
SARS-CoV-2 Infectivity and Severity of COVID-19 According to SARS-CoV-2 Variants: Current Evidence

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
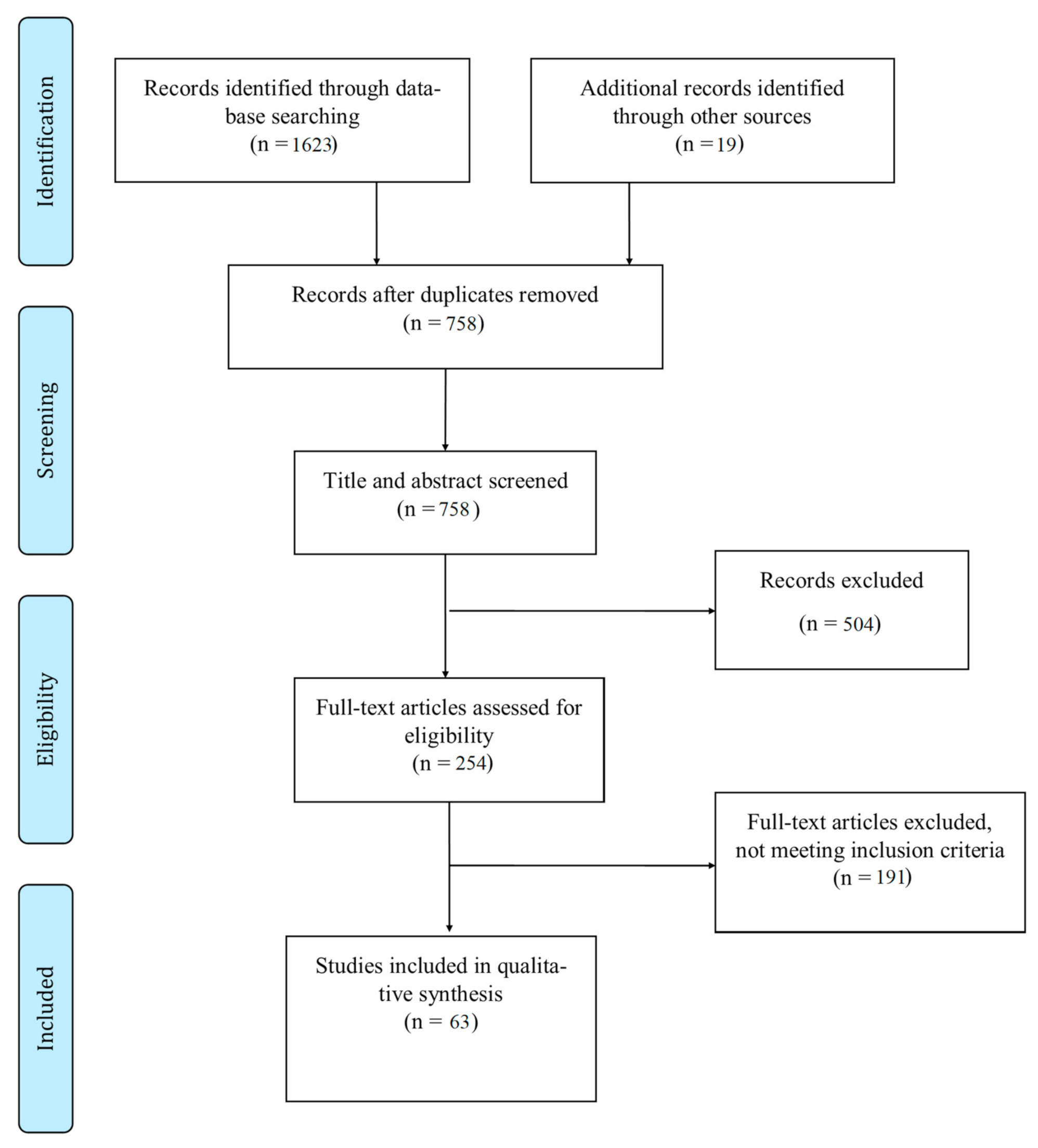
2.1. Search Strategy and Selection Criteria
2.2. Data Collection Process
2.3. Data Synthesis and Analysis
3. Results
3.1. Study Selection and Types of Studies
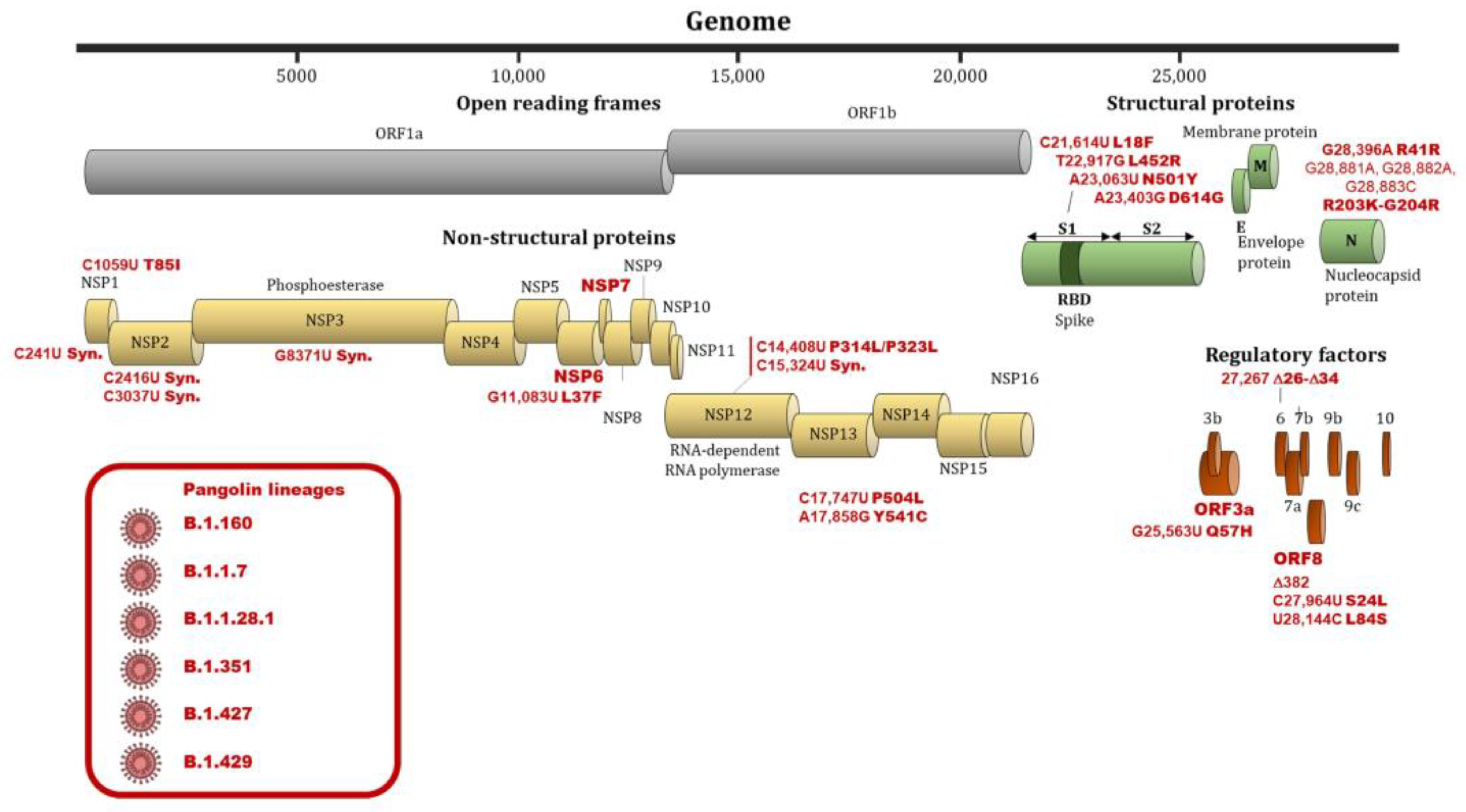
3.2. Relation between Viral Mutation and Infectivity of SARS-CoV-2
3.3. Relation between Viral Mutations and Severity of COVID-19 Infection
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO|Coronavirus. Available online: https://www.who.int/westernpacific/health-topics/coronavirus (accessed on 20 February 2021).
- Coronavirus Update (Live): 111,437,607 Cases and 2,467,436 Deaths from COVID-19 Virus Pandemic-Worldometer N.D. Available online: https://www.worldometers.info/coronavirus/ (accessed on 20 February 2021).
- Daoud, A.; Laktineh, A.; Macrander, C.; Mushtaq, A.; Soubani, A.O. Pulmonary Complications of Influenza Infection: A Targeted Narrative Review. Postgrad Med. 2019, 131, 299–308. [Google Scholar] [CrossRef]
- Hayden, F.G. Rhinovirus and the Lower Respiratory Tract. Rev. Med. Virol. 2004, 14, 17–31. [Google Scholar] [CrossRef]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Chidambaram, V.; Tun, N.L.; Haque, W.Z.; Majella, M.G.; Sivakumar, R.K.; Kumar, A.; Hsu, A.T.-W.; Ishak, I.A.; Nur, A.A.; Ayeh, S.K.; et al. Factors Associated with Disease Severity and Mortality among Patients with COVID-19: A Systematic Review and Meta-Analysis. PLoS ONE 2020, 15, e0241541. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-C.; Ko, W.-C.; Lee, P.-I.; Jean, S.-S.; Hsueh, P.-R. Extra-Respiratory Manifestations of COVID-19. Int. J. Antimicrob. Agents 2020, 56, 106024. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 Genomic Variations Associated with Mortality Rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, P.; Niyogi, S. ORF3a mutation associated higher mortality rate in SARS-CoV-2 infection. Epidemiol. Infect. 2020, 148, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Flores, M.; Cardozo, T. SARS-CoV-2 Viral Spike G614 Mutation Exhibits Higher Case Fatality Rate. Int. J. Clin. Pr. 2020, 74, e13525. [Google Scholar] [CrossRef]
- Eaaswarkhanth, M.; Al Madhoun, A.; Al-Mulla, F. Could the D614G Substitution in the SARS-CoV-2 Spike (S) Protein Be Associated with Higher COVID-19 Mortality? Int. J. Infect. Dis. 2020, 96, 459–460. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO|SARS-CoV-2 Variants. Available online: http://www.who.int/csr/don/31-december-2020-sars-cov2-variants/en/ (accessed on 20 February 2021).
- CDC. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (accessed on 31 May 2021).
- Koyama, T.; Platt, D.; Parida, L. Variant Analysis of SARS-CoV-2 Genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Levasseur, A.; Delerce, J.; Caputo, A.; Brechard, L.; Colson, P.; Lagier, J.-C.; Fournier, P.-E.; Raoult, D. Genomic Diversity and Evolution of Coronavirus (SARS-CoV-2) in France from 309 COVID-19-Infected Patients. bioRxiv 2020. [Google Scholar] [CrossRef]
- Colson, P.; Levasseur, A.; Delerce, J.; Chaudet, H.; Bossi, V.; Ben Khedher, M.; Fournier, P.E.; Lagier, J.C.; Raoult, D. Dramatic increase in the SARS-CoV-2 mutation rate and low mortality rate during the second epidemic in summer in Marseille. Preprints. Available online: https://doi.org/10.35088/68c3-ew82 (accessed on 31 May 2021).
- Stauft, C.B.; Lien, C.Z.; Selvaraj, P.; Liu, S.; Wang, T.T. The G614 Pandemic SARS-CoV-2 Variant Is Not More Pathogenic than the Original D614 Form in Adult Syrian Hamsters. Virology 2021, 556, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Nie, J.; Wu, J.; Zhang, L.; Ding, R.; Wang, H.; Zhang, Y.; Li, T.; Liu, S.; Zhang, M.; et al. SARS-CoV-2 501Y.V2 Variants Lack Higher Infectivity but Do Have Immune Escape. Cell 2021, 184, 2362–2371.e9. [Google Scholar] [CrossRef]
- Van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No Evidence for Increased Transmissibility from Recurrent Mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef] [PubMed]
- Young, B.E.; Wei, W.E.; Fong, S.-W.; Mak, T.-M.; Anderson, D.E.; Chan, Y.-H.; Pung, R.; Heng, C.S.; Ang, L.W.; Zheng, A.K.E.; et al. Association of SARS-CoV-2 Clades with Clinical, Inflammatory and Virologic Outcomes: An Observational Study. EBioMedicine 2021, 66, 103319. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 Spike-Protein D614G Mutation Increases Virion Spike Density and Infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, Q.; Wei, P.; Chen, Z.; Aviszus, K.; Yang, J.; Downing, W.; Jiang, C.; Liang, B.; Reynoso, L.; et al. The Basis of a More Contagious 501Y.V1 Variant of SARS-CoV-2. Cell Res. 2021. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike Mutation D614G Alters SARS-CoV-2 Fitness. Nature 2020, 1–9. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G Variant Exhibits Enhanced Replication Ex Vivo and Earlier Transmission in Vivo. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yao, H.; Lu, X.; Chen, Q.; Xu, K.; Chen, Y.; Cheng, L.; Liu, F.; Wu, Z.; Wu, H.; Jin, C.; et al. Patient-derived SARS-CoV-2 mutations impact viral replication dynamics and infectivity in vitro and with clinical implications in vivo. Cell Discov. 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, Infectivity, and Neutralization of a Spike L452R SARS-CoV-2 Variant. Cell 2021. [Google Scholar] [CrossRef]
- Pokhrel, S.; Kraemer, B.R.; Lee, L.; Samardzic, K.; Mochly-Rosen, D. Increased Elastase Sensitivity and Decreased Intramolecular Interactions in the More Transmissible 501Y.V1 and 501Y.V2 SARS-CoV-2 Variants’ Spike Protein–an in Silico Analysis. PLoS ONE 2021, 16, e0251426. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jang, U.S.; Soh, S.M.; Lee, J.Y.; Lee, H.R. The Impact on Infectivity and Neutralization Efficiency of SARS-CoV-2 Lineage, B.1.351 Pseudovirus. Viruses 2021, 13, 633. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, J.; Gao, K.; Hozumi, Y.; Yin, C.; Wei, G.-W. Analysis of SARS-CoV-2 Mutations in the United States Suggests Presence of Four Substrains and Novel Variants. Commun. Biol. 2021, 4, 1–14. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage, B.1.1.7 in England. Science 2021, 372. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lou, J.; Cao, L.; Zheng, H.; Chong, M.K.C.; Chen, Z.; Chan, R.W.Y.; Zee, B.C.Y.; Chan, P.K.S.; Wang, M.H. Quantifying the Transmission Advantage Associated with N501Y Substitution of SARS-CoV-2 in the UK: An Early Data-Driven Analysis. J. Travel Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Krutikov, M.; Hayward, A.; Shallcross, L. Spread of a Variant SARS-CoV-2 in Long-Term Care Facilities in England. New Engl. J. Med. 2021, 384, 1671–1673. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Redondo, R.; Nam, H.H.; Roberts, S.C.; Simons, L.M.; Jennings, L.J.; Qi, C.; Achenbach, C.J.; Hauser, A.R.; Ison, M.G.; Hultquist, J.F.; et al. A Clade of SARS-CoV-2 Viruses Associated with Lower Viral Loads in Patient Upper Airways. EBioMedicine 2020, 62, 103112. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M.; et al. Genomic Characteristics and Clinical Effect of the Emergent SARS-CoV-2 B.1.1.7 Lineage in London, UK: A Whole-Genome Sequencing and Hospital-Based Cohort Study. Lancet Infect. Dis. 2021, 0, S1473–S3099. [Google Scholar]
- Zhao, S.; Lou, J.; Chong, M.K.C.; Cao, L.; Zheng, H.; Chen, Z.; Chan, R.W.Y.; Zee, B.C.Y.; Chan, P.K.S.; Wang, M.H. Inferring the Association between the Risk of COVID-19 Case Fatality and N501Y Substitution in SARS-CoV-2. Viruses 2021, 13, 638. [Google Scholar] [CrossRef] [PubMed]
- Long, S.W.; Olsen, R.J.; Christensen, P.A.; Bernard, D.W.; Davis, J.J.; Shukla, M.; Nguyen, M.; Saavedra, M.O.; Yerramilli, P.; Pruitt, L.; et al. Molecular Architecture of Early Dissemination and Massive Second Wave of the SARS-CoV-2 Virus in a Major Metropolitan Area. mBio 2020, 11, e02707-20. [Google Scholar] [CrossRef]
- Zhang, X.; Tan, Y.; Ling, Y.; Lu, G.; Liu, F.; Yi, Z.; Jia, X.; Wu, M.; Shi, B.; Xu, S.; et al. Viral and Host Factors Related to the Clinical Outcome of COVID-19. Nature 2020, 583, 437. [Google Scholar] [CrossRef]
- Nakamichi, K.; Shen, J.Z.; Lee, C.S.; Lee, A.; Roberts, E.A.; Simonson, P.D.; Roychoudhury, P.; Andriesen, J.; Randhawa, A.K.; Mathias, P.C.; et al. Hospitalization and Mortality Associated with SARS-CoV-2 Viral Clades in COVID-19. Sci. Rep. 2021, 11, 4802. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Pham, T.N.; Van, T.D.; Nguyen, T.T.; Nguyen, D.T.N.; Le, H.N.M.; Eden, J.-S.; Rockett, R.J.; Nguyen, T.T.H.; Vu, B.T.N.; et al. Genetic Diversity of SARS-CoV-2 and Clinical, Epidemiological Characteristics of COVID-19 Patients in Hanoi, Vietnam. PLoS ONE 2020, 15, e0242537. [Google Scholar] [CrossRef]
- Elizondo, V.; Harkins, G.W.; Mabvakure, B.; Smidt, S.; Zappile, P.; Marier, C.; Maurano, M.T.; Perez, V.; Mazza, N.; Beloso, C.; et al. SARS-CoV-2 Genomic Characterization and Clinical Manifestation of the COVID-19 Outbreak in Uruguay. Emerg. Microbes Infect. 2021, 10, 51–65. [Google Scholar] [CrossRef]
- Colson, P.; Levasseur, A.; Gautret, P.; Fenollar, F.; Thuan Hoang, V.; Delerce, J.; Bitam, I.; Saile, R.; Maaloum, M.; Padane, A.; et al. Introduction into the Marseille Geographical Area of a Mild SARS-CoV-2 Variant Originating from Sub-Saharan Africa: An Investigational Study. Travel Med. Infect. Dis. 2021, 40, 101980. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.-E.; Colson, P.; Levasseur, A.; Devaux, C.A.; Gautret, P.; Bedotto, M.; Delerce, J.; Brechard, L.; Pinault, L.; Lagier, J.-C.; et al. Emergence and Outcomes of the SARS-CoV-2 “Marseille-4” Variant. Int J. Infect. Dis. 2021, 106, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Quéromès, G.; Destras, G.; Bal, A.; Regue, H.; Burfin, G.; Brun, S.; Fanget, R.; Morfin, F.; Valette, M.; Trouillet-Assant, S.; et al. Characterization of SARS-CoV-2 ORF6 Deletion Variants Detected in a Nosocomial Cluster during Routine Genomic Surveillance, Lyon, France. Emerg. Microbes Infect. 2021, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.; Hokama, P.; Roche, A.M.; Reddy, S.; Hwang, Y.; Kessler, L.; Glascock, A.; Li, Y.; Whelan, J.N.; Weiss, S.R.; et al. SARS-CoV-2 Genomic Variation in Space and Time in Hospitalized Patients in Philadelphia. mBio 2021, 12. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Hozumi, Y.; Yin, C.; Wei, G.-W. Decoding Asymptomatic COVID-19 Infection and Transmission. J. Phys. Chem. Lett. 2020, 11, 10007–10015. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.-W.; Chan, Y.-H.; Mak, T.-M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a Major Deletion in the SARS-CoV-2 Genome on the Severity of Infection and the Inflammatory Response: An Observational Cohort Study. The Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Wongtrakoongate, P.; Thawornwattana, Y.; Hongeng, S.; Thitithanyanont, A. SARS-CoV-2 Genetic Variations Associated with COVID-19 Severity. medRxiv 2020. [Google Scholar] [CrossRef]
- Nagy, Á.; Pongor, S.; Győrffy, B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int J. Antimicrob Agents 2021, 57, 106272. [Google Scholar] [CrossRef]
- Verma, P.; Elkaffas, R.; Shodunke, O.C.; Hrab, P.; Adebayo, O.H.; Alam, H.M.; Gbedema, W.; Agbonyin, M.A.; Osman, A.M.; Igbokwe, J.O.; et al. COVID-19 Mortality Risk Assessment among Various Age Groups Using Phylogenetic Analysis. Preprints 2020. [Google Scholar] [CrossRef]
- Hu, J.; Li, C.; Wang, S.; Li, T.; Zhang, H. Genetic variants are identified to increase risk of COVID-19 related mortality from UK Biobank data. Hum. Genomics. 2021, 15, 10. [Google Scholar] [CrossRef]
- Bhadra, B. Significance of Mutation Rate of Structural and Non-Structural Proteins of SARS-Cov-2 Showed with Lower Death Rate of COVID-19. J. Biomed. Res. Environ. Sci. 2020, 6, 017–022. [Google Scholar]
- Biswas, S.K.; Mudi, S.R. Spike Protein D614G and RdRp P323L: The SARS-CoV-2 Mutations Associated with Severity of COVID-19. Genom. Inform. 2020, 18. [Google Scholar] [CrossRef]
- Dumonteil, E.; Fusco, D.; Drouin, A.; Herrera, C. Genomic Signatures of SARS-CoV-2 Associated with Patient Mortality. Viruses 2021, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.; De Bruyne, K.; van Belkum, A.; West, B. Different SARS-CoV-2 Haplotypes Associate with Geographic Origin and Case Fatality Rates of COVID-19 Patients. Infect. Genet. Evol. 2021, 90, 104730. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chong, K.C.; Wong, M.C.S.; Boon, S.S.; Huang, J.; Wang, M.H.; Ng, R.W.Y.; Lai, C.K.C.; Chan, P.K.S. A Global Analysis on Replacement of Genetic Variants of SARS-CoV-2 in Associate with Containment Capacity and Changes in Disease Severity. Clin. Microbiol. Infect. 2021. [Google Scholar] [CrossRef] [PubMed]
- Challen, R.; Brooks-Pollock, E.; Read, J.M.; Dyson, L.; Tsaneva-Atanasova, K.; Danon, L. Risk of Mortality in Patients Infected with SARS-CoV-2 Variant of Concern 202012/1: Matched Cohort Study. BMJ 2021, 372. [Google Scholar] [CrossRef]
- Grint, D.J.; Wing, K.; Williamson, E.; McDonald, H.I.; Bhaskaran, K.; Evans, D.; Evans, S.J.; Walker, A.J.; Hickman, G.; Nightingale, E.; et al. Case Fatality Risk of the SARS-CoV-2 Variant of Concern B.1.1.7 in England, 16 November to 5 February. Eurosurveillance 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Jarvis, C.I.; CMMID COVID-19 Working Group; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased Mortality in Community-Tested Cases of SARS-CoV-2 Lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Cao, C.; He, L.; Tian, Y.; Qin, Y.; Sun, H.; Ding, W.; Gui, L.; Wu, P. Molecular Epidemiology Analysis of Early Variants of SARS-CoV-2 Reveals the Potential Impact of Mutations P504L and Y541C (NSP13) in the Clinical COVID-19 Outcomes. Infect. Genet. Evol. 2021, 92, 104831. [Google Scholar] [CrossRef]
- Pandit, B.; Bhattacharjee, S.; Bhattacharjee, B. Association of Clade-G SARS-CoV-2 Viruses and Age with Increased Mortality Rates across 57 Countries and India. Infect. Genet. Evol. 2021, 90, 104734. [Google Scholar] [CrossRef]
- Patone, M.; Thomas, K.; Hatch, R.; Tan, P.S.; Coupland, C.; Liao, W.; Mouncey, P.; Harrison, D.; Rowan, K.; Horby, P.; et al. Analysis of Severe Outcomes Associated with the SARS-CoV-2 Variant of Concern 202012/01 in England Using ICNARC Case Mix Programme and QResearch Databases. medRxiv 2021. [Google Scholar] [CrossRef]
- Flores-Alanis, A.; Cruz-Rangel, A.; Rodríguez-Gómez, F.; González, J.; Torres-Guerrero, C.A.; Delgado, G.; Cravioto, A.; Morales-Espinosa, R. Molecular Epidemiology Surveillance of SARS-CoV-2: Mutations and Genetic Diversity One Year after Emerging. Pathogens 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.N.; Hughes, S.; Ngai, S.; Baumgartner, J.; Wang, J.C.; McGibbon, E.; Devinney, K.; Luoma, E.; Bertolino, D.; Hwang, C.; et al. Rapid Emergence and Epidemiologic Characteristics of the SARS-CoV-2 B.1.526 Variant—New York City, New York, January 1–April 5, 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Martin Webb, L.; Matzinger, S.; Grano, C.; Kawasaki, B.; Stringer, G.; Bankers, L.; Herlihy, R. Identification of and Surveillance for the SARS-CoV-2 Variants B.1.427 and B.1.429—Colorado, January–March 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.L.; Hoang, V.T.; Nguyen, N.N.; Delerce, J.; Chaudet, H.; Levasseur, A.; Lagier, J.C.; Raoult, D.; Colson, P.; Gautret, P. Clinical Outcomes in COVID-19 Patients Infected with Different SARS-Cov-2 Variants in Marseille, France. Clin. Microbiol. Infect. 2021. [Google Scholar] [CrossRef] [PubMed]
- Esper, F.P.; Cheng, Y.-W.; Adhikari, T.M.; Tu, Z.J.; Li, D.; Li, E.A.; Farkas, D.H.; Procop, G.W.; Ko, J.S.; Chan, T.A.; et al. Genomic Epidemiology of SARS-CoV-2 Infection During the Initial Pandemic Wave and Association with Disease Severity. JAMA Netw. Open 2021, 4, e217746. [Google Scholar] [CrossRef] [PubMed]
- Loney, T.; Khansaheb, H.; Ramaswamy, S.; Harilal, D.; Deesi, Z.O.; Varghese, R.M.; Ali, A.B.A.; Khadeeja, A.; Suwaidi, H.A.; Alkhajeh, A.; et al. Genotype-Phenotype Correlation Identified a Novel SARS-CoV-2 Variant Possibly Linked to Severe Disease. Transbound Emerg. Dis. 2021. [Google Scholar] [CrossRef]
- Martins, A.F.; Zavascki, A.P.; Wink, P.L.; Volpato, F.C.Z.; Monteiro, F.L.; Rosset, C.; De-Paris, F.; Ramos, Á.K.; Barth, A.L. Detection of SARS-CoV-2 Lineage P.1 in Patients from a Region with Exponentially Increasing Hospitalisation Rate, February 2021, Rio Grande Do Sul, Southern Brazil. Eurosurveillance 2021, 26. [Google Scholar] [CrossRef]
- Sardar, R.; Satish, D.; Birla, S.; Gupta, D. Integrative Analyses of SARS-CoV-2 Genomes from Different Geographical Locations Reveal Unique Features Potentially Consequential to Host-Virus Interaction, Pathogenesis and Clues for Novel Therapies. Heliyon 2020, 6, e04658. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The Establishment of Reference Sequence for SARS-CoV-2 and Variation Analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef] [PubMed]
- Groves, D.C.; Rowland-Jones, S.L.; Angyal, A. The D614G Mutations in the SARS-CoV-2 Spike Protein: Implications for Viral Infectivity, Disease Severity and Vaccine Design. Biochem Biophys Res. Commun. 2021, 538, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Risk Assessment: Risk Related to the Spread of New SARS-CoV-2 Variants of Concern in the EU/EEA–First Update. Available online: https://www.ecdc.europa.eu/en/publications-data/covid-19-risk-assessment-spread-new-variants-concern-eueea-first-update (accessed on 2 June 2021).
- NERVTAG. Paper on COVID-19 Variant of Concern B.1.1.7. Available online: https://www.gov.uk/government/publications/nervtag-paper-on-covid-19-variant-of-concern-b117 (accessed on 2 June 2021).
- Grabowski, F.; Preibisch, G.; Giziński, S.; Kochańczyk, M.; Lipniacki, T. SARS-CoV-2 Variant of Concern 202012/01 Has about Twofold Replicative Advantage and Acquires Concerning Mutations. Viruses 2021, 13, 392. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Lineage | Country First Detected (Community) | Mutation/Deletion | Year and Month First Detected | Number of Affected Countries * |
|---|---|---|---|---|
| B.1.1.7 | United Kingdom | 69del, 70del, 144del, (E484K), (S494P), N501Y, A570D, D614G, P681H, T716I, S982A, D1118H (K1191N) | September 2020 | 136 |
| B.1.351 | South Africa | D80A, D215G, 241del, 242del, 243del, K417N, E484K, N501Y, D614G, A701V | September 2020 | 92 |
| P.1 | Brazil | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I | December 2020 | 54 |
| B.1.617.2 ¥ | India | T19R, (G142D), 156del, 157del, R158G, L452R, T478K, D614G, P681R, D950N | December 2020 | 57 |
| B.1.427 ⁋ | United States (California) | L452R, D614G | January 2021 | − |
| B.1.429 ⁋ | United States (California) | S13I, W152C, L452R, D614G | January 2021 | − |
| Ref. | Country Where Patients Were Sampled | Period of Time | Number of Patients | Type of Samples | Sequencing Methods/Data Availability | Variants | Outcome |
|---|---|---|---|---|---|---|---|
| 1.1. Studies showing no impact | |||||||
| 1.1.1. In vitro and/or animal model studies | |||||||
| [18] | NA | NA | Animal model study with a mutant virus and a wild-type virus | NA | NA | SARS-CoV2 D614 and G614 variants in the Spike glycoprotein | Hamsters infected with the two variants exhibited comparable viral loads in lung tissues as well as similar amounts of virus shedding in nasal washes |
| [19] | NA | NA | In vitro study | NA | NA | 501Y.V2 variant | No significant increase in infectivity was observed in these cell lines for any of the pseudotyped viruses with 501Y.V2-related mutations compared to the reference 614G variant |
| 1.1.2. Studies based on SARS-CoV-2 genomes downloaded from electronic databases or based on community-based testing dataset | |||||||
| [20] | Various | 46,723 | ND | 46,723 complete SARS-CoV-2 genomes downloaded from GISAID | 12,706 variable positions | None of the recurrent SARS-CoV-2 mutations were associated with increased viral transmission | |
| [21] | Singapore | 22 January to 22 April 2020 | The first 10,000 COVID-19 cases were extracted from the Ministry of Health database. 319 patients had SARS-CoV-2 sequences available | Nasopharyngeal swabs | Sequencing of SARS-CoV-2 was performed in one of four laboratories in Singapore together with GISAID submission and case matching. Pangolin COVID-19 Lineage Assigner and CoVsurver were used to assign lineage and clade to each sequence, respectively | 29 were infected with clade S, 90 with clade L/V, 96 with clade G (containing D614G variant), and 104 with other clades ‘O’ | No significant difference in the transmissibility of clade G infections was observed |
| 1.1.3. Clinical studies | |||||||
| [16] | France | 29 February to 4 April 2020 | 309 | Nasopharyngeal swabs | Sequencing by Illumina protocols on MiSeq platform (Illumina) | A total of 321 mutational events were reported in the SARS-CoV-2 genomes divided into 5 clusters. Cluster 1 (44 patients, 14.2%, positions (28,881-28,882-28,883)) with two non-synonymous mutations in protein N (R203K; G204R). Cluster 2 (39 patients, 12.6%, position 15,324) contains a synonymous mutation (C15324U). Cluster 3 (126, 100 and 211 patients, at positions 2416, 8371, 25,563, respectively) includes one synonymous mutation (C2416U), and two non-synonymous mutations (nsp3: Q1884H; ORF3a: Q57H). Cluster 4 (68 patients, 22%, position 1059) contains one non-synonymous mutation (nsp2: T85I). Finally, cluster 5 (from 297 to 303 patients, 96–98%, positions 241, 3037, 14,408, 23,403) displays one mutation in 5′UTR (C241U), one synonymous mutation (C3037U) and two non-synonymous mutations (nsp12b: P314L, S protein 133 D614G) | Coronavirus genome isolates from 38 patients’ isolates with PVirO were widely distributed across the groups, including diverse mutational events meaning that there is no correlation between higher viral loads |
| 1.2. Studies showing increased infectivity | |||||||
| 1.2.1. In vitro—In silico and/or animal model studies | |||||||
| [15] | NA | NA | In vitro study | NA | NA | S mutants reported in the public domain or mutants at putative N-linked glycosylation sites | Pseudotyped viruses expressing either the D614G single mutation or a combination of mutations that included D614G are more infectious than the reference strain, whereas no difference was found between single D614G and D614G combination variants |
| [22] | NA | NA | In vitro study | NA | NA | SARS-CoV2 D614G mutation in the Spike glycoprotein | Pseudovirus G614 infected hACE2-293T cells with approximately 9-fold higher efficiency than did Pseudovirus D614 |
| [23] | NA | NA | In vitro study | NA | NA | New 501Y variant | 501Y variant binds to human Angiotensin Converting Enzyme 2 (ACE2) approximetely10 times more tightly than the native version |
| [24] | NA | NA | In vitro and animal model study | NA | NA | SARS-CoV2 D614G mutation in the Spike glycoprotein | D614G mutation increases the infectivity of SARS-CoV-2 produced from a human lung cell line. Hamsters infected with the G614 variant produced higher infectious titers in the nasal washes and trachea, but not lungs, confirming clinical evidence that the D614G mutation enhances viral loads in the upper respiratory tract of COVID-19 patients |
| [25] | NA | NA | In vitro and animal model study with a mutant virus and a wild-type virus | NA | NA | SARS-CoV-2 D614G mutation in the Spike glycoprotein | SARS-CoV-2 variants harboring the D614G substitution replicated more efficiently in some immortalized epithelial cell lines and exhibited significantly faster droplet transmission between infected hamsters than the wild-type virus |
| [26] | China | 22 January to 4 February 2020 | In vitro and clinical study among 11 patients | Sputum, stool and nasopharyngeal swabs | Deep sequencing by the Novaseq 6000 platform (Illumina) | 33 mutations were identified in 11 isolates | Different viral isolates exhibit significant variations of viral load when infecting Vero-E6 cells. ZJU-1, which clusters with the S-D614G clade, has a viral load 19 times higher than ZJU-2 and ZJU-8. A near 270-fold difference in viral load was observed between ZJU-10 and ZJU-2 at 24 h post infection. In addition, there was a higher viral load leads to a higher cell death ratio |
| [27] | USA | 1 September 2020 to 29 January 2021 | In vivo and in vitro study | Nasopharyngeal swabs | 2172 SARS-CoV-2 genomes were sequenced | B.1.427/B.1.429 and spike L452R variants | B.1.427/B.1.429 variant was 20% more transmissible with 2-fold increased shedding in vivo. The spike L452R mutation conferred increased infectivity in vitro |
| [28] | − | − | In silico study | − | Computational methods, including Molecular Operating Environment (MOE) analysis and software, were used to predict the outcome of substitutions with regard to the protein structure to examine the features acquired by the new variants that enable them to increase the rate of infection and spreading without increasing the severity of COVID-19, the pathology resulting from the infection | B.1.1.7 and B.1.351 variants | B.1.1.7 and B.1.351 variants acquired an increased transmissibility |
| [29] | NA | NA | In vitro study | NA | NA | B.1.351 variant | The three different pseudoviruses of B.1.351 lineage had significantly increased infectivity compared with other mutants that indicated Wuhan strains |
| 1.2.2. Studies analyzing SARS-CoV-2 genomes downloaded from electronic databases or based on community-based testing dataset | |||||||
| [9] | 23 countries | Approximately 20,000 case reports | ND | SARS-CoV-2 strains for each country were extracted from NextStrain open-source project. Amino acid sequences of ORF3a protein were downloaded from NCBI protein database | 218 viral strains from 15 countries were further analyzed for amino acid mutations from NextStrain database | Mutation in ORF3a protein was associated with increased infection of SARS-CoV-2 | |
| [30] | UK | March to May 2020 | 999 | Throat or combined nose/throat swabs | Long-read whole genomesequencing (Oxford Nanopore Technologies (ONT), Oxford, UK) using the ARTIC network protocol | SARS-CoV2 D614G mutation in the Spike glycoprotein | SARS-CoV-2 variants harboring the D614G substitution were associated with potentially higher viral loads in COVID-19 patients |
| [31] | USA | 7823 | 28,726 complete SARS-CoV-2 genome sequences downloaded from GISAID | 4968 single mutations were detected with the top eight missense mutations (i.e., 14,408C > U-(P323L), 23,403A > G-(D614G), 25,563G > U-(Q57H), 1059C > U-(T85I), 28,144U > C-(L84S), 17,858A > G-(Y541C), 17,747C > U-(P504L), and 27,964C > U-(S24L)) identified | Based on co-mutation and time evolution analysis, three concurrent mutations 17,747C > U-(P504L), 17,858A > G-(Y541C), and 28,144U > C tend to fade out, while the other five concurrent mutations can enhance the infectivity of SARS-CoV-2 | ||
| [32] | 17 countries | 24,175 | ND | 24,175 complete SARS-CoV-2 genomes downloaded from GISAID | 11,904 single mutations found in 6 distinct clusters | Mutations on the RBD strengthen the binding of S protein and ACE2, leading to more infectious SARS-CoV-2 | |
| [33] | UK | 29 January to 16 June 2020 | ND | ND | 21,231 614G and 5755 614D de-duplicated whole-genome sequences were downloaded from The COVID-19 Genomics UK consortium dataset | 245 and 62 clusters of 614G and 614D variants containing UK virus genomes from 10 or more different patients were identified, respectively | 614G variant was associated with higher viral load and younger age of patient. |
| [34] | United Kingdom | − | − | − | Bigdata analysis from COVID-19 Genomes UK dataset | VOC 202012/01 variant | VOC 202012/01 variant was 43–82% more transmissible than pre-existing variants |
| [35] | UK | 1 August to 31 December 2020 | 87,830 | − | 94,934 sequences originating from UK were downloaded from GISAID database. The surveillance data of daily number of COVID-19 cases in UK were collected from the World Health Organization (WHO) COVID-19 surveillance platform | VOC 202012/01 variant | VOC 202012/01 variant associated with 46–58% increase in infectivity (reproduction number) |
| [36] | UK | 16 November to 13 December 2020 | 143,994 samples obtained from staff and residents of long-term care facilities throughout England | − | B.1.1.7 variant was identified in samples with S gene target failure on PCR | B.1.1.7 and non-B.1.1.7 variants | B.1.1.7 variant was significantly associated with decrease in median Ct values |
| 1.2.3. Clinical studies | |||||||
| [37] | USA | Mid-March 2020 | 88 | Nasopharyngeal swabs | Library sequencing performed on the Nanopore MinION device using FLO-MIN106D Type R9.4.1 flow cells | Most of the sequences (93%) clustered in three main clades (Clade 1, 2 and 3), defining mutations at the US level | Patients infected with Clade 1 viruses had significantly higher average viral loads in their upper airways relative to patients infected with Clade 2 viruses |
| [38] | Brazil | November to December 2020 | 147 samples | − | Sequencing was conducted using ARTIC V3 multiplexed amplicon scheme and the MinION sequencing platform | P.1 variant | P.1 variant was associated with increased transmissibility |
| [39] | UK | 09 November 2020 to 20 December 2020 | 496 with 341 patients had sequenced samples | Nose and throat samples | Illumina MiSeq 500v2 kits and MiSeq reagent cartridge V2 were used for sequencing. The COG-UK Mutation Explorer was used to identify potential mutations of concern | B.1.1.7 variant (198 patients) and non-B.1.1.7 variant (143 patients) | Significantly lower Ct values associated with B.1.1.7 compared with non-B.1.1.7 |
| Ref. | Country Where Patients Were Sampled | Period of Time | Number of Patients | Type of Samples | Sequencing Methods/Data Availability | Variants | Outcome |
|---|---|---|---|---|---|---|---|
| 2.1. Studies showing no impact | |||||||
| 2.1.1. Studies analyzing SARS-CoV-2 genomes downloaded from electronic databases or based on community-based testing dataset | |||||||
| [40] | − | 1 September 2020 to 31 January 2021 | 182,982 | − | 182,982 complete SARS-CoV-2 strains were retrieved from GISAID | N501Y variant | No statistically significant evidence of change in COVID-19 mortality risk associated with 501Y variants was observed |
| [21] | Singapore | 22 January to 22 April 2020 | The first 10,000 COVID-19 cases were extracted from the Ministry of Health database. 319 patients had SARS-CoV-2 sequences available | Nasopharyngeal swabs | Sequencing of SARS-CoV-2 was performed in one of four laboratories in Singapore together with GISAID submission and case matching. Pangolin COVID-19 Lineage Assigner and CoVsurver were used to assign lineage and clade to each sequence, respectively | 29 were infected with clade S, 90 with clade L/V, 96 with clade G (containing D614G variant), and 104 with other clades, “O” | Infections with clade S or clade O were associated with lower odds of developing hypoxia requiring supplemental oxygen compared with clade L/V. No significant difference in the severity of clade G infections was observed |
| [30] | UK | March to May 2020 | 999 | Throat or combined nose/throat swabs | Long-read whole genome sequencing (Oxford Nanopore Technologies (ONT), Oxford, UK) using the ARTIC network protocol | SARS-CoV2 D614G mutation in the Spike glycoprotein | SARS-CoV-2 variants harboring the D614G substitution were not associated with disease severity |
| [33] | UK | 29 January to 16 June 2020 | ND | ND | 21,231 614G and 5755 614D de-duplicated whole-genome sequences were downloaded from The COVID-19 Genomics UK consortium dataset | 245 and 62 clusters of 614G and 614D variants containing UK virus genomes from 10 or more different patients were identified, respectively | 614G variant was not associated with mortality or severity of COVID-19 |
| [34] | United Kingdom | − | − | − | Bigdata analysis from COVID-19 Genomes UK dataset | VOC 202012/01 variant | No difference in severity of disease (hospitalization, transfer to ICU and death) was observed, compared to VOC 202012/01 and pre-existing variants |
| 2.1.2. Clinical studies | |||||||
| [41] | USA | 5 March to 11 May 2020 (first wave) and 12 May to 7 July 2020 (second wave) | 1026 (first wave) and 4059 (second wave) | Nasopharyngeal swabs | Long reads were generated with the LSK-109 sequencing kit, 24 native barcodes (NBD104 and NBD114 kits), and a GridION instrument (Oxford Nanopore). Short reads were generated with a NexteraXT kit and a NextSeq 550 instrument (Illumina) | SARS-CoV2 D614G mutation in the Spike glycoprotein | No relationship between virus clades and disease severity (overall mortality, transfer to ICU, mechanical ventilation and length of stay) |
| [42] | China | 20 January–25 February 2020 | 112 | Sputum or nasopharyngeal swabs | Sequencing by Illumina protocols on MiSeq platform (Illumina) | Clade I (ORF3a: p.251G > V (subclade V), or S: p.614D > G (subclade G)). Clade II (ORF8: p.84L > S (28,144U > C) and ORF1ab: p.2839S (8782C > U) | There were no significant differences between variants regarding disease severity, leukocytes, lymphocyte and platelet count, CD3 T cell count, hemoglobin, C-reactive protein, Lactose dehydrogenase, complement C3,D-dimer or IL-6 and IL-8 level, or the duration of virus shedding after onset |
| [43] | USA | 1 March to 15 April 2020 | 190 | Nasopharyngeal samples | Samples were sequenced on MiSeq, NextSeq or NovaSeq instruments (Illumina) using 1 × 185, 1 × 75, or 1 × 100 runs, respectively | 97 samples corresponded to what we refer to as “Clade 1” and 91 corresponded to “Clade 2”. Two of 190 samples did not fall into either of the two major clades. When mapped onto GISAID and NextStrain clades, in clade 1, 89 corresponded to clades GH/20C, 6 were mapped to G/20A, and 2 were mapped to G/20B. In clade 2, 86 corresponded to S/19B, and 5 were mapped onto L/19A. 2 of the 190 samples that did not fall into either of the major clades corresponded to GH/20C and S/19B | A trend toward higher rates of hospitalization of patients with Clade 2 virus was observed (p = 0.06). Mortality was not significantly different in patients infected with Clade 1 and 2 viruses |
| [44] | Vietnam | 6 March to 15 April 2020 | 44 | Nasopharyngeal and oropharyngeal swabs | Sequencing was performed on an Illumina Miseq platform (Nextera XT Library preparation kit) | 85 mutations covering 67 variant types among the 44 SARS-CoV-2 genomes. The most ubiquitous modifications were C3037U, C14,408U (P323L) and A23,403G (D614G) occurring in 40/44 samples. Two other variants C241U and GGG to AAC at 28881-3 were detected in 39 and 33 sequences, respectively | These mutations were not associated with differences in phenotype of illness |
| [45] | Uruguay | March to May 2020 | 44 | Naso-oropharyngeal swabs | Whole SARS-CoV-2 genomes were sequenced using Illumina NovaSeq 6000 | D614G mutation | The spike D614G mutation and clade G-related viruses were not associated with any clinical parameters, severity, or lethality of COVID-19 infection |
| [46] | France | March to 14 August 2020 | 417 | Nasopharyngeal swabs | Positive samples for SARS-CoV-2 with Ct < 30 were sequenced using next-generation sequencing and Illumina technology. Positive samples with Ct > 30 or with Ct < 30 but genome sequences that were not obtained were tested for Marseille 1 variant by RT-qPCR | Marseille-1 variant and Clade 20A strains | Compared to Clade 20A strains, the Marseille-1 variant was associated with lower frequency of dyspnea, hypoxemia and hospitalization. However, no significant differences of transfer to ICU and death frequency were observed |
| [47] | France | March to November 2020 | 759 | Nasopharyngeal swabs | Positive samples for SARS-CoV-2 with Ct < 30 were sequenced using next-generation sequencing and Illumina technology | Marseille-4 variant and Clade 20A strains | Compared to Clade 20A strains, the Marseille-4 variant was associated with a lower frequency of cough, rhinitis and olfactory and gustatory disorders but with a higher frequency of hypoxemia. No significant differences of hospitalization, transfer to ICU and death frequency were observed |
| [48] | France | February to April 2020 | 229 | Nasopharyngeal swabs | Positive samples for SARS-CoV-2 with Ct < 20 were sequenced using an RNA metagenomic next-generation sequencing on an Illumina NextSeqTM 550 with mid-output 2 × 150 flow cell | Two frameshifting deletions were detected in the ORF6 protein at the same position (27,267): D26 and D34 | No significant difference in clinical presentation could be observed between hospitalized patients harboring or not harboring the ORF6 deletion |
| [39] | UK | 9 November 2020 to 20 December 2020 | 496 with 341 patients had sequenced samples | Nose and throat samples | Illumina MiSeq 500v2 kits and MiSeq reagent cartridge V2 were used for sequencing. The COG-UK Mutation Explorer was used to identify potential mutations of concern | B.1.1.7 variant (198 patients) and non-B.1.1.7 variant (143 patients) | No association of the B.1.1.7 variant with severe disease was observed in hospitalized patients |
| [49] | USA | 30 March to 17 July 2020 | 27 hospitalized patients | Swab eluate or neat endotracheal aspirate or saliva | Genomes were analyzed by reverse transcription of the viral RNA to make a cDNA copy, PCR amplification of genome segments, Nextera library preparation, and Illumina sequencing | All genomes were found to encode the D614G spike polymorphism | No significant associations were found between SARS-CoV-2 variants and clinical outcomes |
| 2.2. Studies showing a decreased severity of COVID-19 | |||||||
| 2.2.1. Studies analyzing SARS-CoV-2 downloaded from electronic databases or based on community-based testing dataset | |||||||
| [50] | Various | − | 75,775 | ND | 75,775 SARS-CoV-2 complete genome sequences were downloaded from GISAID database. 9912 samples have patient status information recorded as asymptomatic, symptomatic, hospitalized, intensive care unit, deceased. Of which, 537 samples are labeled as asymptomatic (76) and symptomatic (461) cases | 11,083G > U mutation changes leucine to phenylalanine residue at position 37 of NSP6 protein | 11,083G > U mutation was significantly associated with asymptomatic patients (OR = 33.4, p = 8.45.10−35) |
| 2.2.2. Clinical studies | |||||||
| [17] | France | June to September 2020 | 691 | Nasopharyngeal swabs | Next-generation sequencing using Illumina technology with the Illumina Nextera XT Paired end strategy on a MiSeq instrument | Marseille-1 to Marseille-7, located in most SARS-CoV-2 genes including structural and non-structural genes among which nsp2, nsp3 (predicted phosphoesterase), nsp5 (membrane glycoprotein), nsp12 (RNA-dependent RNA polymerase), S (Spike glycoprotein), ORF3a, E (membrane glycoprotein), M (membrane glycoprotein), ORF8 and N (Nucleocapsid phosphoprotein) | SARS-CoV-2 mutation rate was negatively associated with mortality rate |
| [51] | Singapore | 22 January to 21 March 2020 | 131 | Respiratory sample | 2 specific PCRs were used to detect the 382-nucleotide deletion in SARS-CoV-2 | 92 (70%) were infected with the wild-type virus, ten (8%) had a mix of wild-type and ∆382-variant viruses, and 29 (22%) had only the ∆382 variant | Infection with the ∆382 variant was only associated with lower odds of developing hypoxia requiring supplemental oxygen (adjusted odds ratio 0·07 (95% CI 0·00–0·48)) compared with infection with wild-type virus only |
| 2.3. Studies showing an increased severity of COVID-19 | |||||||
| 2.3.1. Animal model studies | |||||||
| [18] | NA | NA | Animal model study with a mutant virus and a wild-type virus | NA | NA | SARS-CoV2 D614 and G614 variants in the Spike glycoprotein | Hamsters infected with the two variants exhibited comparable pathologies in lung tissues |
| 2.3.2. Studies analyzing SARS-CoV-2 downloaded from electronic databases or based on community-based testing dataset | |||||||
| [8] | 50 countries from six geographic areas | 12,343 | ND | 12,343 SARS-CoV-2 sequences isolated in 50 different countries from six geographic areas obtained from GISAID database | 1234 mutations, including 57Q > H, 251G > V (ORF3 protein), 265U > I, 378V > I, 5865Y > C, 5828P > L, 4489A > V, 2016U > K, 3606L > F, 4715P > L (ORF1ab protein), 614D > G (S protein), 204G > R, 203R > K, 13P > L (N protein), 175U > M (M protein), 84L > S (ORF8 protein) | ORF1ab 4715L and S protein 614G variants were significantly more frequent in patients from countries where high fatality rates were reported | |
| [9] | 23 countries | Approximately 20,000 case reports | ND | SARS-CoV-2 strains for each country were extracted from NextStrain open-source project. Amino acid sequences of ORF3a protein were downloaded from NCBI protein database | 218 viral strains from 15 countries were further analyzed for amino acid mutations from NextStrain database | Mutation in ORF3a protein was associated with increased mortality rate of SARS-CoV-2 | |
| [10] | Various | ND | ND | SARS-CoV-2 viral spike sequences were accessed from the GISAID database | D614G variant | Both the average and median case fatality rates correlate strongly (p < 0.02) with the proportion of G614 variant | |
| [11] | Various | 4246 | ND | 4246 SARS-CoV-2 genomes downloaded from GISAID | D614G variant | D614G variant was associated with high mortality related to COVID-19 in European populations | |
| [52] | Various | 152 | Not documented | Genomes of SARS-CoV-2 with patient status. Criteria for selection were full-length sequences and high sequencing coverage (downloaded from the GISAID database) | Two genetic variations were observed at the nucleotide position 11,083, namely thymine (11,083U, 75/152 = 49.34%) and guanine (11,083G, 72/152 = 47.37%) | Viruses causing symptomatic cases tended to have 11,083G (N(11,083U)/N(11,083G) = 15/65). The relative risk ratio of developing symptoms given 11,083G to 11,083U was (65/72)/(15/75) = 4.51 times (95% confidence interval = 2.85–7.14), and the odds ratio was estimated to be 37.14 by the Wald method (95% confidence interval = 14.17–97.33) | |
| [53] | Various | 73,020 | ND | 72,331 viral sequences downloaded from GISAID database. Clinical data were available for 5094 patients, and 3184 of them also had follow-up data | 2121 different mutations affecting the protein structure were identified | Mutations leading to severe outcome with low prevalence were found in the surface (S) glycoprotein and in NSP7 | |
| [54] | Various | 3608 | ND | 3068 SARS-CoV-2 genomes downloaded from GISAID | 7 different variants (Clade G, GH, GR, L, O, S and V) | Patients infected with virus clades L, G and O are exposed to higher risk than the base level | |
| [55] | UK | − | 1096 | ND | 1096 SARS-CoV-2 complete sequences were downloaded from UK Biobank | 216 different verified super-variants across 10 repetitions of the discovery-validation procedure were found. Two super-variants, chr6_148 and chr7_23, were identified in 4 out of 10 repetitions. Six other super-variants, chr2_197, chr2_221, chr8_99, chr10_57, chr16_4 and chr17_26 were identified in 3 out of 10 repetitions | Eight genetic variants are identified to significantly increased risk of COVID-19 mortality |
| [56] | Various | April to July 2020 | 41,304 | ND | 41,304 SARS-CoV-2 protein sequences from 49 different countries were downloaded from NCBI GenBank | Mutation at NSP6, ORF8, S, M, E and N proteins | A relationship of positive tendency between the death rate and the mutation rate was noted in the cases of NSP6 and S proteins |
| [57] | − | − | 2443 | − | 45,000 genomes were downloaded from GISAID database. However, 2443 sequences with patient status were available and only 102 were analyses (56 in severe group (defined as “severe”, “ICU”, “died”) and 46 in mild group (defined as “mild”, “asymptomatic” and “not hospitalized”)) | 103 mutations in the mildly affected group (37 silent and 66 missense mutations) and 111 mutations (40 silent and 71 missense mutations) in the severely affected group were identified, including A1812D, L3606F, P4715L, D614G, A879S, Q57H, L84S, S194L, S202N, R203K and G204R variants | Spike protein D614G and RdRp P323L mutations in SARS-CoV-2 were associated with severity of COVID-19 |
| [58] | − | December 2019 to 26 June 2020 | 3205 whole-genome sequences | − | 3205 whole-genome sequences were collected from GISAID database | Phylogenetic analysis revealed four well-resolved clades (G, GH, GR and L.S.O.V) | Clade GR associated with a high mortality rate. Clades G and GH have intermediate mortality rates |
| [59] | − | − | 90,000 genome sequences | − | 692 SARS-CoV-2 genomic sequences originating from the USA, India, Italy, France and Spain downloaded from GISAID database | D614G variant | D614G variant was positively associated with case severity |
| [60] | − | − | 69,571 | 69,571 SARS-CoV-2 sequences isolated in 100 different countries from six geographic areas obtained from GISAID database | Lineage G (G, GH and GR), characterized by the D614G mutation of Spike protein | The SARS-CoV-2 variant lineage G (S-D614G) was associated with increased disease severity of COVID-19 | |
| [61] | UK | 1 October 2020 to 29 January 2021 | 54,906 paired patients from community-based COVID-19 testing centers | − | SARS-CoV-2 positive test results as S gene positive (compatible with previous variants) when cycle threshold values were: S gene <30, N gene <30, and ORF1ab gene <30. B.1.1.7 variant was classified as S gene negative, when cycle threshold values were: S gene not detected, N gene <30, and ORF1ab gene <30 | B.1.1.7 variant | The B.1.1.7 variant was associated with 1.64-fold increase in mortality hazard ratio |
| [62] | UK | 16 November 2020 to 11 January 2021 | 184,786 patients with clinical data retired from OpenSAFELY electronic health records | − | − | B.1.1.7 and non-B.1.1.7 variants | Relative hazard of death was higher in the patients infected with B.1.1.7 variant. This variant was significantly associated with higher absolute risk of death 28 days after a SARS-CoV-2 positive test |
| [63] | UK | 1 November 2020 to 14 February 2021 | 1,146,534 patients retired from community dataset | − | Bigdata analysis from datasets provided by Public Health England | B.1.1.7 and non-B.1.1.7 variants | Hazard of death associated with B.1.1.7 is 61% (42–82%) higher than with pre-existing variants |
| [64] | − | 23 December 2019 to 21 March 2020 | 1962 SARS-CoV-2 genomes | − | SARS-CoV-2 genomes were retired from 2019nCoVR. The COVID-19 infection, mortality, and recovery rates were collected from the USA Centers for Disease Control and Prevention (CDC) and virusncov | ORF1ab (p.5828P > L and p.5865Y > C), NSP13 (P504L and Y541C) | ORF1ab variation (p.5828P > L and p.5865Y > C) and NSP13 variation (P504L and Y541C) were associated with high infection and fatality rates |
| [65] | − | − | 27,304 | − | 27,304 SARS-CoV-2 sequences were downloaded from GISAID database | Clade G A23403G (S:D614G) variant | Clade G viruses was associated with higher death rates |
| [66] | UK | 1 November 2020 to 27 January 2021 | 198,420 patients extracted from a large primary care (QResearch), the national critical care (ICNARC CMP) and the COVID-19 testing (PHE) database | B.1.1.7 variant was classified as S-gene molecular diagnostic assay failure | B.1.1.7 variant | Patients with B.1.1.7 variant were at increased risk of critical care admission and mortality compared with patients without. For patients receiving critical care, mortality appears independent of virus strain | |
| [67] | − | Up to 13 November 2020 | 2213 | 2213 complete genomes were downloaded from NCBI and GISAID databases. Clinical data were available for 118 patients | Seven frequent mutations resulting in dN substitutions were identified | P323L, D614G, R203K and G204R substitutions were associated with disease severity | |
| [68] | USA | 1 January to 5 April 2021 | 9765 SARS-CoV-2 specimens | SARS-CoV-2 specimens were sequenced at the Public Health Laboratory or the Pandemic Response Laboratory | B.1.526 (3679 patients), B.1.1.7 (1815 patients) and non-variant of concern (VOCs)/variant of interest (VOIs) variants | No difference in hospitalization or death rate was observed between B.1.526 and non-VOI/VOC variants. Patients infected with the B.1.1.7 variant were more likely to be hospitalized than those with non-VOI/VOC infections | |
| [69] | USA | 4 January to 20 March 2021 | 327 | Data analyzed 327 COVID-19 B.1.427/B.1.429 cases retired from Colorado Department of Public Health and Environment database | B.1.427/B.1.429 | B.1.427/B.1.429 more frequently cause discernible and severe illness than nationally circulating lineages do overall | |
| 2.3.3. Clinical studies | |||||||
| [70] | France | March 2020 to January 2021 | 740 | Nasopharyngeal swabs | Next-generation sequencing using Illumina technology with the Illumina Nextera XT Paired end strategy on a MiSeq instrument (for Clade 20A, Marseille-1 and Marseille-4 variants identification) and real-time PCR for N501Y variant identification | Clade 20A (254 patients), Marseille-1 (85 patients), Marseille-4 (190 patients) and N501Y (211 patients) variants | A lower rate of hospitalization associated with N501Y variant infection as compared to Clade 20A and Marseille-4 variant. A higher hospitalization rate was associated with Marseille-4 variant and Clade 20A infection as compared to Marseille-1 variant |
| [71] | USA | 11 March to 22 April 2020 | 302 | Nasopharyngeal swabs | Genomic sequences were constructed for each isolate according to variants called from sequence reads and the reference sequence (NC_045512.2). Multiple sequence alignments were performed using MAFFT software version 7.0. SARS-CoV-2 clade assignment followed GISAID clade guidelines and lineage nomenclature | 6 different viral clades circulated; G, GR, and GH (clade group 2) represented 84.4% (255 of 302) of all identified isolates. The remainder included V, S, and Wuhan clades (clade group 1) | Clade group 1 infection was associated with higher mortality than clade group 2 |
| [72] | UAE | 29 January to 30 June 2020 | 256 | Nasopharyngeal swabs | RNA libraries were prepared using the TruSeq Stranded Total RNA Library kit from Illumina. Libraries were sequenced using the NovaSeq SP Reagent kit from Illumina | 115 patients had SARS-CoV-2 sequences. A total of 986 mutations were identified in 115 genomes, 272 were unique (majority were missense, n = 134) and 20/272 mutations were novel | A missense (Q271R) and synonymous (R41R) mutation in the S and N proteins, respectively, were identified in 2/27 patients with severe COVID-19 but not in patients with mild or moderate disease (0/86); p = 0.05 |
| [73] | Brazil | January to February 2021 | 68 | Oro/nasopharyngeal swab | Sequencing libraries were prepared using the CleanPlex SARS-CoV-2 panel (Paragon Genomics, Hayward, CA, USA) protocol. The resulting libraries were pooled in equimolar amounts and sequenced in Illumina MiSeq (Illumina, San Diego, CA, USA) | P.1 variant | P.1 variant was associated with increase in COVID-19 cases and hospitalization rate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dao, T.L.; Hoang, V.T.; Colson, P.; Lagier, J.C.; Million, M.; Raoult, D.; Levasseur, A.; Gautret, P. SARS-CoV-2 Infectivity and Severity of COVID-19 According to SARS-CoV-2 Variants: Current Evidence. J. Clin. Med. 2021, 10, 2635. https://doi.org/10.3390/jcm10122635
Dao TL, Hoang VT, Colson P, Lagier JC, Million M, Raoult D, Levasseur A, Gautret P. SARS-CoV-2 Infectivity and Severity of COVID-19 According to SARS-CoV-2 Variants: Current Evidence. Journal of Clinical Medicine. 2021; 10(12):2635. https://doi.org/10.3390/jcm10122635
Chicago/Turabian StyleDao, Thi Loi, Van Thuan Hoang, Philippe Colson, Jean Christophe Lagier, Matthieu Million, Didier Raoult, Anthony Levasseur, and Philippe Gautret. 2021. "SARS-CoV-2 Infectivity and Severity of COVID-19 According to SARS-CoV-2 Variants: Current Evidence" Journal of Clinical Medicine 10, no. 12: 2635. https://doi.org/10.3390/jcm10122635
APA StyleDao, T. L., Hoang, V. T., Colson, P., Lagier, J. C., Million, M., Raoult, D., Levasseur, A., & Gautret, P. (2021). SARS-CoV-2 Infectivity and Severity of COVID-19 According to SARS-CoV-2 Variants: Current Evidence. Journal of Clinical Medicine, 10(12), 2635. https://doi.org/10.3390/jcm10122635