
RBM20-Related Cardiomyopathy: Current Understanding and Future Options



Abstract
:1. RBM20 Mutations Cause Highly Penetrant Cardiomyopathies


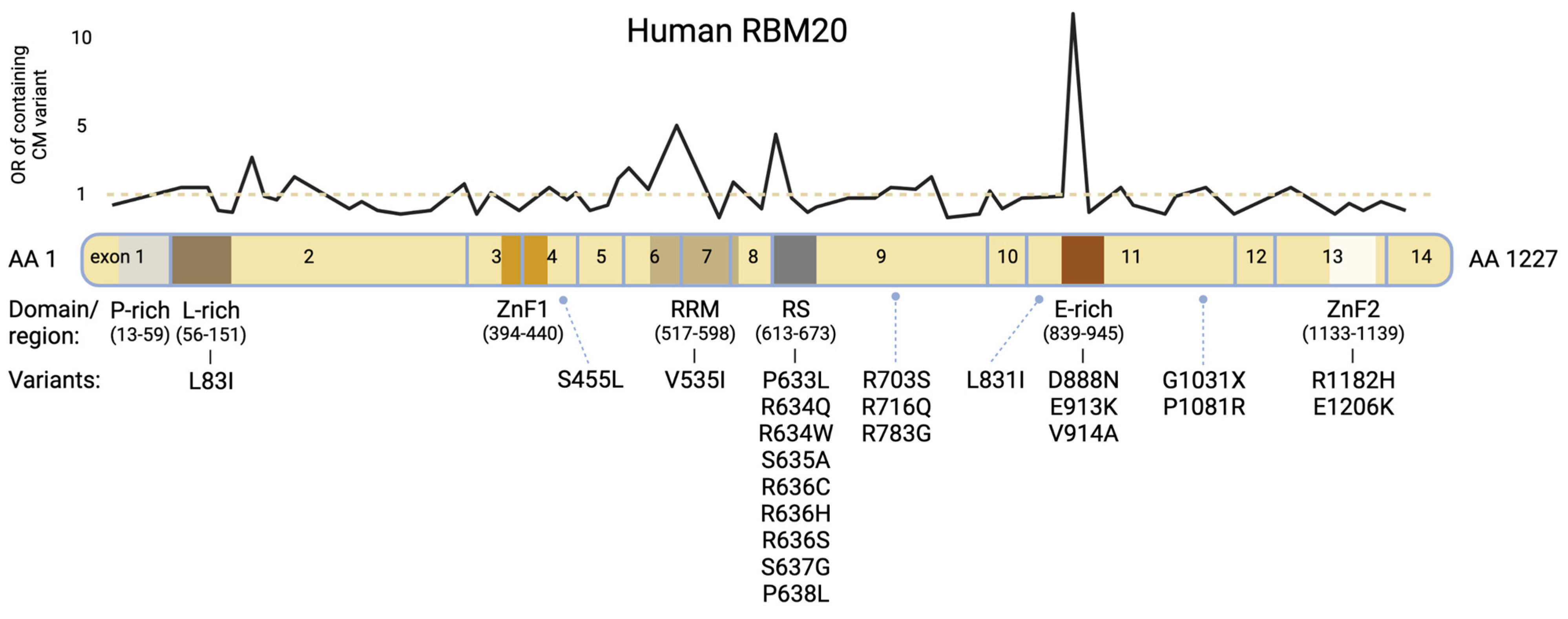{kind=link}
{kind=link}
{kind=link}
| Domain | Mutation | Exon | Pathogenicity | Reference |
|---|---|---|---|---|
| Leu-rich-region | L83I | 2 | unknown | [18] |
| Other | S455L | 4 | unknown | [18] |
| RRM-domain | V535I | 6 | pathogenic | [6,16] |
| RS-domain | P633L | 9 | pathogenic | [19] |
| RS-domain | R634Q | 9 | pathogenic | [1,2,6,16] |
| RS-domain | R634W | 9 | pathogenic | [10,16] |
| RS-domain | S635A | 9 | pathogenic | [3,4,6,10] |
| RS-domain | R636C | 9 | pathogenic | [16] |
| RS-domain | R636H | 9 | pathogenic | [1,2,16,20,21] |
| RS-domain | R636S | 9 | pathogenic | [1,2,6,22] |
| RS-domain | S637G | 9 | pathogenic | [1,3,6,23] |
| RS-domain | P638L | 9 | pathogenic | [1,2,6,15,18] |
| Other | R703S | 9 | unknown | [18] |
| Other | R716Q | 9 | unknown | [6,16] |
| Other | R783G | 9 | pathogenic | [24] |
| Other | L831I | 11 | unknown | [18] |
| Glu-rich-region | D888N | 11 | unknown | [18] |
| Glu-rich-region | E913K | 11 | pathogenic | [2,25] |
| Glu-rich-region | V914A | 11 | pathogenic | [15] |
| Other | G1031X * | 11 | pathogenic | [10,18] |
| Other | P1081R | 11 | unknown | [18] |
| ZnF-2 | R1182H | 13 | unknown | [13] |
| ZnF-2 | E1206K | 13 | unknown | [18] |
2. Model Systems to Dissect the Pathophysiology and Enable Therapeutic Studies
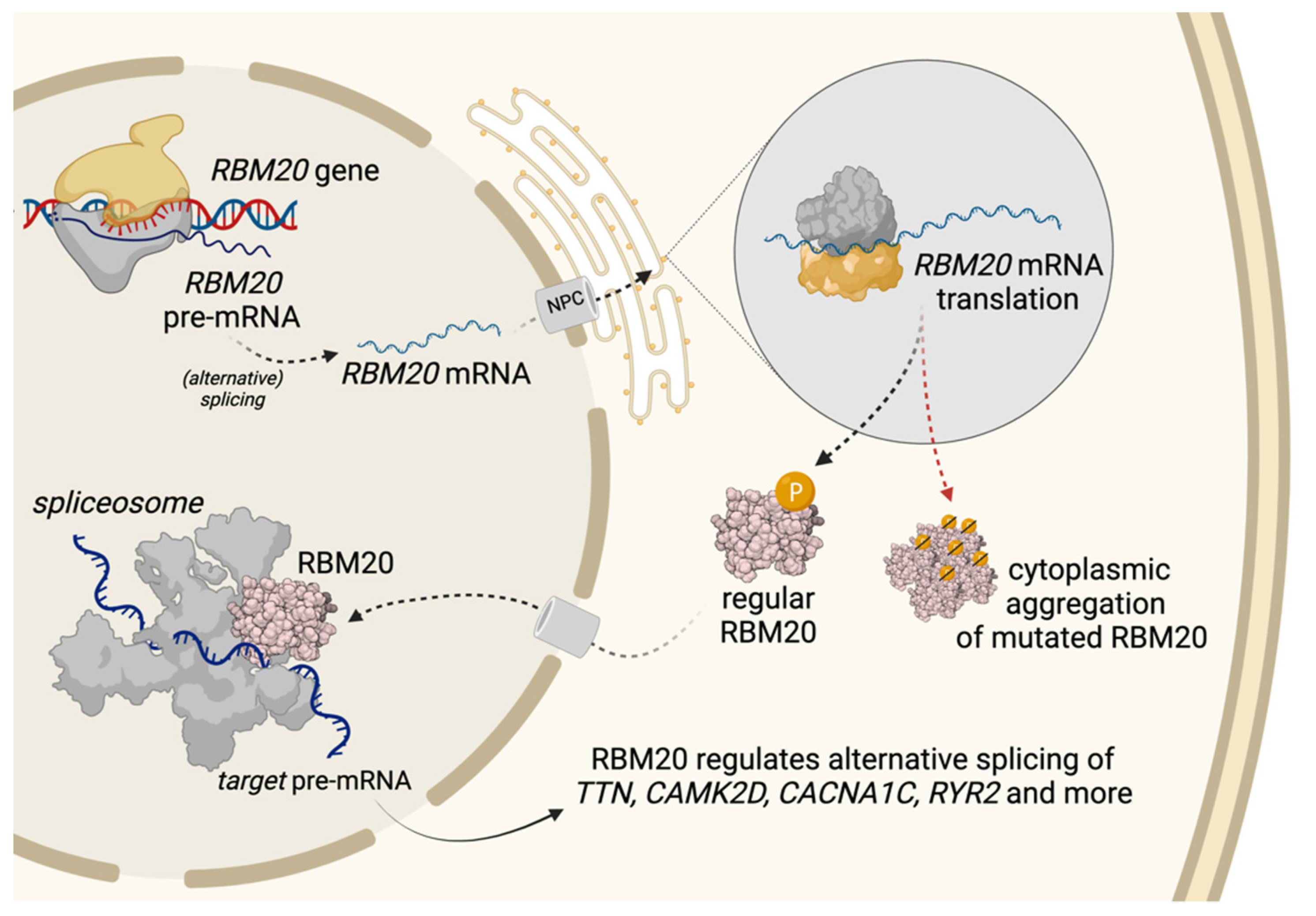
3. Trafficking of RBM20 and Aggregation Formation
4. Splicing Targets and Their Function
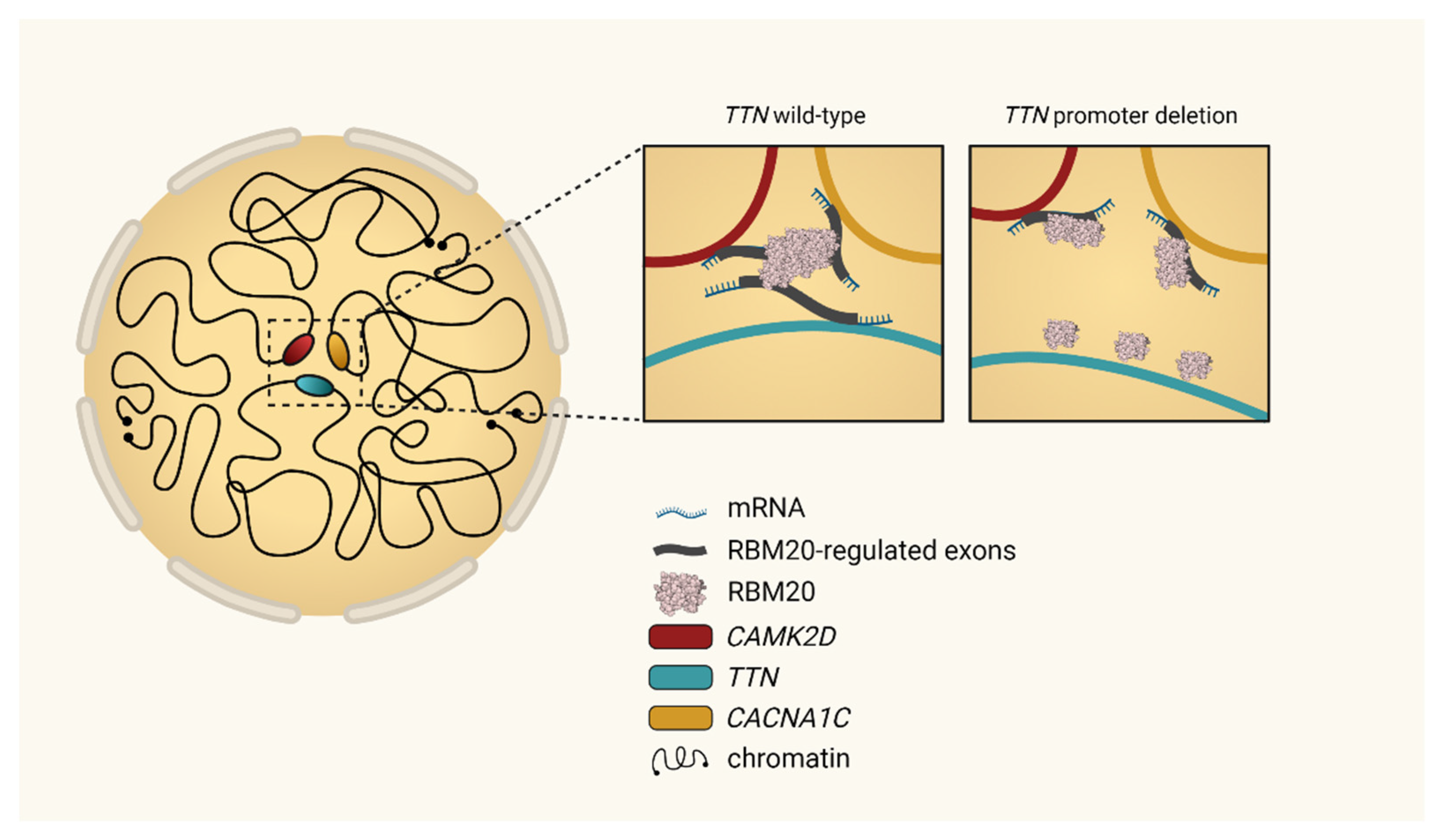
4.1. TTN
4.2. RYR2
4.3. CAMK2D
4.4. Identification of Novel Targets
4.5. Interactions between RBM20s Targets
4.6. Interactions with Other Splice Regulators
5. Clinical Presentation and Risk Management
6. Current Treatment Concepts in RBM20 Patients
6.1. Treatment of Heart Failure
6.2. ICD-Therapy
6.3. Heart Transplantation and Assist Devices
7. Future Therapeutic Options
7.1. RBM20 Upregulation
7.2. RBM20 Downregulation
7.3. Ca2+-Modulation
7.4. Gene Editing
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in Ribonucleic Acid Binding Protein Gene Cause Familial Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Hey, T.M.; Rasmussen, T.B.; Madsen, T.; Aagaard, M.M.; Harbo, M.; Mølgaard, H.; Møller, J.E.; Eiskjær, H.; Mogensen, J. Pathogenic RBM20-Variants Are Associated With a Severe Disease Expression in Male Patients With Dilated Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, K.; Sasano, T.; Hiraoka, Y.; Togo-Ohno, M.; Soejima, Y.; Sawabe, M.; Tsuchiya, M.; Ogawa, H.; Furukawa, T.; Kuroyanagi, H. A missense mutation in the RSRSP stretch of Rbm20 causes dilated cardiomyopathy and atrial fibrillation in mice. Sci. Rep. 2020, 10, 17894. [Google Scholar] [CrossRef] [PubMed]
- Streckfuss-Bömeke, K.; Tiburcy, M.; Fomin, A.; Luo, X.; Li, W.; Fischer, C.; Özcelik, C.; Perrot, A.; Sossalla, S.; Haas, J.; et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell Cardiol. 2017, 113, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-phenotype associations in dilated cardiomyopathy: Meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, W.; Dewey, C.N.; Greaser, M.L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013, 41, 2659–2672. [Google Scholar] [CrossRef] [Green Version]
- Maatz, H.; Jens, M.; Liss, M.; Schafer, S.; Heinig, M.; Kirchner, M.; Adami, E.; Rintisch, C.; Dauksaite, V.; Radke, M.H.; et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Investig. 2014, 124, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kimura, A.; Kuroyanagi, H. Alternative Splicing Regulator RBM20 and Cardiomyopathy. Front. Mol. Biosci. 2018, 5, 105. [Google Scholar] [CrossRef]
- Murayama, R.; Kimura-Asami, M.; Togo-Ohno, M.; Yamasaki-Kato, Y.; Naruse, T.K.; Yamamoto, T.; Hayashi, T.; Ai, T.; Spoonamore, K.G.; Kovacs, R.J. Phosphorylation of the RSRSP stretch is critical for splicing regulation by RNA-Binding Motif Protein 20 (RBM20) through nuclear localization. Sci. Rep. 2018, 8, 8970. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [PubMed]
- Parikh, V.N.; Caleshu, C.; Reuter, C.; Lazzeroni, L.C.; Ingles, J.; Garcia, J.; McCaleb, K.; Adesiyun, T.; Sedaghat-Hamedani, F.; Kumar, S.; et al. Regional Variation in RBM20 Causes a Highly Penetrant Arrhythmogenic Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005371. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2014, 36, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Gi, W.T.; Tugrul, O.F.; Amr, A.; Haas, J.; Zhu, F.; Ehlermann, P.; Uhlmann, L.; Katus, H.A.; et al. Clinical and genetic insights into non-compaction: A meta-analysis and systematic review on 7598 individuals. Clin. Res. Cardiol. 2019, 108, 1297–1308. [Google Scholar] [CrossRef]
- Gaertner, A.; Klauke, B.; Felski, E.; Kassner, A.; Brodehl, A.; Gerdes, D.; Stanasiuk, C.; Ebbinghaus, H.; Schulz, U.; Dubowy, K.O.; et al. Cardiomyopathy-associated mutations in the RS domain affect nuclear localization of RBM20. Hum. Mutat. 2020, 41, 1931–1943. [Google Scholar] [CrossRef]
- Li, D.; Morales, A.; Gonzalez-Quintana, J.; Norton, N.; Siegfried, J.D.; Hofmeyer, M.; Hershberger, R.E. Identification of Novel Mutations in RBM20 in Patients with Dilated Cardiomyopathy. Clin. Transl. Sci. 2010, 3, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef]
- Refaat, M.M.; Lubitz, S.A.; Makino, S.; Islam, Z.; Frangiskakis, J.M.; Mehdi, H.; Gutmann, R.; Zhang, M.L.; Bloom, H.L.; MacRae, C.A. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm 2012, 9, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Briganti, F.; Sun, H.; Wei, W.; Wu, J.; Zhu, C.; Liss, M.; Karakikes, I.; Rego, S.; Cipriano, A.; Snyder, M.; et al. iPSC Modeling of RBM20-Deficient DCM Identifies Upregulation of RBM20 as a Therapeutic Strategy. Cell Rep. 2020, 32, 108117. [Google Scholar] [CrossRef]
- Wells, Q.S.; Becker, J.R.; Su, Y.R.; Mosley, J.D.; Weeke, P.; D’Aoust, L.; Ausborn, N.L.; Ramirez, A.H.; Pfotenhauer, J.P.; Naftilan, A.J.; et al. Whole Exome Sequencing Identifies a Causal RBM20 Mutation in a Large Pedigree With Familial Dilated Cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chami, N.; Tadros, R.; Lemarbre, F.; Lo, K.S.; Beaudoin, M.; Robb, L.; Labuda, D.; Tardif, J.-C.; Racine, N.; Talajic, M.; et al. Nonsense Mutations in BAG3 are Associated With Early-Onset Dilated Cardiomyopathy in French Canadians. Can. J. Cardiol. 2014, 30, 1655–1661. [Google Scholar] [CrossRef]
- Schneider, J.W.; Oommen, S.; Qureshi, M.Y.; Goetsch, S.C.; Pease, D.R.; Sundsbak, R.S.; Guo, W.; Sun, M.; Sun, H.; Kuroyanagi, H.; et al. Dysregulated ribonucleoprotein granules promote cardiomyopathy in RBM20 gene-edited pigs. Nat. Med. 2020, 26, 1788–1800. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Bouvagnet, P.; Chevalier, P.; Sebbag, L.; Dulac, A.; Dauphin, C.; Jouk, P.-S.; Delrue, M.-A.; Thambo, J.-B.; Le Metayer, P.; et al. Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur. J. Med. Genet. 2011, 54, e570–e575. [Google Scholar] [CrossRef] [PubMed]
- Vakhrushev, Y.; Kozyreva, A.; Semenov, A.; Sokolnikova, P.; Lubimtseva, T.; Lebedev, D.; Smolina, N.; Zhuk, S.; Mitrofanova, L.; Vasichkina, E.; et al. RBM20-Associated Ventricular Arrhythmias in a Patient with Structurally Normal Heart. Genes 2021, 12, 94. [Google Scholar] [CrossRef]
- Beqqali, A.; Bollen, I.A.; Rasmussen, T.B.; van den Hoogenhof, M.M.; van Deutekom, H.W.; Schafer, S.; Haas, J.; Meder, B.; Sørensen, K.E.; van Oort, R.J.; et al. A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc. Res. 2016, 112, 452–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahr, H.C.; Jaalouk, D.E. Exploring the Crosstalk Between LMNA and Splicing Machinery Gene Mutations in Dilated Cardiomyopathy. Front. Genet. 2018, 9, 231. [Google Scholar] [CrossRef]
- Lin, S.; Fu, X.-D. SR proteins and related factors in alternative splicing. Adv. Exp. Med. Biol. 2007, 623, 107–122. [Google Scholar]
- Guo, W.; Sun, M. RBM20, a potential target for treatment of cardiomyopathy via titin isoform switching. Biophys. Rev. 2018, 10, 15–25. [Google Scholar] [CrossRef]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Methawasin, M.; Hutchinson, K.R.; Lee, E.-J.; Smith III, J.E.; Saripalli, C.; Hidalgo, C.G.; Ottenheijm, C.A.; Granzier, H. Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation 2014, 129, 1924–1936. [Google Scholar] [CrossRef]
- Dauksaite, V.; Gotthardt, M. Molecular basis of titin exon exclusion by RBM20 and the novel titin splice regulator PTB4. Nucleic Acids Res. 2018, 46, 5227–5238. [Google Scholar] [CrossRef] [PubMed]
- Greaser, M.L.; Warren, C.M.; Esbona, K.; Guo, W.; Duan, Y.; Parrish, A.M.; Krzesinski, P.R.; Norman, H.S.; Dunning, S.; Fitzsimons, D.P.; et al. Mutation that dramatically alters rat titin isoform expression and cardiomyocyte passive tension. J. Mol. Cell Cardiol. 2008, 44, 983–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Methawasin, M.; Strom, J.G.; Slater, R.E.; Fernandez, V.; Saripalli, C.; Granzier, H. Experimentally increasing the compliance of titin through RNA binding motif-20 (RBM20) inhibition improves diastolic function in a mouse model of heart failure with preserved ejection fraction. Circulation 2016, 134, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Rebs, S.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Meder, B.; Streckfuss-Bömeke, K. Generation of pluripotent stem cell lines and CRISPR/Cas9 modified isogenic controls from a patient with dilated cardiomyopathy harboring a RBM20 p.R634W mutation. Stem Cell Res. 2020, 47, 101901. [Google Scholar] [CrossRef]
- Wyles, S.P.; Hrstka, S.C.; Reyes, S.; Terzic, A.; Olson, T.M.; Nelson, T.J. Pharmacological modulation of calcium homeostasis in familial dilated cardiomyopathy: An in vitro analysis from an RBM20 patient-derived iPSC model. Clin. Transl. Sci. 2016, 9, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Wu, J.; Sun, H.; Briganti, F.; Meder, B.; Wei, W.; Steinmetz, L.M. Single-molecule, full-length transcript isoform sequencing reveals disease-associated RNA isoforms in cardiomyocytes. Nat. Commun. 2021, 12, 4203. [Google Scholar] [CrossRef]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef]
- Schweingruber, C.; Rufener, S.C.; Zünd, D.; Yamashita, A.; Mühlemann, O. Nonsense-mediated mRNA decay—Mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2013, 1829, 612–623. [Google Scholar] [CrossRef]
- Lennermann, D.; Backs, J.; van den Hoogenhof, M.M.G. New Insights in RBM20 Cardiomyopathy. Curr. Heart Fail. Rep. 2020, 17, 234–246. [Google Scholar] [CrossRef]
- Kowalczyk, M.S.; Hughes, J.R.; Babbs, C.; Sanchez-Pulido, L.; Szumska, D.; Sharpe, J.A.; Sloane-Stanley, J.A.; Morriss-Kay, G.M.; Smoot, L.B.; Roberts, A.E. Nprl3 is required for normal development of the cardiovascular system. Mamm. Genome 2012, 23, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Iijima, M.; Yoshimoto, N.; Niimi, T.; Kuroda, S.i.; Maturana, A.D. RBM 20 and RBM 24 cooperatively promote the expression of short enh splice variants. FEBS Lett. 2016, 590, 2262–2274. [Google Scholar] [CrossRef]
- Chauveau, C.; Rowell, J.; Ferreiro, A. A Rising Titan: TTN Review and Mutation Update. Hum. Mutat. 2014, 35, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- LeWinter, M.M.; Granzier, H.L. Cardiac titin and heart disease. J. Cardiovasc. Pharmacol. 2014, 63, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Deo, R.C. Alternative splicing, internal promoter, nonsense-mediated decay, or all three: Explaining the distribution of truncation variants in titin. Circ. Cardiovasc. Genet. 2016, 9, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Tunwell, R.E.; Wickenden, C.; Bertrand, B.M.; Shevchenko, V.I.; Walsh, M.B.; Allen, P.D.; Lai, F.A. The human cardiac muscle ryanodine receptor-calcium release channel: Identification, primary structure and topological analysis. Biochem. J. 1996, 318, 477–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laitinen, P.J.; Brown, K.M.; Piippo, K.; Swan, H.; Devaney, J.M.; Brahmbhatt, B.; Donarum, E.A.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the Cardiac Ryanodine Receptor (RyR2) Gene in Familial Polymorphic Ventricular Tachycardia. Circulation 2001, 103, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E. Calmodulin kinase signaling in heart: An intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol. Ther. 2005, 106, 39–55. [Google Scholar] [CrossRef]
- Zhang, M.; Gao, H.; Liu, D.; Zhong, X.; Shi, X.; Yu, P.; Jin, L.; Liu, Y.; Tang, Y.; Song, Y. CaMKII-δ9 promotes cardiomyopathy through disrupting UBE2T-dependent DNA repair. Nat. Cell Biol. 2019, 21, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Bertero, A.; Fields, P.A.; Ramani, V.; Bonora, G.; Yardimci, G.G.; Reinecke, H.; Pabon, L.; Noble, W.S.; Shendure, J.; Murry, C.E. Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory. Nat. Commun. 2019, 10, 1538. [Google Scholar] [CrossRef] [Green Version]
- Fochi, S.; Lorenzi, P.; Galasso, M.; Stefani, C.; Trabetti, E.; Zipeto, D.; Romanelli, M.G. The Emerging Role of the RBM20 and PTBP1 Ribonucleoproteins in Heart Development and Cardiovascular Diseases. Genes 2020, 11, 402. [Google Scholar] [CrossRef] [Green Version]
- Lorenzi, P.; Sangalli, A.; Fochi, S.; Dal Molin, A.; Malerba, G.; Zipeto, D.; Romanelli, M.G. RNA-binding proteins RBM20 and PTBP1 regulate the alternative splicing of FHOD3. Int. J. Biochem. Cell Biol. 2019, 106, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Takeya, R.; Suetsugu, S.; Kan-o, M.; Narusawa, M.; Shiose, A.; Tominaga, R.; Sumimoto, H. Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles. J. Biol. Chem. 2009, 284, 29873–29881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimura, T.; Takeya, R.; Ishikawa, T.; Yamano, T.; Matsuo, A.; Tatsumi, T.; Nomura, T.; Sumimoto, H.; Kimura, A. Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ. J. 2013, 77, 2990–2996. [Google Scholar] [CrossRef] [Green Version]
- Wooten, E.C.; Hebl, V.B.; Wolf, M.J.; Greytak, S.R.; Orr, N.M.; Draper, I.; Calvino, J.E.; Kapur, N.K.; Maron, M.S.; Kullo, I.J. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Ushijima, T.; Fujimoto, N.; Matsuyama, S.; Kan-o, M.; Kiyonari, H.; Shioi, G.; Kage, Y.; Yamasaki, S.; Takeya, R.; Sumimoto, H. The actin-organizing formin protein Fhod3 is required for postnatal development and functional maintenance of the adult heart in mice. J. Biol. Chem. 2018, 293, 148–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Waning, J.I.; Caliskan, K.; Hoedemaekers, Y.M.; van Spaendonck-Zwarts, K.Y.; Baas, A.F.; Boekholdt, S.M.; van Melle, J.P.; Teske, A.J.; Asselbergs, F.W.; Backx, A.P. Genetics, clinical features, and long-term outcome of noncompaction cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 711–722. [Google Scholar] [CrossRef]
- Miszalski-Jamka, K.; Jefferies, J.L.; Mazur, W.; Głowacki, J.; Hu, J.; Lazar, M.; Gibbs, R.A.; Liczko, J.; Kłyś, J.; Venner, E. Novel genetic triggers and genotype–phenotype correlations in patients with left ventricular noncompaction. Circ. Cardiovasc. Genet. 2017, 10, e001763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure. J. Am. Coll. Cardiol. 2017, 70, 776–803. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA Guideline for the Management of Heart Failure. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, R.; Mason, P.K. Lamin A/C deficiency as a cause of familial dilated cardiomyopathy. Curr. Opin. Cardiol. 2009, 24, 203–208. [Google Scholar] [CrossRef]
- Anselme, F.; Moubarak, G.; Savouré, A.; Godin, B.; Borz, B.; Drouin-Garraud, V.; Gay, A. Implantable cardioverter-defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm 2013, 10, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2017, 39, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Liss, M.; Radke, M.H.; Eckhard, J.; Neuenschwander, M.; Dauksaite, V.; von Kries, J.P.; Gotthardt, M. Drug discovery with an RBM20 dependent titin splice reporter identifies cardenolides as lead structures to improve cardiac filling. PLoS ONE 2018, 13, e0198492. [Google Scholar] [CrossRef] [PubMed]
- Beraldi, R.; Li, X.; Martinez Fernandez, A.; Reyes, S.; Secreto, F.; Terzic, A.; Olson, T.M.; Nelson, T.J. Rbm20-deficient cardiogenesis reveals early disruption of RNA processing and sarcomere remodeling establishing a developmental etiology for dilated cardiomyopathy. Hum. Mol. Genet. 2014, 23, 3779–3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Gene | Encoded Protein | Function | References |
|---|---|---|---|
| APTX | Aprataxin | DNA repair | [6] |
| CACNA1C | Calcium channel, voltage-dependent, L-type, alpha 1C sub-unit | sub-unit of the L-type calcium channel | [6] |
| CAMK2D | Calcium/calmodulin-dependent protein kinase II delta | Serine/threonine kinase; regulates many cardiac proteins through phosphorylation | [6,33] |
| CAMK2G | Calcium/calmodulin-dependent protein kinase II Gamma | Serine/threonine kinase; regulates many cardiac proteins through phosphorylation | [6] |
| DAB1 | Disabled-1 | Neuronal development | [6] |
| DNM3 | Dynamin-3 | Actin-membrane budding | [6] |
| DST | Dystonin | Adhesion junction plaque protein | [8] |
| DTNA | Dystrobrevin alpha | Part of the dystrophin-associated complex linking ECM and cytoskeleton | [6] |
| ENAH | Protein-enabled homolog | Actin-associated | [8] |
| FHOD3 | Formin homology 2 domain-containing 3 | Sarcomeric assembly | [6] |
| FNBP1 | Formin-binding protein 1 | Actin cytoskeleton regulation | [6] |
| GIT2 | G protein-coupled receptor kinase interactor 2 | Cytoskeletal dynamics | [6] |
| IMMT | Inner membrane mitochondrial protein | Part of the mitochondrial inner membrane complex | [8,37] |
| KALRN | Kalirin | Serine/threonine protein kinase | [6] |
| KCNIP2 | KV channel-interacting protein 2 | Sub-unit of voltage-gated potassium channel complex | [6] |
| LDB3 | LIM domain binding 3 | Sarcomeric stabilization | [6,8] |
| LMO7 | LIM domain only protein 7 | - | [8] |
| LRRFIP1 | Leucine-rich repeat flightless-interacting protein 1 | Transcriptional repressor | [8] |
| MECP2 | Methyl CpG–binding protein 2 | Transcriptional regulator; highly expressed in neuronal cells | [6] |
| MLIP | Muscular-enriched A-type laminin-interacting protein | Interacts with lamin A/C; potentially involved in cardiac homeostasis | [8] |
| MTMR1 | Myotubularin-related protein 1 | - | [6] |
| MYH7 | Myosin heavy chain 7 | Cardiac slow twitch myosin heavy chain beta isoform; muscle contraction | [8] |
| MYOM1 | Myomesin-1 | Sarcomeric; links titin and thick filament | [8] |
| NEXN | Nexilin | Actin-associated; DCM-associated | [8] |
| NFIA | Nuclear factor I A | Transcription factor | [6] |
| NPRL3 | Nitrogen permease regulator-like 3 | Inhibits mTORC1; necessary for cardiovascular development | [6,41] |
| NTRK3 | Tropomyosin receptor kinase C | Neutrophin-3-receptor | [6] |
| OBSCN | Obscurin | Sarcomeric signaling | [8] |
| PDLIM3 | PDZ and LIM domain protein 3 | Binds alpha actinin-2; relevant for right ventricular function | [8] |
| PDLIM5 | PDZ and LIM domain protein 5 | LIM domain protein; protein-protein interaction | [6,42] |
| PLEKHA5 | Pleckstrin homology domain-containing family A member 5 | - | [6] |
| RALGPS1 | Ral GEF with PH domain- and SH3-binding motif 1 | - | [6] |
| RTN4 | Reticulon 4 | Neurite outgrowth inhibitor in the central nervous system | [8] |
| RYR2 | Ryanodine receptor 2 | Calcium receptor in the SR; allows release of Ca2+ into the cytosol | [8] |
| SEMA6D | Semaphorin 6D | Neuronal regulation | [6] |
| SH3KBP1 | SH3 domain-containing kinase-binding protein 1 | - | [6] |
| SLC38A10 | Putative sodium-coupled neutral amino acid transporter 10 | Sodium-dependent amino acid/proton antiporter | [6] |
| SORBS1 | Sorbin and SH3 domain-containing 1 | Cytoskeletal formation | [6] |
| SPEN | Msx2-interacting protein | Hormone inducible transcriptional repressor | [6] |
| TNNT2 | Cardiac troponin T | Part of the cardiac troponin complex regulating muscle contraction dependent on calcium | [8] |
| TPM1 | Tropomyosin alpha-1 chain | Cytoskeletal; contraction | [6] |
| TRDN | Triadin | Forms a complex with RyR and CASQ2; calcium release from the SR | [6] |
| TTN | Titin | Sarcomeric spring; compliance of the heart | [6,8] |
| UBE2F | Ubiquitin-conjugating enzyme E2 F (putative) | - | [6] |
| ZNF451 | E3 SUMO-protein ligase ZNF451 | Protein sumoylation | [6] |
| RBM20-CM vs. DCM Odds Ratio (CI; p-Value) | RBM20-CM vs. TTNtv-CM Odds Ratio (CI; p-Value) | RBM20-CM vs. LMNA-CM Odds Ratio (CI; p-Value) | |
|---|---|---|---|
| Evidence of sustained VA * | 14.7 (6.0–36.0; p < 0.001) | 27.3 (3.4–223.0; p < 0.001) | 1.2 (0.6–2.4; p = 0.65) |
| Family history of SCA ** | 5.9 (3.1–11.2; p < 0.001) | 6.2 (2.6–14.5; p < 0.001) | 1.4 (0.6–2.8; p = 0.46) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koelemen, J.; Gotthardt, M.; Steinmetz, L.M.; Meder, B. RBM20-Related Cardiomyopathy: Current Understanding and Future Options. J. Clin. Med. 2021, 10, 4101. https://doi.org/10.3390/jcm10184101
Koelemen J, Gotthardt M, Steinmetz LM, Meder B. RBM20-Related Cardiomyopathy: Current Understanding and Future Options. Journal of Clinical Medicine. 2021; 10(18):4101. https://doi.org/10.3390/jcm10184101
Chicago/Turabian StyleKoelemen, Jan, Michael Gotthardt, Lars M. Steinmetz, and Benjamin Meder. 2021. "RBM20-Related Cardiomyopathy: Current Understanding and Future Options" Journal of Clinical Medicine 10, no. 18: 4101. https://doi.org/10.3390/jcm10184101
APA StyleKoelemen, J., Gotthardt, M., Steinmetz, L. M., & Meder, B. (2021). RBM20-Related Cardiomyopathy: Current Understanding and Future Options. Journal of Clinical Medicine, 10(18), 4101. https://doi.org/10.3390/jcm10184101