
Biologic and Therapeutic Implications of Genomic Alterations in Acute Lymphoblastic Leukemia



Abstract
:1. Introduction
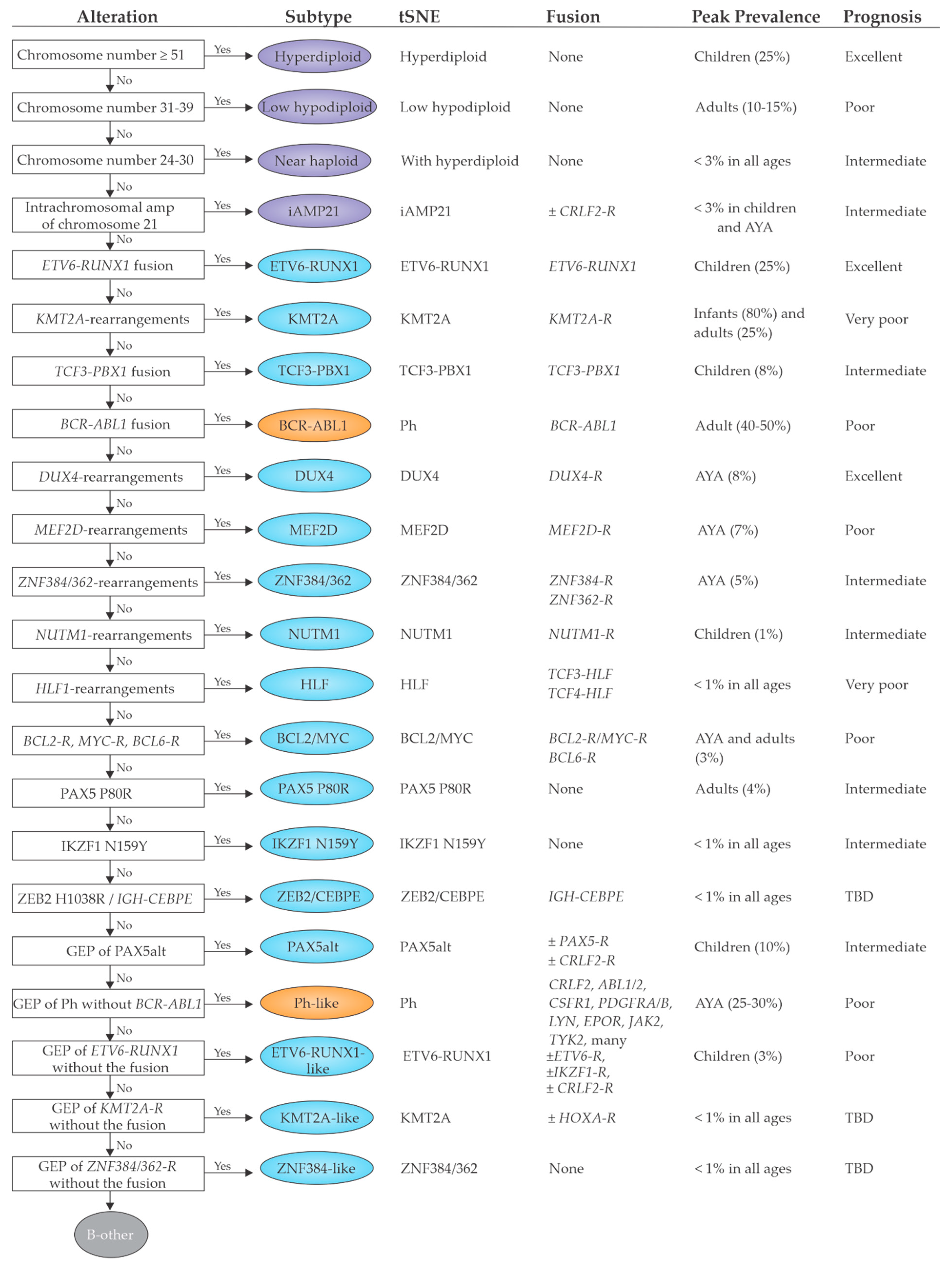
2. B-Cell Precursor Acute Lymphoblastic Leukemia
2.1. Previously Established Subtypes with Recurring Chromosomal Abnormalities
2.1.1. Subtypes with Chromosomal Aneuploidy
2.1.2. iAMP21
2.1.3. Subtypes with Recurrent Chromosomal Translocations and/or Gene Fusions
2.2. Emerging B-ALL Subtypes Defined by Genome Sequencing Studies
2.2.1. DUX4, MEF2D, ZNF384 and NUTM1 Gene Fusions
2.2.2. Subtypes That Phenocopy Established Subtypes
Ph-Like ALL
ETV6-RUNX1-Like ALL
2.2.3. Subtypes Defined by a Single Point Mutation
PAX5 P80R and PAX5alt
IKZF1 N159Y
ZEB2 H1038R and IGH-CEBPE
2.3. Prognostic Implications
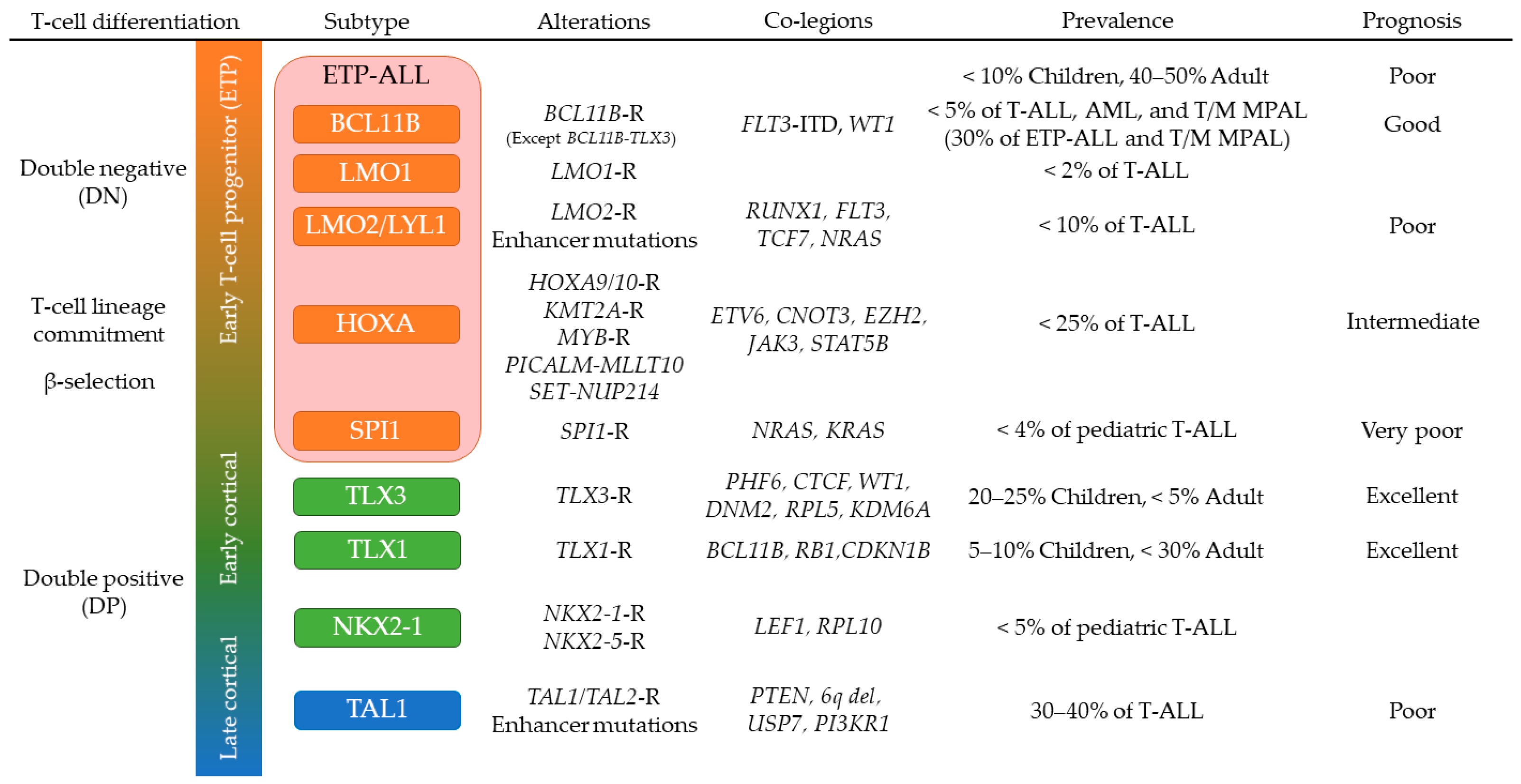
3. T-Cell Acute Lymphoblastic Leukemia (T-ALL)
3.1. Genomic Overview of T-ALL
3.2. T-ALL in Early Stages of Cortical Thymocyte Maturation
3.3. TAL1-Driven T-ALL with Late Stages of Cortical Thymocyte Maturation
3.4. Early T-Cell Precursor (ETP) ALL and Mixed Phenotype Acute Leukemia
3.5. NOTCH1 Activating Mutations in T-ALL
4. Implications for Diagnosis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tran, T.H.; Hunger, S.P. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Pui, C.-H. Precision medicine in acute lymphoblastic leukemia. Front. Med. 2020, 14, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Mullighan, C.G. Pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 2524. [Google Scholar] [CrossRef] [PubMed]
- Stock, W.; Luger, S.M.; Advani, A.S.; Yin, J.; Harvey, R.C.; Mullighan, C.G.; Willman, C.L.; Fulton, N.; Laumann, K.M.; Malnassy, G.; et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: Results of CALGB 10403. Blood 2019, 133, 1548–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montefiori, L.E.; Bendig, S.; Gu, Z.; Chen, X.; Polonen, P.; Ma, X.; Murison, A.; Zeng, A.; Garcia-Prat, L.; Dickerson, K.; et al. Enhancer hijacking drives oncogenic BCL11B expression in lineage ambiguous stem cell leukemia. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Pui, C.-H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2018, 16, 227–240. [Google Scholar] [CrossRef]
- Schwab, C.; Harrison, C.J. Advances in B-cell Precursor Acute Lymphoblastic Leukemia Genomics. HemaSphere 2018, 2, e53. [Google Scholar] [CrossRef]
- Li, J.; Dai, Y.; Wu, L.; Zhang, M.; Ouyang, W.; Huang, J.; Chen, S. Emerging molecular subtypes and therapeutic targets in B-cell precursor acute lymphoblastic leukemia. Front. Med. 2021, 15, 347–371. [Google Scholar] [CrossRef]
- Mullighan, C.G. How advanced are we in targeting novel subtypes of ALL? Best Pract. Res. Clin. Haematol. 2019, 32, 101095. [Google Scholar] [CrossRef]
- Kimura, S.; Mullighan, C.G. Molecular markers in ALL: Clinical implications. Best Pract. Res. Clin. Haematol. 2020, 33, 101193. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-F.; Dai, Y.-T.; Lilljebjörn, H.; Shen, S.-H.; Cui, B.-W.; Bai, L.; Liu, Y.-F.; Qian, M.-X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1223 cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, K.G.; Morin, R.D.; Zhang, J.; Hirst, M.; Zhao, Y.; Su, X.; Chen, S.-C.; Payne-Turner, D.; Churchman, M.L.; Harvey, R.; et al. Genetic Alterations Activating Kinase and Cytokine Receptor Signaling in High-Risk Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Klco, J.M.; Mullighan, C.G. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat. Rev. Cancer 2020, 21, 122–137. [Google Scholar] [CrossRef]
- Perez-Andreu, V.; Roberts, K.G.; Harvey, R.; Yang, W.; Cheng, C.; Pei, D.; Xu, H.; Gastier-Foster, J.; Lim, J.Y.-S.; Chen, I.-M.; et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat. Genet. 2013, 45, 1494–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinghaus, E.; Stanulla, M.; Richter, G.; Kronnie, G.T.; Cario, G.; Cazzaniga, G.; Horstmann, M.; Grümayer, R.P.; Cavé, H.; Trka, J.; et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia 2011, 26, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Zhao, X.; Devidas, M.; Yang, W.; Gocho, Y.; Smith, C.; Gastier-Foster, J.M.; Li, Y.; Xu, H.; Zhang, S.; et al. Genome-Wide Association Study of Susceptibility Loci for T-Cell Acute Lymphoblastic Leukemia in Children. J. Natl. Cancer Inst. 2019, 111, 1350–1357. [Google Scholar] [CrossRef]
- Qian, M.; Xu, H.; Perez-Andreu, V.; Roberts, K.G.; Zhang, H.; Yang, W.; Zhang, S.; Zhao, X.; Smith, C.; Devidas, M.; et al. Novel susceptibility variants at the ERG locus for childhood acute lymphoblastic leukemia in Hispanics. Blood 2019, 133, 724–729. [Google Scholar] [CrossRef] [Green Version]
- Molina, O.; Abad, M.A.; Solé, F.; Menéndez, P. Aneuploidy in Cancer: Lessons from Acute Lymphoblastic Leukemia. Trends Cancer 2020, 7, 37–47. [Google Scholar] [CrossRef]
- Greaves, M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat. Rev. Cancer 2018, 18, 471–484. [Google Scholar] [CrossRef]
- Paulsson, K.; Lilljebjörn, H.; Biloglav, A.; Olsson, L.; Rissler, M.; Castor, A.; Barbany, G.; Fogelstrand, L.; Nordgren, A.; Sjögren, H.; et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat. Genet. 2015, 47, 672–676. [Google Scholar] [CrossRef]
- Paulsson, K.; Johansson, B. High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosom. Cancer 2009, 48, 637–660. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Zhang, J.; Kasper, L.H.; Lerach, S.; Payne-Turner, D.; Phillips, L.A.; Heatley, S.; Holmfeldt, L.; Collins-Underwood, J.R.; Ma, J.; et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, O.; Vinyoles, M.; Granada, I.; Roca-Ho, H.; Gutierrez-Agüera, F.; Valledor, L.; López, C.M.L.; Rodríguez-González, P.; Trincado, J.L.; Tirados-Menéndez, S.; et al. Impaired Condensin Complex and Aurora B kinase underlie mitotic and chromosomal defects in hyperdiploid B-cell ALL. Blood 2020, 136, 313–327. [Google Scholar] [CrossRef]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Hagström-Andersson, A.; Chen, S.-C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comeaux, E.Q.; Mullighan, C.G. TP53Mutations in Hypodiploid Acute Lymphoblastic Leukemia. Cold Spring Harb. Perspect. Med. 2016, 7, a026286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, A.J.; Shago, M.; Mikhail, F.M.; Raimondi, S.C.; Hirsch, B.A.; Loh, M.L.; Raetz, E.A.; Borowitz, M.J.; Wood, B.L.; Maloney, K.W.; et al. Masked hypodiploidy: Hypodiploid acute lymphoblastic leukemia (ALL) mimicking hyperdiploid ALL in children: A report from the Children’s Oncology Group. Cancer Genet. 2019, 238, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Jeha, S.; Pei, D.; Payne-Turner, D.; Coustan-Smith, E.; Roberts, K.G.; Waanders, E.; Choi, J.K.; Ma, X.; Raimondi, S.C.; et al. Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood 2015, 126, 2896–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pui, C.-H.; Rebora, P.; Schrappe, M.; Attarbaschi, A.; Baruchel, A.; Basso, G.; Cavé, H.; Elitzur, S.; Koh, K.; Liu, H.-C.; et al. Outcome of Children with Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J. Clin. Oncol. 2019, 37, 770–779. [Google Scholar] [CrossRef] [PubMed]
- McNeer, J.L.; Devidas, M.; Dai, Y.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; Kahwash, S.; Borowitz, M.J.; Wood, B.L.; Larsen, E.; et al. Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children with Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Ribera, J.; Granada, I.; Morgades, M.; Vives, S.; Genescà, E.; González, C.; Nomdedeu, J.; Escoda, L.; Montesinos, P.; Mercadal, S.; et al. The poor prognosis of low hypodiploidy in adults with B-cell precursor acute lymphoblastic leukaemia is restricted to older adults and elderly patients. Br. J. Haematol. 2019, 186, 263–268. [Google Scholar] [CrossRef]
- Diaz-Flores, E.; Comeaux, E.Q.; Kim, K.L.; Melnik, E.M.; Beckman, K.; Davis, K.L.; Wu, K.; Akutagawa, J.; Bridges, O.; Marino, R.; et al. Bcl-2 Is a Therapeutic Target for Hypodiploid B-Lineage Acute Lymphoblastic Leukemia. Cancer Res. 2019, 79, 2339–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C.J. Blood Spotlight on iAMP21 acute lymphoblastic leukemia (ALL), a high-risk pediatric disease. Blood 2015, 125, 1383–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C.J.; Schwab, C. Constitutional abnormalities of chromosome 21 predispose to iAMP21-acute lymphoblastic leukaemia. Eur. J. Med. Genet. 2016, 59, 162–165. [Google Scholar] [CrossRef]
- Moorman, A.V.; Robinson, H.; Schwab, C.; Richards, S.M.; Hancock, J.; Mitchell, C.D.; Goulden, N.; Vora, A.; Harrison, C. Risk-Directed Treatment Intensification Significantly Reduces the Risk of Relapse among Children and Adolescents with Acute Lymphoblastic Leukemia and Intrachromosomal Amplification of Chromosome 21: A Comparison of the MRC ALL97/99 and UKALL2003 Trials. J. Clin. Oncol. 2013, 31, 3389–3396. [Google Scholar] [CrossRef]
- Golub, T.; McLean, T.; Stegmaier, K.; Ritz, J.; Sallan, S.; Neuberg, D.; Gilliland, D.G. TEL-AML1: The most common gene rearrangement in childhood ALL. Blood 1995, 86, 2377. [Google Scholar]
- Sundaresh, A.; Williams, O. Mechanism of ETV6-RUNX1 Leukemia. Adv. Exp. Med. Biol. 2017, 962, 201–216. [Google Scholar] [CrossRef]
- Shurtleff, S.A.; Buijs, A.; Behm, F.G.; Rubnitz, J.; Raimondi, S.C.; Hancock, M.L.; Chan, G.C.F.; Pui, C.H.; Grosveld, G.; Downing, J.R. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia 1995, 9, 1985–1989. [Google Scholar]
- Ford, A.M.; Greaves, M. ETV6-RUNX1 + Acute Lymphoblastic Leukaemia in Identical Twins. Adv. Exp. Med. Biol. 2017, 962, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, R.P.; Schoenmakers, E.F.P.M.; Van Reijmersdal, S.V.; Hehir-Kwa, J.Y.; Van Kessel, A.G.; van Leeuwen, F.N.; Hoogerbrugge, P.M. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia 2007, 21, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Rapado, I.; Li, Y.; Potter, N.E.; Wedge, D.; Tubio, J.; Alexandrov, L.B.; Van Loo, P.; Cooke, S.L.; Marshall, J.; et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmeister, T.; Gökbuget, N.; Schwartz, S.; Fischer, L.; Hubert, D.; Sindram, A.; Hoelzer, D.; Thiel, E. Clinical features and prognostic implications of TCF3-PBX1 and ETV6-RUNX1 in adult acute lymphoblastic leukemia. Haematologica 2009, 95, 241–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeha, S.; Pei, D.; Choi, J.; Cheng, C.; Sandlund, J.T.; Coustan-Smith, E.; Campana, D.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia without Cranial Irradiation: St Jude Total Therapy Study 16. J. Clin. Oncol. 2019, 37, 3377–3391. [Google Scholar] [CrossRef]
- Pui, C.-H.; Tang, J.-Y.; Yang, J.J.; Chen, S.-J.; Chen, Z. International Collaboration to Save Children with Acute Lymphoblastic Leukemia. J. Glob. Oncol. 2019, 5, 1–2. [Google Scholar] [CrossRef]
- Buchner, M.; Müschen, M. Targeting the B-cell receptor signaling pathway in B lymphoid malignancies. Curr. Opin. Hematol. 2014, 21, 341–349. [Google Scholar] [CrossRef]
- Van der Veer, A.; van der Velden, V.H.; Willemse, M.E.; Hoogeveen, P.G.; Petricoin, E.F.; Beverloo, H.B.; Escherich, G.; Horstmann, M.A.; Pieters, R.; den Boer, M.L. Interference with pre-B-cell receptor signaling offers a therapeutic option for TCF3-rearranged childhood acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bicocca, V.; Chang, B.; Masouleh, B.K.; Muschen, M.; Loriaux, M.M.; Druker, B.J.; Tyner, J.W. Crosstalk between ROR1 and the Pre-B Cell Receptor Promotes Survival of t(1;19) Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.-J.; Bornhauser, B.; Gombert, M.; Kratsch, C.; Stütz, A.M.; et al. Genomics and drug profiling of fatal TCF3-HLF−positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Frismantas, V.; Dobay, M.P.; Rinaldi, A.; Tchinda, J.; Dunn, S.H.; Kunz, J.; Richter-Pechanska, P.; Marovca, B.; Pail, O.; Jenni, S.; et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood 2017, 129, e26–e37. [Google Scholar] [CrossRef] [Green Version]
- Glover, J.M.; Loriaux, M.; Tyner, J.; Druker, B.; Chang, B.H. In vitro sensitivity to dasatinib in lymphoblasts from a patient with t(17;19)(q22;p13) gene rearrangement pre-B acute lymphoblastic leukemia. Pediatr. Blood Cancer 2011, 59, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Mouttet, B.; Vinti, L.; Ancliff, P.; Bodmer, N.; Brethon, B.; Cario, G.; Chen-Santel, C.; Elitzur, S.; Hazar, V.; Kunz, J.; et al. Durable remissions in TCF3-HLF positive acute lymphoblastic leukemia with blinatumomab and stem cell transplantation. Haematologica 2019, 104, e244–e247. [Google Scholar] [CrossRef] [Green Version]
- Leonard, J.; Wolf, J.S.; Degnin, M.; Eide, C.A.; LaTocha, D.; Lenz, K.; Wilmot, B.; Mullighan, C.G.; Loh, M.; Hunger, S.P.; et al. Aurora A kinase as a target for therapy in TCF3-HLF rearranged acute lymphoblastic leukemia. Haematologica 2021. [Google Scholar] [CrossRef] [PubMed]
- El Chaer, F.; Keng, M.; Ballen, K.K. MLL-Rearranged Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2020, 15, 83–89. [Google Scholar] [CrossRef]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; Boer, M.D.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2001, 30, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, E.-J.; Ross, M.E.; Shurtleff, S.A.; Williams, W.; Patel, D.; Mahfouz, R.; Behm, F.G.; Raimondi, S.C.; Relling, M.V.; Patel, A.; et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002, 1, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Valentine, M.C.; Linabery, A.; Chasnoff, S.; Hughes, A.E.O.; Mallaney, C.; Sanchez, N.; Giacalone, J.; Heerema, N.A.; Hilden, J.M.; Spector, L.; et al. Excess congenital non-synonymous variation in leukemia-associated genes in MLL−infant leukemia: A Children’s Oncology Group report. Leukemia 2013, 28, 1235–1241. [Google Scholar] [CrossRef]
- Perner, F.; Gadrey, J.Y.; Xiong, Y.; Hatton, C.; Eschle, B.K.; Weiss, A.; Stauffer, F.; Gaul, C.; Tiedt, R.; Perry, J.A.; et al. Novel inhibitors of the histone methyltransferase DOT1L show potent antileukemic activity in patient-derived xenografts. Blood 2020, 136, 1983–1988. [Google Scholar] [CrossRef]
- Chen, C.-W.; Koche, R.; Sinha, A.U.; Deshpande, A.J.; Zhu, N.; Eng, R.; Doench, J.; Xu, H.; Chu, S.H.; Qi, J.; et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat. Med. 2015, 21, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.; Linhares, B.M.; Chen, D.; Jih, G.; Perkey, E.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2019, 130, 981–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Kohler, M.E.; Fry, T.; Ernst, P. Does lineage plasticity enable escape from CAR-T cell therapy? Lessons from MLL-r leukemia. Exp. Hematol. 2021, 100, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.; Chen, I.-M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome–Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef]
- Bernt, K.; Hunger, S.P.M. Current Concepts in Pediatric Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Front. Oncol. 2014, 4, 54. [Google Scholar] [CrossRef] [PubMed]
- Slayton, W.B.; Schultz, K.R.; Kairalla, J.A.; Devidas, M.; Mi, X.; Pulsipher, M.A.; Chang, B.H.; Mullighan, C.; Iacobucci, I.; Silverman, L.B.; et al. Dasatinib Plus Intensive Chemotherapy in Children, Adolescents, and Young Adults with Philadelphia Chromosome–Positive Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0622. J. Clin. Oncol. 2018, 36, 2306–2314. [Google Scholar] [CrossRef]
- Foà, R.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, M.-C.; Canichella, M.; Viero, P.; Ferrara, F.; Lunghi, M.; et al. Dasatinib–Blinatumomab for Ph-Positive Acute Lymphoblastic Leukemia in Adults. N. Engl. J. Med. 2020, 383, 1613–1623. [Google Scholar] [CrossRef]
- Foà, R.; Vitale, A.; Vignetti, M.; Meloni, G.; Guarini, A.; De Propris, M.S.; Elia, L.; Paoloni, F.; Fazi, P.; Cimino, G.; et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome–positive acute lymphoblastic leukemia. Blood 2011, 118, 6521–6528. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.; Miller, C.B.; Radtke, I.; Phillips, L.A.; Dalton, J.T.; Ma, J.; White, D.; Hughes, T.; Le Beau, M.M.; Pui, C.-H.; et al. BCR–ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008, 453, 110–114. [Google Scholar] [CrossRef]
- Iacobucci, I.; Storlazzi, C.T.; Cilloni, D.; Lonetti, A.; Ottaviani, E.; Soverini, S.; Astolfi, A.; Chiaretti, S.; Vitale, A.; Messa, F.; et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1–positive acute lymphoblastic leukemia patients: On behalf of Gruppo Italiano Malattie Ematologiche dell’Adulto Acute Leukemia Working Party (GIMEMA AL WP). Blood 2009, 114, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, G.; Iacobucci, I.; Storlazzi, C.T.; Vignetti, M.; Paoloni, F.; Cilloni, D.; Soverini, S.; Vitale, A.; Chiaretti, S.; Cimino, G.; et al. IKZF1 (Ikaros) Deletions in BCR-ABL1–Positive Acute Lymphoblastic Leukemia Are Associated with Short Disease-Free Survival and High Rate of Cumulative Incidence of Relapse: A GIMEMA AL WP Report. J. Clin. Oncol. 2009, 27, 5202–5207. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-F.; Wang, B.-Y.; Zhang, W.-N.; Huang, J.-Y.; Li, B.-S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.-J.; Dai, Y.-J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilljebjörn, H.; Henningsson, R.; Wittsten, A.H.; Olsson, L.; Orsmark-Pietras, C.; Von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, M.S.G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Tsuzuki, S.; Kawazu, M.; Hayakawa, F.; Kojima, S.; Ueno, T.; Imoto, N.; Kohsaka, S.; Kunita, A.; Doi, K.; et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat. Genet. 2016, 48, 569–574. [Google Scholar] [CrossRef]
- Zhang, J.; McCastlain, K.; Yoshihara, H.; Xu, B.; Chang, Y.; Churchman, M.L.; Wu, G.; Li, Y.; Wei, L.; Iacobucci, I.; et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat. Genet. 2016, 48, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Dib, C.; Zakharova, V.; Popova, E.; Kiseleva, E.; Chernyak, B.; Lipinski, M.; Vassetzky, Y.S. DUX4 Pathological Expression: Causes and Consequences in Cancer. Trends Cancer 2019, 5, 268–271. [Google Scholar] [CrossRef]
- Himeda, C.L.; Jones, P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu. Rev. Genom. Hum. Genet. 2019, 20, 265–291. [Google Scholar] [CrossRef]
- Miettinen, M.; Felisiak-Golabek, A.; Contreras, A.L.; Glod, J.; Kaplan, R.N.; Killian, J.K.; Lasota, J. New fusion sarcomas: Histopathology and clinical significance of selected entities. Hum. Pathol. 2019, 86, 57–65. [Google Scholar] [CrossRef]
- Sirvent, N.; Trassard, M.; Ebran, N.; Attias, R.; Pedeutour, F. Fusion of EWSR1 with the DUX4 facioscapulohumeral muscular dystrophy region resulting from t(4;22)(q35;q12) in a case of embryonal rhabdomyosarcoma. Cancer Genet. Cytogenet. 2009, 195, 12–18. [Google Scholar] [CrossRef]
- Jeha, S.; Choi, J.; Roberts, K.G.; Pei, D.; Coustan-Smith, E.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; Gruber, T.A.; Raimondi, S.C.; et al. Clinical Significance of Novel Subtypes of Acute Lymphoblastic Leukemia in the Context of Minimal Residual Disease–Directed Therapy. Blood Cancer Discov. 2021, 2, 326–337. [Google Scholar] [CrossRef]
- Paietta, E.; Roberts, K.G.; Wang, V.; Gu, Z.; Buck, G.; Pei, D.; Cheng, C.; Levine, R.L.; Abdel-Wahab, O.; Cheng, Z.; et al. Molecular Classification Improves Risk Assessment in Adult BCR-ABL1-negative B-ALL. Blood 2021. [Google Scholar] [CrossRef]
- Schinnerl, D.; Mejstrikova, E.; Schumich, A.; Zaliova, M.; Fortschegger, K.; Nebral, K.; Attarbaschi, A.; Fiser, K.; Kauer, M.O.; Popitsch, N.; et al. CD371 cell surface expression: A unique feature of DUX4-rearranged acute lymphoblastic leukemia. Haematologica 2019, 104, e352–e355. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Okuno, Y.; Kawashima, N.; Muramatsu, H.; Okuno, T.; Wang, X.; Kataoka, S.; Sekiya, Y.; Hamada, M.; Murakami, N.; et al. MEF2D-BCL9 Fusion Gene Is Associated With High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Adolescents. J. Clin. Oncol. 2016, 34, 3451–3459. [Google Scholar] [CrossRef] [PubMed]
- Ohki, K.; Kiyokawa, N.; Saito, Y.; Hirabayashi, S.; Nakabayashi, K.; Ichikawa, H.; Momozawa, Y.; Okamura, K.; Yoshimi, A.; Ogata-Kawata, H.; et al. Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica 2018, 104, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, H.; Yoshida, K.; Shiozawa, Y.; Nannya, Y.; Iijima-Yamashita, Y.; Kiyokawa, N.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Isobe, T.; et al. Landscape of driver mutations and their clinical impacts in pediatric B-cell precursor acute lymphoblastic leukemia. Blood Adv. 2020, 4, 5165–5173. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, S.; Yasuda, T.; Kojima, S.; Kawazu, M.; Akahane, K.; Inukai, T.; Imaizumi, M.; Morishita, T.; Miyamura, K.; Ueno, T.; et al. Targeting MEF2D-fusion Oncogenic Transcriptional Circuitries in B-cell Precursor Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2020, 1, 82–95. [Google Scholar] [CrossRef]
- Tange, N.; Hayakawa, F.; Yasuda, T.; Odaira, K.; Yamamoto, H.; Hirano, D.; Sakai, T.; Terakura, S.; Tsuzuki, S.; Kiyoi, H. Staurosporine and venetoclax induce the caspase-dependent proteolysis of MEF2D-fusion proteins and apoptosis in MEF2D-fusion (+) ALL cells. Biomed. Pharmacother. 2020, 128, 110330. [Google Scholar] [CrossRef]
- Zaliova, M.; Stuchly, J.; Winkowska, L.; Musilova, A.; Fiser, K.; Slamova, M.; Starkova, J.; Vaskova, M.; Hrusak, O.; Sramkova, L.; et al. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica 2019, 104, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Alexander, T.B.; Gu, Z.; Iacobucci, I.; Dickerson, K.; Choi, J.K.; Xu, B.; Payne-Turner, D.; Yoshihara, H.; Loh, M.L.; Horan, J.; et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018, 562, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, S.; Butler, E.R.; Ohki, K.; Kiyokawa, N.; Bergmann, A.K.; Möricke, A.; Boer, J.M.; Cavé, H.; Cazzaniga, G.; Yeoh, A.E.J.; et al. Clinical characteristics and outcomes of B-ALL with ZNF384 rearrangements: A retrospective analysis by the Ponte di Legno Childhood ALL Working Group. Leukemia 2021, 1–6. [Google Scholar] [CrossRef]
- Hirabayashi, S.; Ohki, K.; Nakabayashi, K.; Ichikawa, H.; Momozawa, Y.; Okamura, K.; Yaguchi, A.; Terada, K.; Saito, Y.; Yoshimi, A.; et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 2016, 102, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Janet, N.B.; Kulkarni, U.; Arun, A.K.; Bensega, B.; Devasia, A.J.; Korula, A.; Abraham, A.; George, B.; Mathews, V.; Balasubramanian, P. Systematic application of fluorescence in situ hybridization and immunophenotype profile for the identification of ZNF384 gene rearrangements in B cell acute lymphoblastic leukemia. Int. J. Lab. Hematol. 2021, 43, 658–663. [Google Scholar] [CrossRef]
- Bueno, C.; Tejedor, J.R.; Bashford-Rogers, R.; González-Silva, L.; Valdés-Mas, R.; Agraz-Doblás, A.; de la Guardia, R.D.; Ribera, J.; Zamora, L.; Bilhou-Nabera, C.; et al. Natural history and cell of origin of TCF3-ZNF384 and PTPN11 mutations in monozygotic twins with concordant BCP-ALL. Blood 2019, 134, 900–905. [Google Scholar] [CrossRef]
- Griffith, M.; Griffith, O.L.; Krysiak, K.; Skidmore, Z.; Christopher, M.J.; Klco, J.; Ramu, A.; Lamprecht, T.L.; Wagner, A.H.; Campbell, K.; et al. Comprehensive genomic analysis reveals FLT3 activation and a therapeutic strategy for a patient with relapsed adult B-lymphoblastic leukemia. Exp. Hematol. 2016, 44, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Boer, J.M.; Valsecchi, M.G.; Hormann, F.M.; Antic, Z.; Zaliova, M.; Schwab, C.; Cazzaniga, G.; Arfeuille, C.; Cave, H.; Attarbaschi, A.; et al. Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 2021, 1–5. [Google Scholar] [CrossRef]
- Hormann, F.M.; Hoogkamer, A.Q.; Beverloo, H.B.; Boeree, A.; Dingjan, I.; Wattel, M.M.; Stam, R.W.; Escherich, G.; Pieters, R.; Boer, M.L.D.; et al. NUTM1 is a recurrent fusion gene partner in B-cell precursor acute lymphoblastic leukemia associated with increased expression of genes on chromosome band 10p12.31-12.2. Haematologica 2019, 104, e455–e459. [Google Scholar] [CrossRef] [Green Version]
- McEvoy, C.R.; Fox, S.B.; Prall, O.W.J. Emerging entities in NUTM1-rearranged neoplasms. Genes Chromosom. Cancer 2020, 59, 375–385. [Google Scholar] [CrossRef]
- French, C.A. NUT Carcinoma: Clinicopathologic features, pathogenesis, and treatment. Pathol. Int. 2018, 68, 583–595. [Google Scholar] [CrossRef]
- Boer, M.L.D.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.; Buijs-Gladdines, J.G.; Peters, S.T.; Van Zutven, L.J.; Beverloo, H.B.; Van der Spek, P.J.; Escherich, G.; et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1and Prognosis in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Iacobucci, I.; Roberts, K. Genetic Alterations and Therapeutic Targeting of Philadelphia-Like Acute Lymphoblastic Leukemia. Genes 2021, 12, 687. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Reshmi, S.C.; Harvey, R.; Chen, I.-M.; Patel, K.; Stonerock, E.; Jenkins, H.; Dai, Y.; Valentine, M.; Gu, Z.; et al. Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: A report from the Children’s Oncology Group. Blood 2018, 132, 815–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshmi, S.C.; Harvey, R.C.; Roberts, K.G.; Stonerock, E.; Smith, A.; Jenkins, H.; Chen, I.-M.; Valentine, M.; Liu, Y.; Li, Y.; et al. Targetable kinase gene fusions in high-risk B-ALL: A study from the Children’s Oncology Group. Blood 2017, 129, 3352–3361. [Google Scholar] [CrossRef] [Green Version]
- Tasian, S.K.; Hurtz, C.; Wertheim, G.B.; Bailey, N.; Lim, M.; Harvey, R.; Chen, I.-M.; Willman, C.L.; Astles, R.; Zebrowski, A.; et al. High incidence of Philadelphia chromosome-like acute lymphoblastic leukemia in older adults with B-ALL. Leukemia 2016, 31, 981–984. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Roberts, K.G.; Jabbour, E.; Patel, K.; Eterovic, A.K.; Chen, K.; Zweidler-McKay, P.; Lu, X.; Fawcett, G.; Wang, S.A.; et al. Ph-like acute lymphoblastic leukemia: A high-risk subtype in adults. Blood 2017, 129, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.G. The biology of Philadelphia chromosome-like ALL. Best Pract. Res. Clin. Haematol. 2017, 30, 212–221. [Google Scholar] [CrossRef]
- Chiaretti, S.; Messina, M.; Foà, R. BCR/ABL1-like acute lymphoblastic leukemia: How to diagnose and treat? Cancer 2018, 125, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.G.; Collins-Underwood, J.R.; Phillips, L.A.; Loudin, M.G.; Liu, W.; Zhang, J.; Ma, J.; Coustan-Smith, E.; Harvey, R.C.; Willman, C.L.; et al. Rearrangement of CRLF2 in B-progenitor–and Down syndrome–associated acute lymphoblastic leukemia. Nat. Genet. 2009, 41, 1243–1246. [Google Scholar] [CrossRef] [Green Version]
- Yoda, A.; Yoda, Y.; Chiaretti, S.; Bar-Natan, M.; Mani, K.; Rodig, S.J.; West, N.; Xiao, Y.; Brown, J.R.; Mitsiades, C.; et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2009, 107, 252–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, L.J.; Jones, L.; Enshaei, A.; Tonin, S.; Ryan, S.L.; Eswaran, J.; Nakjang, S.; Papaemmanuil, E.; Tubio, J.M.C.; Fielding, A.K.; et al. Characterisation of the genomic landscape ofCRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosom. Cancer 2017, 56, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertzberg, L.; Vendramini, E.; Ganmore, I.; Cazzaniga, G.; Schmitz, M.; Chalker, J.; Shiloh, R.; Iacobucci, I.; Shochat, C.; Zeligson, S.; et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: A report from the International BFM Study Group. Blood 2010, 115, 1006–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobucci, I.; Li, Y.; Roberts, K.G.; Dobson, S.M.; Kim, J.C.; Payne-Turner, D.; Harvey, R.C.; Valentine, M.; McCastlain, K.; Easton, J.; et al. Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia. Cancer Cell 2016, 29, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Möricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; Kronnie, G.T.; Bourquin, J.-P.; Bornhauser, B.; et al. IKZF1plus Defines a New Minimal Residual Disease–Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar] [CrossRef] [Green Version]
- Tanasi, I.; Ba, I.; Sirvent, N.; Braun, T.; Cuccuini, W.; Ballerini, P.; Duployez, N.; Tanguy-Schmidt, A.; Tamburini, J.; Maury, S.; et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood 2019, 134, 1351–1355. [Google Scholar] [CrossRef]
- Schewe, D.M.; Lenk, L.; Vogiatzi, F.; Winterberg, D.; Rademacher, A.V.; Buchmann, S.; Henry, D.; Bergmann, A.K.; Cario, G.; Cox, M.C. Larotrectinib in TRK fusion–positive pediatric B-cell acute lymphoblastic leukemia. Blood Adv. 2019, 3, 3499–3502. [Google Scholar] [CrossRef]
- Niswander, L.M.; Loftus, J.P.; Lainey, E.; Caye-Eude, A.; Pondrom, M.; Hottman, D.A.; Iacobucci, I.; Mullighan, C.G.; Jain, N.; Konopleva, M.; et al. Therapeutic potential of ruxolitinib and ponatinib in patients with EPOR-rearranged Philadelphia chromosome-like acute lymphoblastic leukemia. Haematologica 2021. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Min, J.; Jarusiewicz, J.; Actis, M.; Bradford, S.Y.-C.; Mayasundari, A.; Yang, L.; Chepyala, D.; Alcock, L.J.; Roberts, K.G.; et al. Degradation of Janus kinases in CRLF2-rearranged acute lymphoblastic leukemia. Blood 2021. [Google Scholar] [CrossRef]
- Qin, H.; Cho, M.; Haso, W.; Zhang, L.; Tasian, S.K.; Oo, H.Z.; Negri, G.L.; Lin, Y.; Zou, J.; Mallon, B.S.; et al. Eradication of B-ALL using chimeric antigen receptor–expressing T cells targeting the TSLPR oncoprotein. Blood 2015, 126, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Kotrová, M.; Bresolin, S.; Stuchly, J.; Stary, J.; Hrusak, O.; Kronnie, G.T.; Trka, J.; Zuna, J.; Vaskova, M. ETV6/RUNX1-like acute lymphoblastic leukemia: A novel B-cell precursor leukemia subtype associated with the CD27/CD44 immunophenotype. Genes Chromosom. Cancer 2017, 56, 608–616. [Google Scholar] [CrossRef]
- Zaliova, M.; Moorman, A.V.; Cazzaniga, G.; Stanulla, M.; Harvey, R.C.; Roberts, K.G.; Heatley, S.L.; Loh, M.L.; Konopleva, M.; Chen, I.-M.; et al. Characterization of leukemias with ETV6-ABL1 fusion. Haematologica 2016, 101, 1082–1093. [Google Scholar] [CrossRef] [Green Version]
- Nishii, R.; Baskin-Doerfler, R.; Yang, W.; Oak, N.; Zhao, X.; Yang, W.; Hoshitsuki, K.; Bloom, M.; Verbist, K.C.; Burns, M.A.; et al. Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood 2021, 137, 364–373. [Google Scholar] [CrossRef]
- Bastian, L.; Schroeder, M.P.; Eckert, C.; Schlee, C.; Tanchez, J.O.; Kämpf, S.; Wagner, D.L.; Schulze, V.; Isaakidis, K.; Lázaro-Navarro, J.; et al. PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 2019, 33, 1895–1909. [Google Scholar] [CrossRef]
- Passet, M.; Boissel, N.; Sigaux, F.; Saillard, C.; Bargetzi, M.; Ba, I.; Thomas, X.; Graux, C.; Chalandon, Y.; Leguay, T.; et al. PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood 2019, 133, 280–284. [Google Scholar] [CrossRef]
- Shah, S.; Schrader, K.; Waanders, E.; Timms, A.E.; Vijai, J.; Miething, C.; Wechsler, J.; Yang, J.; Hayes, J.; Klein, R.; et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 1226–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vairy, S.; Tran, T.H. IKZF1 alterations in acute lymphoblastic leukemia: The good, the bad and the ugly. Blood Rev. 2020, 44, 100677. [Google Scholar] [CrossRef] [PubMed]
- Churchman, M.L.; Mullighan, C.G. Ikaros: Exploiting and targeting the hematopoietic stem cell niche in B-progenitor acute lymphoblastic leukemia. Exp. Hematol. 2016, 46, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Churchman, M.L.; Low, J.; Qu, C.; Paietta, E.M.; Kasper, L.H.; Chang, Y.; Payne-Turner, D.; Althoff, M.J.; Song, G.; Chen, S.-C.; et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Potuckova, E.; Lukes, J.; Winkowska, L.; Starkova, J.; Janotova, I.; Sramkova, L.; Stary, J.; Zuna, J.; Stanulla, M.; et al. Frequency and prognostic impact of ZEB2 H1038 and Q1072 mutations in childhood B-other acute lymphoblastic leukemia. Haematologica 2020, 106, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Aifantis, I.; Raetz, E.A.; Buonamici, S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat. Rev. Immunol. 2008, 8, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Yui, M.; Rothenberg, E. Developmental gene networks: A triathlon on the course to T cell identity. Nat. Rev. Immunol. 2014, 14, 529–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teachey, D.T.; Pui, C.-H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019, 20, e142–e154. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Seki, M.; Kimura, S.; Isobe, T.; Yoshida, K.; Ueno, H.; Nakajima-Takagi, Y.; Wang, C.; Lin, L.; Kon, A.; Suzuki, H.; et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Mansour, M.; Abraham, B.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Qian, M.; Zhang, H.; Guo, Y.; Yang, J.; Zhao, X.; He, H.; Lu, J.; Pan, J.; Chang, M.; et al. Whole-genome noncoding sequence analysis in T-cell acute lymphoblastic leukemia identifies oncogene enhancer mutations. Blood 2017, 129, 3264–3268. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.R.; Duke, V.; Foroni, L.; Patel, B.; Allen, C.; Ancliff, P.J.; Gale, R.E.; Linch, D.C. Notch-1 Mutations Are Secondary Events in Some Patients with T-Cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2007, 13, 6964–6969. [Google Scholar] [CrossRef] [Green Version]
- Gianni, F.; Belver, L.; Ferrando, A. The Genetics and Mechanisms of T-Cell Acute Lymphoblastic Leukemia. Cold Spring Harb. Perspect. Med. 2019, 10, a035246. [Google Scholar] [CrossRef]
- Belver, L.; Ferrando, L.B.A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef]
- Kimura, S.; Seki, M.; Kawai, T.; Goto, H.; Yoshida, K.; Isobe, T.; Sekiguchi, M.; Watanabe, K.; Kubota, Y.; Nannya, Y.; et al. DNA methylation-based classification reveals difference between pediatric T-cell acute lymphoblastic leukemia and normal thymocytes. Leukemia 2019, 34, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Roels, J.; Thénoz, M.; Szarzyńska, B.; Landfors, M.; De Coninck, S.; Demoen, L.; Provez, L.; Kuchmiy, A.; Strubbe, S.; Reunes, L.; et al. Aging of Preleukemic Thymocytes Drives CpG Island Hypermethylation in T-cell Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2020, 1, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.-H.; Downing, J.R.; Gilliland, D.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Soulier, J.; Clappier, E.; Cayuela, J.-M.; Regnault, A.; García-Peydró, M.; Dombret, H.; Baruchel, A.; Toribio, M.L.; Sigaux, F. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood 2005, 106, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Dadi, S.; Le Noir, S.; Bornet, D.P.; Lhermitte, L.; Zacarias-Cabeza, J.; Bergeron, J.; Villarèse, P.; Vachez, E.; Dik, W.A.; Millien, C.; et al. TLX Homeodomain Oncogenes Mediate T Cell Maturation Arrest in T-ALL via Interaction with ETS1 and Suppression of TCRα Gene Expression. Cancer Cell 2012, 21, 563–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giacomo, D.; La Starza, R.; Gorello, P.; Pellanera, F.; Atak, Z.K.; De Keersmaecker, K.; Pierini, V.; Harrison, C.J.; Arniani, S.; Moretti, M.; et al. 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T and myeloid immature acute leukemia. Blood 2021. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Azzato, E.M.; Mullighan, C.G. Integration of Next-Generation Sequencing to Treat Acute Lymphoblastic Leukemia with Targetable Lesions: The St. Jude Children’s Research Hospital Approach. Front. Pediatr. 2017, 5, 258. [Google Scholar] [CrossRef]
- Gocho, Y.; Liu, J.; Hu, J.; Yang, W.; Dharia, N.V.; Zhang, J.; Shi, H.; Du, G.; John, A.; Lin, T.-N.; et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat. Rev. Cancer 2021, 2, 1–16. [Google Scholar] [CrossRef]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.; Li, C.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [Green Version]
- Park, S.T.; Sun, X.-H. The Tal1 Oncoprotein Inhibits E47-mediated Transcription. J. Biol. Chem. 1998, 273, 7030–7037. [Google Scholar] [CrossRef] [Green Version]
- Sanda, T.; Lawton, L.N.; Barrasa, M.I.; Fan, Z.P.; Kohlhammer, H.; Gutierrez, A.; Ma, W.; Tatarek, J.; Ahn, Y.; Kelliher, M.A.; et al. Core Transcriptional Regulatory Circuit Controlled by the TAL1 Complex in Human T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Nottingham, W.T.; Jarratt, A.; Burgess, M.; Speck, C.L.; Cheng, J.F.; Prabhakar, S.; Rubin, E.M.; Li, P.S.; Sloane-Stanley, J.; Kong, A.S.J.; et al. Runx1-mediated hematopoietic stem-cell emergence is controlled by a Gata/Ets/SCL-regulated enhancer. Blood 2007, 110, 4188–4197. [Google Scholar] [CrossRef] [Green Version]
- Choi, A.; Illendula, A.; Pulikkan, J.A.; Roderick, J.E.; Tesell, J.; Yu, J.; Hermance, N.; Zhu, L.J.; Castilla, L.H.; Bushweller, J.H.; et al. RUNX1 is required for oncogenic Myb and Myc enhancer activity in T-cell acute lymphoblastic leukemia. Blood 2017, 130, 1722–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neil, J.; Shank, J.; Cusson, N.; Murre, C.; Kelliher, M. TAL1/SCL induces leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell 2004, 5, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Draheim, K.M.; Hermance, N.; Yang, Y.; Arous, E.; Calvo, J.; Kelliher, M.A. A DNA-binding mutant of TAL1 cooperates with LMO2 to cause T cell leukemia in mice. Oncogene 2010, 30, 1252–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrando, A.A.; Look, A.T. Gene expression profiling in T-cell acute lymphoblastic leukemia. Semin. Hematol. 2003, 40, 274–280. [Google Scholar] [CrossRef]
- Leong, W.Z.; Tan, S.H.; Ngoc, P.C.T.; Amanda, S.; Yam, A.W.Y.; Liau, W.-S.; Gong, Z.; Lawton, L.N.; Tenen, D.G.; Sanda, T. ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 2017, 31, 2343–2360. [Google Scholar] [CrossRef]
- Tan, S.H.; Leong, W.Z.; Ngoc, P.C.T.; Tan, T.K.; Bertulfo, F.C.; Lim, M.C.; An, O.; Li, Z.; Yeoh, A.E.J.; Fullwood, M.J.; et al. The enhancer RNA ARIEL activates the oncogenic transcriptional program in T-cell acute lymphoblastic leukemia. Blood 2019, 134, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Piovan, E.; Yu, J.; Tosello, V.; Herranz, D.; Ambesi-Impiombato, A.; Da Silva, A.C.; Sanchez-Martin, M.; Perez-Garcia, A.; Rigo, I.; Castillo, M.; et al. Direct Reversal of Glucocorticoid Resistance by AKT Inhibition in Acute Lymphoblastic Leukemia. Cancer Cell 2013, 24, 766–776. [Google Scholar] [CrossRef] [Green Version]
- Jena, N.; Sheng, J.; Hu, J.K.; Li, W.; Zhou, W.; Lee, G.; Tsichlis, N.; Pathak, A.; Brown, N.; Deshpande, A.; et al. CDK6-mediated repression of CD25 is required for induction and maintenance of Notch1-induced T-cell acute lymphoblastic leukemia. Leukemia 2015, 30, 1033–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coustan-Smith, E.; Mullighan, C.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.; Basso, G.; et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009, 10, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Dolai, S.; Delgado-Martin, C.; Vincent, T.; Robbins, A.; Selvanathan, A.; Ryan, T.; Hall, J.; Wood, A.C.; Tasian, S.K.; et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood 2015, 125, 1759–1767. [Google Scholar] [CrossRef] [Green Version]
- McCarter, A.C.; Wang, Q.; Chiang, M. Notch in Leukemia. Adv. Exp. Med. Biol. 2018, 1066, 355–394. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.; Ferrando, A.A.; Lee, W.; Iv, J.P.M.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [Green Version]
- Sulis, M.L.; Williams, O.; Palomero, T.; Tosello, V.; Pallikuppam, S.; Real, P.J.; Barnes, K.; Zuurbier, L.; Meijerink, J.P.; Ferrando, A.A. NOTCH1 extracellular juxtamembrane expansion mutations in T-ALL. Blood 2008, 112, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Haydu, J.E.; De Keersmaecker, K.; Duff, M.K.; Paietta, E.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Ferrando, A. An activating intragenic deletion in NOTCH1 in human T-ALL. Blood 2012, 119, 5211–5214. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.J.; Buonamici, S.; Sulis, M.L.; Palomero, T.; Vilimas, T.; Basso, G.; Ferrando, A.; Aifantis, I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J. Exp. Med. 2007, 204, 1825–1835. [Google Scholar] [CrossRef]
- O’Neil, J.; Grim, J.; Strack, P.; Rao, S.; Tibbitts, D.; Winter, C.; Hardwick, J.; Welcker, M.; Meijerink, J.; Pieters, R.; et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. J. Exp. Med. 2007, 204, 1813–1824. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Puente, X.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martin-Subero, J.I.; Munar, M.; Rubio-Perez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- Chiang, M.; Xu, L.; Shestova, O.; Histen, G.; L’Heureux, S.; Romany, C.; Childs, M.E.; Gimotty, P.A.; Aster, J.C.; Pear, W.S. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras–initiated leukemia. J. Clin. Investig. 2008, 118, 3181–3194. [Google Scholar] [CrossRef] [Green Version]
- Wendorff, A.A.; Quinn, S.A.; Rashkovan, M.; Madubata, C.J.; Ambesi-Impiombato, A.; Litzow, M.R.; Tallman, M.S.; Paietta, E.; Paganin, M.; Basso, G.; et al. Phf6 Loss Enhances HSC Self-Renewal Driving Tumor Initiation and Leukemia Stem Cell Activity in T-ALL. Cancer Discov. 2018, 9, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Albertí-Servera, L.; Demeyer, S.; Govaerts, I.; Swings, T.; De Bie, J.; Gielen, O.; Brociner, M.; Michaux, L.; Maertens, J.; Uyttebroeck, A.; et al. Single-cell DNA amplicon sequencing reveals clonal heterogeneity and evolution in T-cell acute lymphoblastic leukemia. Blood 2021, 137, 801–811. [Google Scholar] [CrossRef]
- De Bie, J.; Demeyer, S.; Alberti-Servera, L.; Geerdens, E.; Segers, H.; Broux, M.; De Keersmaecker, K.; Michaux, L.; Vandenberghe, P.; Voet, T.; et al. Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 1358–1369. [Google Scholar] [CrossRef] [Green Version]
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.; et al. Long-range enhancer activity determinesMycsensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [Green Version]
- Reizis, B. Direct induction of T lymphocyte-specific gene expression by the mammalian Notch signaling pathway. Genes Dev. 2002, 16, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Zheng, R.; Li, M.; Wang, S.; Liu, Y. Advances of target therapy on NOTCH1 signaling pathway in T-cell acute lymphoblastic leukemia. Exp. Hematol. Oncol. 2020, 9, 31. [Google Scholar] [CrossRef]
- Papayannidis, C.; De Angelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavai, A.V.; Quesnelle, C.A.; Norris, D.J.; Han, W.-C.; Gill, P.S.; Shan, W.; Balog, A.; Chen, K.; Tebben, A.J.; Rampulla, R.; et al. Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors. ACS Med. Chem. Lett. 2015, 6, 523–527. [Google Scholar] [CrossRef]
- Sanchez-Martin, M.; Ambesi-Impiombato, A.; Qin, Y.; Herranz, D.; Bansal, M.; Girardi, T.; Paietta, E.; Tallman, M.S.; Rowe, J.M.; De Keersmaecker, K.; et al. Synergistic antileukemic therapies inNOTCH1-induced T-ALL. Proc. Natl. Acad. Sci. USA 2017, 114, 2006–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, P.; Walls, M.; Qiu, M.; Ding, R.; Denlinger, R.H.; Wong, A.; Tsaparikos, K.; Jani, J.P.; Hosea, N.A.; Sands, M.; et al. Evaluation of Selective γ-Secretase Inhibitor PF-03084014 for Its Antitumor Efficacy and Gastrointestinal Safety to Guide Optimal Clinical Trial Design. Mol. Cancer Ther. 2010, 9, 1618–1628. [Google Scholar] [CrossRef] [Green Version]
- Real, P.; Tosello, V.; Palomero, T.; Castillo, M.; Hernando, E.; De Stanchina, E.; Sulis, M.L.; Barnes, K.; Sawai, C.; Homminga, I.; et al. γ-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat. Med. 2008, 15, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Cullion, K.; Draheim, K.M.; Hermance, N.; Tammam, J.; Sharma, V.M.; Ware, C.; Nikov, G.; Krishnamoorthy, V.; Majumder, P.K.; Kelliher, M.A. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 2009, 113, 6172–6181. [Google Scholar] [CrossRef] [Green Version]
- Franciosa, G.; Smits, J.G.A.; Minuzzo, S.; Martinez-Val, A.; Indraccolo, S.; Olsen, J.V. Proteomics of resistance to Notch1 inhibition in acute lymphoblastic leukemia reveals targetable kinase signatures. Nat. Commun. 2021, 12, 2507. [Google Scholar] [CrossRef]
- Trinquand, A.; Tanguy-Schmidt, A.; Ben Abdelali, R.; Lambert, J.; Beldjord, K.; Lengliné, E.; De Gunzburg, N.; Bornet, D.P.; Lhermitte, L.; Mossafa, H.; et al. Toward a NOTCH1/FBXW7/RAS/PTEN–Based Oncogenetic Risk Classification of Adult T-Cell Acute Lymphoblastic Leukemia: A Group for Research in Adult Acute Lymphoblastic Leukemia Study. J. Clin. Oncol. 2013, 31, 4333–4342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, S.; Nakitandwe, J.; Kesserwan, C.A.; Azzato, E.M.; Wheeler, D.A.; Rusch, M.; Shurtleff, S.; Hedges, D.J.; Hamilton, K.V.; Foy, S.G.; et al. Genomes for Kids: The scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Jobanputra, V.; Wrzeszczynski, K.O.; Buttner, R.; Caldas, C.; Cuppen, E.; Grimmond, S.; Haferlach, T.; Mullighan, C.; Schuh, A.; Elemento, O. Clinical interpretation of whole-genome and whole-transcriptome sequencing for precision oncology. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Rosenquist, R.; Cuppen, E.; Buettner, R.; Caldas, C.; Dreau, H.; Elemento, O.; Frederix, G.; Grimmond, S.; Haferlach, T.; Jobanputra, V.; et al. Clinical utility of whole-genome sequencing in precision oncology. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Meggendorfer, M.; Jobanputra, V.; Wrzeszczynski, K.O.; Roepman, P.; de Bruijn, E.; Cuppen, E.; Buttner, R.; Caldas, C.; Grimmond, S.; Mullighan, C.G.; et al. Analytical demands to use whole-genome sequencing in precision oncology. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Walter, W.; Shahswar, R.; Stengel, A.; Meggendorfer, M.; Kern, W.; Haferlach, T.; Haferlach, C. Clinical application of whole transcriptome sequencing for the classification of patients with acute lymphoblastic leukemia. BMC Cancer 2021, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Flensburg, C.; Oshlack, A.; Majewski, I.J. Detecting copy number alterations in RNA-Seq using SuperFreq. Bioinformatics 2021. [Google Scholar] [CrossRef]
- He, J.; Abdel-Wahab, O.; Nahas, M.K.; Wang, K.; Rampal, R.K.; Intlekofer, A.; Patel, J.; Krivstov, A.; Frampton, G.; Young, L.E.; et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood 2016, 127, 3004–3014. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Platform | Capability | Cost | Detectable Subtypes | Difficult Subtypes |
|---|---|---|---|---|
| WTS (RNAseq) | Fusion chimeras Gene expression profiling Mutant allele expression Alternative splicing analysis (BCR/TCR rearrangements) (Sequence mutations) (Copy number analysis) | Moderate | B-ALL ETV6-RUNX1; KMT2A; TCF3-PBX1; BCR-ABL1; DUX4; MEF2D; ZNF384/362 NUTM1; HLF; BCL2/MYC; PAX5alt; ZEB2/CEBPE; -like subtypes | B-ALL Aneuploidies |
| T-ALL HOXA (KMT2A-R, PICALM-MLLT10, SET-NUP214); SPI1; NKX2-1; TAL1 (STIL-TAL1) | T-ALL BCL11B; TLX1/3; LMO1/2; HOXA (others); TAL1 (others); T-other | |||
| WGS | Sequence mutations Structural variants Copy number analysis (BCR/TCR rearrangements) (GWAS) | High | B-ALL Aneuploidies; ETV6-RUNX1; KMT2A; TCF3-PBX1; BCR-ABL1; DUX4; MEF2D; ZNF384/362; NUTM1; HLF; BCL2/MYC; PAX5 P80R; IKZF1 N159Y; ZEB2/CEBPE; Sequence and structural alterations in Ph-like ALL | B-ALL -like subtypes; Part of PAX5alt |
| T-ALL BCL11B; TLX1/3; LMO1/2; HOXA; SPI1; NKX2-1; TAL1 | T-ALL T-other | |||
| WES | Sequence mutations (coding) Structural variants (coding) Copy number analysis | Moderate | B-ALL (Aneuploidies) PAX5 P80R IKZF1 N159Y Sequence mutations in Ph-like ALL (e.g., JAK1/2/3, Ras) | Most of other B-ALL and T-ALL subtypes |
| Targeted sequencing (DNA and/or RNA) | Fusion chimeras (targeted) Gene expression (targeted) Sequence mutations (targeted) Structural variants (targeted) (Copy number analysis) | Low | Targeted alterations | Non-targeted alterations |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobucci, I.; Kimura, S.; Mullighan, C.G. Biologic and Therapeutic Implications of Genomic Alterations in Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 3792. https://doi.org/10.3390/jcm10173792
Iacobucci I, Kimura S, Mullighan CG. Biologic and Therapeutic Implications of Genomic Alterations in Acute Lymphoblastic Leukemia. Journal of Clinical Medicine. 2021; 10(17):3792. https://doi.org/10.3390/jcm10173792
Chicago/Turabian StyleIacobucci, Ilaria, Shunsuke Kimura, and Charles G. Mullighan. 2021. "Biologic and Therapeutic Implications of Genomic Alterations in Acute Lymphoblastic Leukemia" Journal of Clinical Medicine 10, no. 17: 3792. https://doi.org/10.3390/jcm10173792
APA StyleIacobucci, I., Kimura, S., & Mullighan, C. G. (2021). Biologic and Therapeutic Implications of Genomic Alterations in Acute Lymphoblastic Leukemia. Journal of Clinical Medicine, 10(17), 3792. https://doi.org/10.3390/jcm10173792