Proc. Natl. Acad. Sci. USA 2011, 108, 18085–18090. [Google Scholar] [CrossRef] [Green Version]
- Shtykova, E.V.; Petoukhov, M.V.; Dadinova, L.A.; Fedorova, N.V.; Tashkin, V.Y.; Timofeeva, T.A.; Ksenofontov, A.L.; Loshkarev, N.A.; Baratova, L.A.; Jeffries, C.M.; et al. Solution Structure, Self-Assembly, and Membrane Interactions of the Matrix Protein from Newcastle Disease Virus at Neutral and Acidic PH. J. Virol. 2018, 93, e01450-18. [Google Scholar] [CrossRef] [Green Version]
- Briggs, J.A.G.; Riches, J.D.; Glass, B.; Bartonova, V.; Zanetti, G.; Krausslich, H.-G. Structure and Assembly of Immature HIV. Proc. Natl. Acad. Sci. USA 2009, 106, 11090–11095. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.R.; Schooler, J.B.; Ding, H.J.; Kieffer, C.; Fillmore, C.; Sundquist, W.I.; Jensen, G.J. Electron Cryotomography of Immature HIV-1 Virions Reveals the Structure of the CA and SP1 Gag Shells. EMBO J. 2007, 26, 2218–2226. [Google Scholar] [CrossRef]
- Battisti, A.J.; Meng, G.; Winkler, D.C.; McGinnes, L.W.; Plevka, P.; Steven, A.C.; Morrison, T.G.; Rossmann, M.G. Structure and Assembly of a Paramyxovirus Matrix Protein. Proc. Natl. Acad. Sci. USA 2012, 109, 13996–14000. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zheng, W.; Toh, Y.; Betancourt-Solis, M.A.; Tu, J.; Fan, Y.; Vakharia, V.N.; Liu, J.; McNew, J.A.; Jin, M.; et al. Crystal Structure of an Orthomyxovirus Matrix Protein Reveals Mechanisms for Self-Polymerization and Membrane Association. Proc. Natl. Acad. Sci. USA 2017, 114, 8550–8555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faaberg, K.S.; Peeples, M.E. Association of Soluble Matrix Protein of Newcastle Disease Virus with Liposomes Is Independent of Ionic Conditions. Virology 1988, 166, 123–132. [Google Scholar] [CrossRef]
- Shi, Z.; Baumgart, T. Membrane Tension and Peripheral Protein Density Mediate Membrane Shape Transitions. Nat. Commun. 2015, 6, 5974. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, T.; Hess, S.T.; Webb, W.W. Imaging Coexisting Fluid Domains in Biomembrane Models Coupling Curvature and Line Tension. Nature 2003, 425, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Veit, M.; Thaa, B. Association of Influenza Virus Proteins with Membrane Rafts. Adv. Virol. 2011, 2011, 370606. [Google Scholar] [CrossRef]
- Chlanda, P.; Mekhedov, E.; Waters, H.; Sodt, A.; Schwartz, C.; Nair, V.; Blank, P.S.; Zimmerberg, J. Palmitoylation Contributes to Membrane Curvature in Influenza A Virus Assembly and Hemagglutinin-Mediated Membrane Fusion. J. Virol. 2017, 91, e00947-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shtykova, E.V.; Baratova, L.A.; Fedorova, N.V.; Radyukhin, V.A.; Ksenofontov, A.L.; Volkov, V.V.; Shishkov, A.V.; Dolgov, A.A.; Shilova, L.A.; Batishchev, O.V.; et al. Structural Analysis of Influenza A Virus Matrix Protein M1 and Its Self-Assemblies at Low PH. PLoS ONE 2013, 8, e82431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shtykova, E.V.; Dadinova, L.A.; Fedorova, N.V.; Golanikov, A.E.; Bogacheva, E.N.; Ksenofontov, A.L.; Baratova, L.A.; Shilova, L.A.; Tashkin, V.Y.; Galimzyanov, T.R.; et al. Influenza Virus Matrix Protein M1 Preserves Its Conformation with PH, Changing Multimerization State at the Priming Stage Due to Electrostatics. Sci. Rep. 2017, 7, 16793. [Google Scholar] [CrossRef] [Green Version]
- Safo, M.K.; Musayev, F.N.; Mosier, P.D.; Zhou, Q.; Xie, H.; Desai, U.R. Crystal Structures of Influenza A Virus Matrix Protein M1: Variations on a Theme. PLoS ONE 2014, 9, e109510. [Google Scholar] [CrossRef]
- Selzer, L.; Su, Z.; Pintilie, G.D.; Chiu, W.; Kirkegaard, K. Full-length three-dimensional structure of the influenza A virus M1 protein and its organization into a matrix layer. PLoS Biol. 2020, 18, e3000827. [Google Scholar] [CrossRef] [PubMed]
- Arzt, S.; Baudin, F.; Barge, A.; Timmins, P.; Burmeister, W.P.; Ruigrok, R.W.H. Combined Results from Solution Studies on Intact Influenza Virus M1 Protein and from a New Crystal Form of Its N-Terminal Domain Show That M1 Is an Elongated Monomer. Virology 2001, 279, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höfer, C.T.; Di Lella, S.; Dahmani, I.; Jungnick, N.; Bordag, N.; Bobone, S.; Huang, Q.; Keller, S.; Herrmann, A.; Chiantia, S. Structural Determinants of the Interaction between Influenza A Virus Matrix Protein M1 and Lipid Membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2019, 1861, 1123–1134. [Google Scholar] [CrossRef]
- Ruigrok, R.W.H.; Barge, A.; Durrer, P.; Brunner, J.; Ma, K.; Whittaker, G.R. Membrane Interaction of Influenza Virus M1 Protein. Virology 2000, 267, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Bobone, S.; Hilsch, M.; Storm, J.; Dunsing, V.; Herrmann, A.; Chiantia, S. Phosphatidylserine Lateral Organization Influences the Interaction of Influenza Virus Matrix Protein 1 with Lipid Membranes. J. Virol. 2017, 91, e00267-17. [Google Scholar] [CrossRef] [Green Version]
- Hilsch, M.; Goldenbogen, B.; Sieben, C.; Höfer, C.T.; Rabe, J.P.; Klipp, E.; Herrmann, A.; Chiantia, S. Influenza A Matrix Protein M1 Multimerizes upon Binding to Lipid Membranes. Biophys. J. 2014, 107, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Dahmani, I.; Ludwig, K.; Chiantia, S. Influenza A Matrix Protein M1 Induces Lipid Membrane Deformation via Protein Multimerization. Biosci. Rep. 2019, 39, BSR20191024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordyukova, L.V.; Konarev, P.V.; Fedorova, N.V.; Shtykova, E.V.; Ksenofontov, A.L.; Loshkarev, N.A.; Dadinova, L.A.; Timofeeva, T.A.; Abramchuk, S.S.; Moiseenko, A.V.; et al. The Cytoplasmic Tail of Influenza A Virus Hemagglutinin and Membrane Lipid Composition Change the Mode of M1 Protein Association with the Lipid Bilayer. Membranes 2021, 11, 772. [Google Scholar] [CrossRef] [PubMed]
- Raut, P.; Obeng, B.; Waters, H.; Zimmerberg, J.; Gosse, J.A.; Hess, S.T. Phosphatidylinositol 4, 5-Bisphosphate Mediates the Co-Distribution of Influenza A Hemagglutinin and Matrix Protein M1 at the Plasma Membrane. Viruses 2022, 14, 2509. [Google Scholar] [CrossRef]
- Gregoriades, A. Interaction of Influenza M Protein with Viral Lipid and Phosphatidylcholine Vesicles. J. Virol. 1980, 36, 470–479. [Google Scholar] [CrossRef]
- Shishkov, A.; Bogacheva, E.; Dolgov, A.; Chulichkov, A.; Knyazev, D.; Fedorova, N.; Ksenofontov, A.; Kordyukova, L.; Lukashina, E.; Mirsky, V.; et al. The In Situ Structural Characterization of the Influenza A Virus Matrix M1 Protein within a Virion. Protein Pept. Lett. 2009, 16, 1407–1413. [Google Scholar] [CrossRef]
- Gregoriades, A.; Frangione, B. Insertion of Influenza M Protein into the Viral Lipid Bilayer and Localization of Site of Insertion. J. Virol. 1981, 40, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Tsfasman, T.; Kost, V.; Markushin, S.; Lotte, V.; Koptiaeva, I.; Bogacheva, E.; Baratova, L.; Radyukhin, V. Amphipathic Alpha-Helices and Putative Cholesterol Binding Domains of the Influenza Virus Matrix M1 Protein Are Crucial for Virion Structure Organisation. Virus Res. 2015, 210, 114–118. [Google Scholar] [CrossRef]
- Takeda, M.; Leser, G.P.; Russell, C.J.; Lamb, R.A. Influenza Virus Hemagglutinin Concentrates in Lipid Raft Microdomains for Efficient Viral Fusion. Proc. Natl. Acad. Sci. USA 2003, 100, 14610–14617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seladi-Schulman, J.; Steel, J.; Lowen, A.C. Spherical Influenza Viruses Have a Fitness Advantage in Embryonated Eggs, While Filament-Producing Strains Are Selected In Vivo. J. Virol. 2013, 87, 13343–13353. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sieben, C.; Ludwig, K.; Höfer, C.T.; Chiantia, S.; Herrmann, A.; Eghiaian, F.; Schaap, I.A.T. PH-Controlled Two-Step Uncoating of Influenza Virus. Biophys. J. 2014, 106, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batishchev, O.V.; Shilova, L.A.; Kachala, M.V.; Tashkin, V.Y.; Sokolov, V.S.; Fedorova, N.V.; Baratova, L.A.; Knyazev, D.G.; Zimmerberg, J.; Chizmadzhev, Y.A. PH-Dependent Formation and Disintegration of the Influenza A Virus Protein Scaffold to Provide Tension for Membrane Fusion. J. Virol. 2016, 90, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, D.P.; Balogun, R.A.; Yamada, H.; Zhou, Z.H.; Barman, S. Influenza Virus Morphogenesis and Budding. Virus Res. 2009, 143, 147–161. [Google Scholar] [CrossRef]
- Walde, P.; Cosentino, K.; Engel, H.; Stano, P. Giant Vesicles: Preparations and Applications. Chembiochem 2010, 11, 848–865. [Google Scholar] [CrossRef]
- Solon, J. Membrane Deformations Induced by the Matrix Protein of Vesicular Stomatitis Virus in a Minimal System. J. Gen. Virol. 2005, 86, 3357–3363. [Google Scholar] [CrossRef]
- Shnyrova, A.V.; Ayllon, J.; Mikhalyov, I.I.; Villar, E.; Zimmerberg, J.; Frolov, V.A. Vesicle Formation by Self-Assembly of Membrane-Bound Matrix Proteins into a Fluidlike Budding Domain. J. Cell Biol. 2007, 179, 627–633. [Google Scholar] [CrossRef]
- Zhirnov, O.P. Isolation of Matrix Protein M1 from Influenza Viruses by Acid-Dependent Extraction with Nonionic Detergent. Virology 1992, 186, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Méléard, P.; Bagatolli, L.A.; Pott, T. Giant Unilamellar Vesicle Electroformation from Lipid Mixtures to Native Membranes under Physiological Conditions. Methods Enzymol. 2009, 465, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Shnyrova, A.V.; Zimmerberg, J. Chapter 4-Reconstitution of Membrane Budding with Unilamellar Vesicles. Methods Enzymol. 2009, 464, 55–75. [Google Scholar] [CrossRef] [Green Version]
- Baudin, F.; Petit, I.; Weissenhorn, W.; Ruigrok, R.W.H. In Vitro Dissection of the Membrane and RNP Binding Activities of Influenza Virus M1 Protein. Virology 2001, 281, 102–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, B.; Luo, M. Structure of a Bifunctional Membrane-RNA Binding Protein, Influenza Virus Matrix Protein M1. Nat. Struct. Mol. Biol. 1997, 4, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Shilova, L.A.; Knyazev, D.G.; Fedorova, N.V.; Shtykova, E.V.; Batishchev, O.V. Study of Adsorption of Influenza Virus Matrix Protein M1 on Lipid Membranes by the Technique of Fluorescent Probes. Biochem. Moscow Suppl. Ser. A 2017, 11, 225–230. [Google Scholar] [CrossRef]
- Sorre, B.; Callan-Jones, A.; Manzi, J.; Goud, B.; Prost, J.; Bassereau, P.; Roux, A. Nature of Curvature Coupling of Amphiphysin with Membranes Depends on Its Bound Density. Proc. Natl. Acad. Sci. USA 2012, 109, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Yokouchi, Y.; Tsunoda, T.; Imura, T.; Yamauchi, H.; Yokoyama, S.; Sakai, H.; Abe, M. Effect of Adsorption of Bovine Serum Albumin on Liposomal Membrane Characteristics. Colloids Surf. B Biointerfaces 2001, 20, 95–103. [Google Scholar] [CrossRef]
- Marukovich, N.; McMurray, M.; Finogenova, O.; Nesterenko, A.; Batishchev, O.; Ermakov, Y. Interaction of Polylysines with the Surface of Lipid Membranes. In Advances in Planar Lipid Bilayers and Liposomes; Elsevier: Amsterdam, The Netherlands, 2013; Volume 17, pp. 139–166. ISBN 978-0-12-411516-3. [Google Scholar]
- Yuan, F.; Alimohamadi, H.; Bakka, B.; Trementozzi, A.N.; Day, K.J.; Fawzi, N.L.; Rangamani, P.; Stachowiak, J.C. Membrane Bending by Protein Phase Separation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017435118. [Google Scholar] [CrossRef]
- Kralj-Iglic, V.; Iglic, A.; Gomiscek, G.; Sevsek, F.; Arrigler, V.; Hägerstrand, H. Microtubes and Nanotubes of a Phospholipid Bilayer Membrane. J. Phys. A Math. Gen. 2002, 35, 1533–1549. [Google Scholar] [CrossRef]
- Boroske, E.; Elwenspoek, M.; Helfrich, W. Osmotic Shrinkage of Giant Egg-Lecithin Vesicles. Biophys. J. 1981, 34, 95–109. [Google Scholar] [CrossRef] [Green Version]
- Galimzyanov, T.R.; Lyushnyak, A.S.; Aleksandrova, V.V.; Shilova, L.A.; Mikhalyov, I.I.; Molotkovskaya, I.M.; Akimov, S.A.; Batishchev, O.V. Line Activity of Ganglioside GM1 Regulates the Raft Size Distribution in a Cholesterol-Dependent Manner. Langmuir 2017, 33, 3517–3524. [Google Scholar] [CrossRef] [PubMed]
- Kordyukova, L.V.; Shtykova, E.V.; Baratova, L.A.; Svergun, D.I.; Batishchev, O.V. Matrix Proteins of Enveloped Viruses: A Case Study of Influenza A Virus M1 Protein. J. Biomol. Struct. Dyn. 2019, 37, 671–690. [Google Scholar] [CrossRef]
- Fujiyoshi, Y.; Kume, N.P.; Sakata, K.; Sato, S.B. Fine Structure of Influenza A Virus Observed by Electron Cryo-Microscopy. EMBO J. 1994, 13, 318–326. [Google Scholar] [CrossRef]
- Tsafrir, I.; Caspi, Y.; Guedeau-Boudeville, M.-A.; Arzi, T.; Stavans, J. Budding and Tubulation in Highly Oblate Vesicles by Anchored Amphiphilic Molecules. Phys. Rev. Lett. 2003, 91, 138102. [Google Scholar] [CrossRef] [Green Version]
- Stachowiak, J.C.; Schmid, E.M.; Ryan, C.J.; Ann, H.S.; Sasaki, D.Y.; Sherman, M.B.; Geissler, P.L.; Fletcher, D.A.; Hayden, C.C. Membrane Bending by Protein–Protein Crowding. Nat. Cell Biol. 2012, 14, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Foteini, P.; Pippa, N.; Naziris, N.; Demetzos, C. Physicochemical Study of the Protein–Liposome Interactions: Influence of Liposome Composition and Concentration on Protein Binding. J. Liposome Res. 2019, 29, 313–321. [Google Scholar] [CrossRef]
- Chlanda, P.; Schraidt, O.; Kummer, S.; Riches, J.; Oberwinkler, H.; Prinz, S.; Kräusslich, H.-G.; Briggs, J.A.G. Structural Analysis of the Roles of Influenza A Virus Membrane-Associated Proteins in Assembly and Morphology. J. Virol. 2015, 89, 8957–8966. [Google Scholar] [CrossRef] [Green Version]
- Protein-Lipid Interactions: Perspectives, Techniques and Challenges; Protein Biochemistry, Synthesis, Structure and Cellular Functions; Catalá, A. (Ed.) Nova Science Publishers: New York, NY, USA, 2018; ISBN 978-1-5361-3125-3. [Google Scholar]
- Pinigin, K.V.; Kondrashov, O.V.; Jiménez-Munguía, I.; Alexandrova, V.V.; Batishchev, O.V.; Galimzyanov, T.R.; Akimov, S.A. Elastic Deformations Mediate Interaction of the Raft Boundary with Membrane Inclusions Leading to Their Effective Lateral Sorting. Sci. Rep. 2020, 10, 4087. [Google Scholar] [CrossRef]
- Rawicz, W.; Olbrich, K.C.; McIntosh, T.; Needham, D.; Evans, E. Effect of Chain Length and Unsaturation on Elasticity of Lipid Bilayers. Biophys. J. 2000, 79, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molotkovsky; Galimzyanov; Batishchev; Akimov the Effect of Transmembrane Protein Shape on Surrounding Lipid Domain Formation by Wetting. Biomolecules 2019, 9, 729. [CrossRef] [PubMed] [Green Version]
- Siche, S.; Brett, K.; Möller, L.; Kordyukova, L.; Mintaev, R.; Alexeevski, A.; Veit, M. Two Cytoplasmic Acylation Sites and an Adjacent Hydrophobic Residue, but No Other Conserved Amino Acids in the Cytoplasmic Tail of HA from Influenza A Virus Are Crucial for Virus Replication. Viruses 2015, 7, 6458–6475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrich, A.; Dunsing, V.; Bobone, S.; Chiantia, S. Influenza A M2 Recruits M1 to the Plasma Membrane: A Fluorescence Fluctuation Microscopy Study. Biophys. J. 2021, 120, 5478–5490. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loshkareva, A.S.; Popova, M.M.; Shilova, L.A.; Fedorova, N.V.; Timofeeva, T.A.; Galimzyanov, T.R.; Kuzmin, P.I.; Knyazev, D.G.; Batishchev, O.V. Influenza A Virus M1 Protein Non-Specifically Deforms Charged Lipid Membranes and Specifically Interacts with the Raft Boundary. Membranes 2023, 13, 76. https://doi.org/10.3390/membranes13010076
Loshkareva AS, Popova MM, Shilova LA, Fedorova NV, Timofeeva TA, Galimzyanov TR, Kuzmin PI, Knyazev DG, Batishchev OV. Influenza A Virus M1 Protein Non-Specifically Deforms Charged Lipid Membranes and Specifically Interacts with the Raft Boundary. Membranes. 2023; 13(1):76. https://doi.org/10.3390/membranes13010076
Chicago/Turabian StyleLoshkareva, Anna S., Marina M. Popova, Liudmila A. Shilova, Natalia V. Fedorova, Tatiana A. Timofeeva, Timur R. Galimzyanov, Petr I. Kuzmin, Denis G. Knyazev, and Oleg V. Batishchev. 2023. "Influenza A Virus M1 Protein Non-Specifically Deforms Charged Lipid Membranes and Specifically Interacts with the Raft Boundary" Membranes 13, no. 1: 76. https://doi.org/10.3390/membranes13010076
APA StyleLoshkareva, A. S., Popova, M. M., Shilova, L. A., Fedorova, N. V., Timofeeva, T. A., Galimzyanov, T. R., Kuzmin, P. I., Knyazev, D. G., & Batishchev, O. V. (2023). Influenza A Virus M1 Protein Non-Specifically Deforms Charged Lipid Membranes and Specifically Interacts with the Raft Boundary. Membranes, 13(1), 76. https://doi.org/10.3390/membranes13010076