
Genetic Insights into Primary Restrictive Cardiomyopathy



Abstract
:1. Introduction
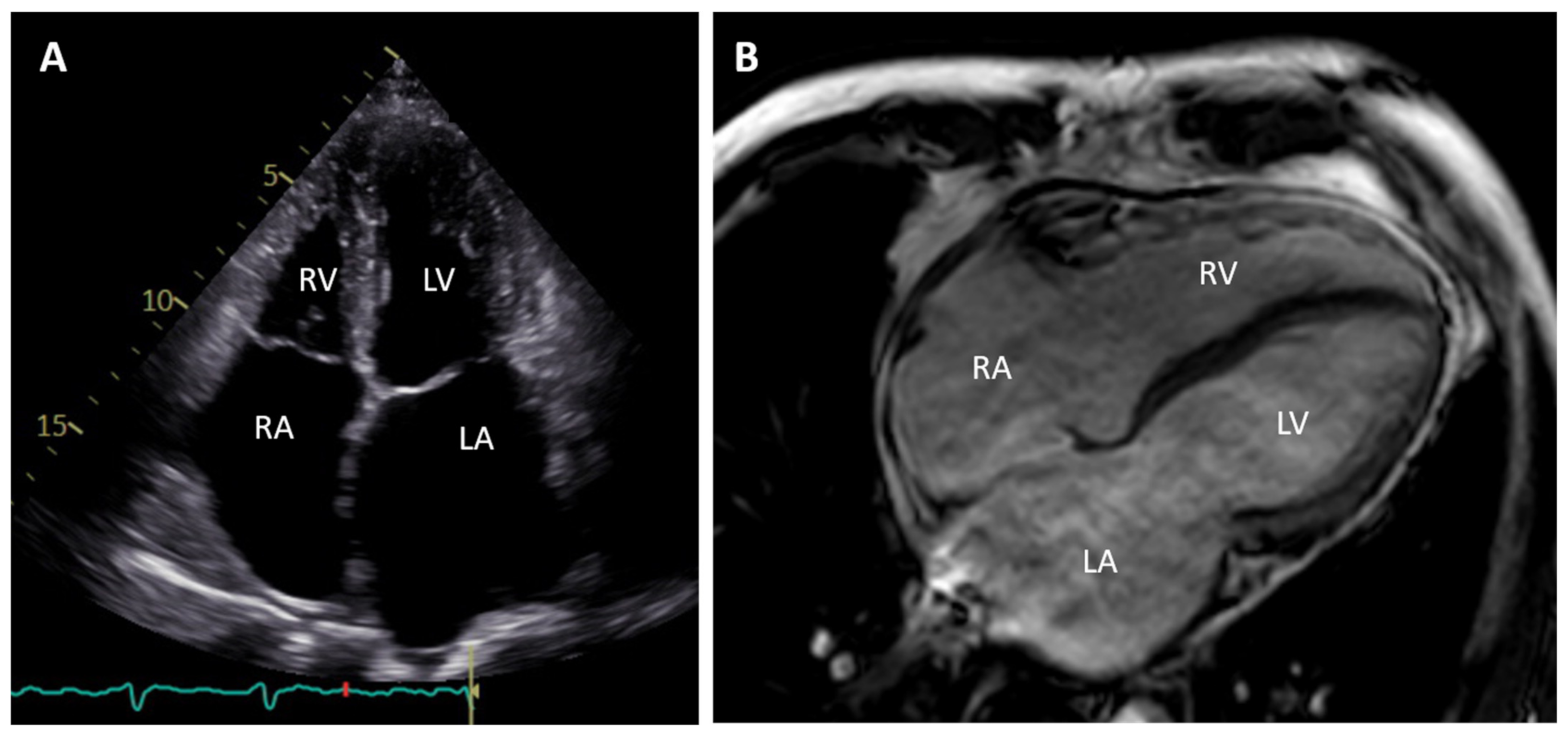
2. Clinical Description
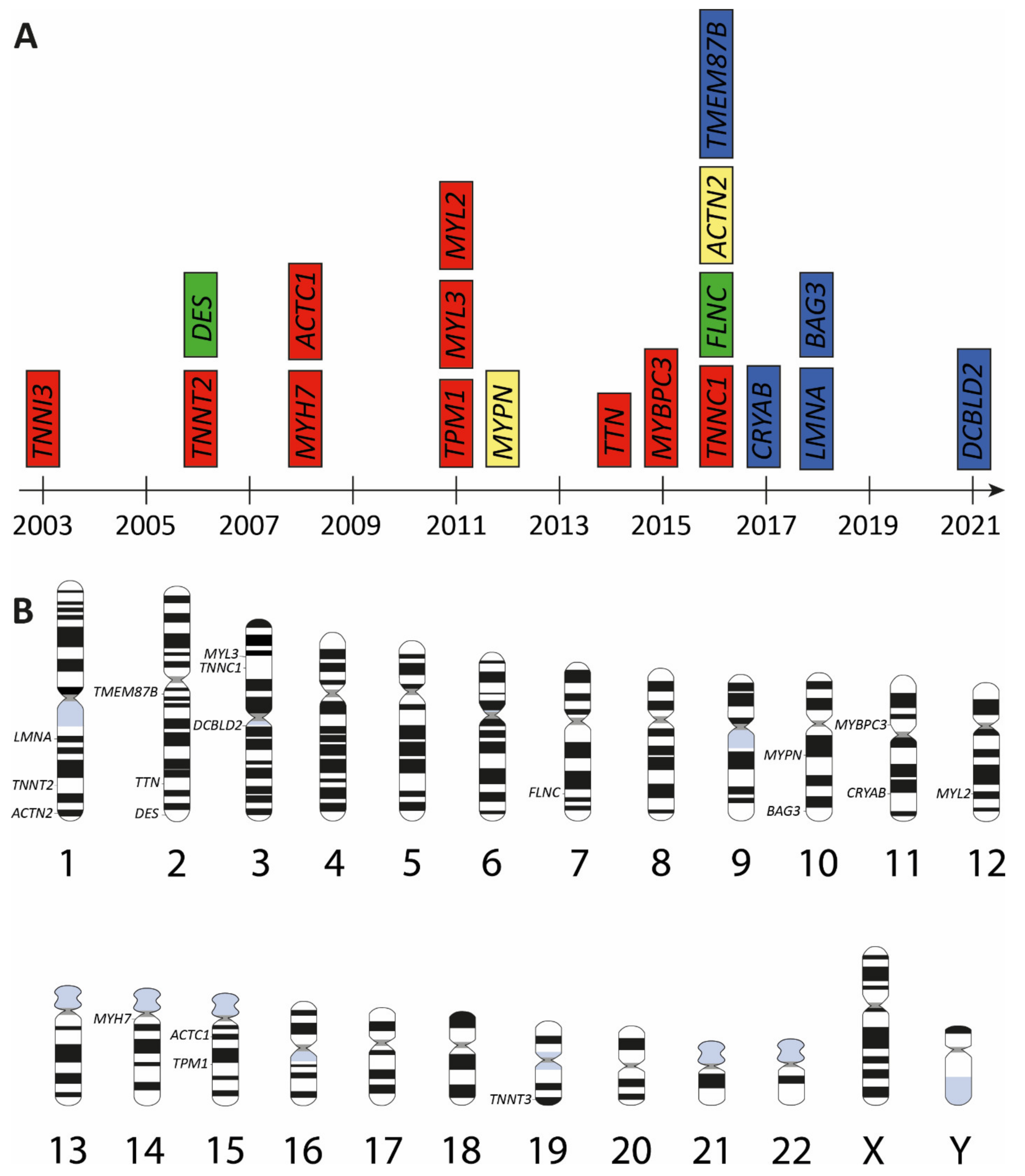
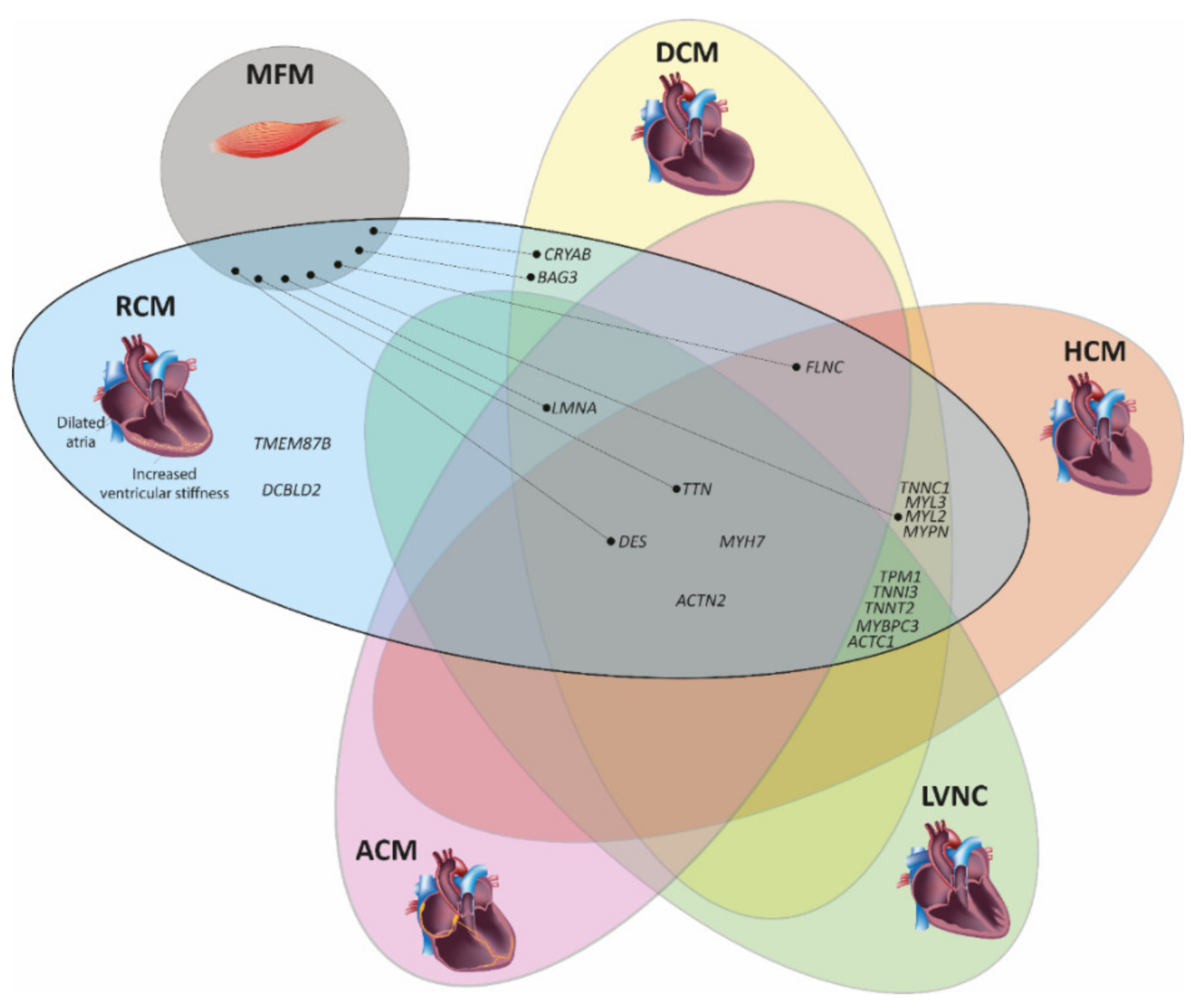
3. Genetic Landscape of Restrictive Cardiomyopathy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Cytogenetic Location | Encoded Protein | Subcellular Protein Localization | First Description | References |
|---|---|---|---|---|---|
| TNNI3 | 19q13.42 | cardiac troponin I | Sarcomere | 2003 | [27] |
| TNNT2 | 1q32.1 | cardiac troponin T | Sarcomere | 2006 | [28] |
| DES | 2q35 | desmin | Intermediate filament | 2006 | [29] |
| ACTC1 | 15q14 | cardiac actin | Sarcomere | 2008 | [30] |
| MYH7 | 14q11.2 | β myosin heavy chain | Sarcomere | 2008 | [31] |
| TPM1 | 15q22.2 | tropomyosin 1 | Sarcomere | 2011 | [32] |
| MYL3 | 3p21.31 | essential myosin light chain 3 | Sarcomere | 2011 | [32] |
| MYL2 | 12q24.11 | cardiac regulatory myosin light chain | Sarcomere | 2011 | [32] |
| MYPN | 10q21.3 | myopalladin | Sarcomere, Z-disc | 2012 | [33] |
| TTN | 2q31.2 | titin | Sarcomere | 2014 | [34] |
| MYBPC3 | 11p11.2 | cardiac myosin binding protein C | Sarcomere | 2015 | [35] |
| TNNC1 | 3p21.1 | cardiac troponin C | Sarcomere | 2016 | [36] |
| FLNC | 7q32.1 | filamin C | Intercalated disc, Z-disc, sarcolemma | 2016 | [37] |
| TMEM87B | 2q13 | transmembrane protein 87 B | Membrane | 2016 | [38] |
| ACTN2 | 1q43 | α actinin 2 | Z-disc | 2016 | [39] |
| CRYAB | 11q23.1 | αB crystallin | IF associated protein, intercalated disc, Z-disc | 2017 | [40] 1 |
| LMNA | 1q22 | lamin A/C | Nuclear lamina | 2018 | [41] |
| BAG3 | 10q26.11 | bcl2 associated athanogene 3 | Cytosol | 2018 | [42] |
| DCBLD2 | 3q12.1 | discoidin cub and lccl domain containing protein 2 | Membrane | 2021 | [43] 2 |
3.1. Mutations in Genes Encoding for Sarcomere Proteins
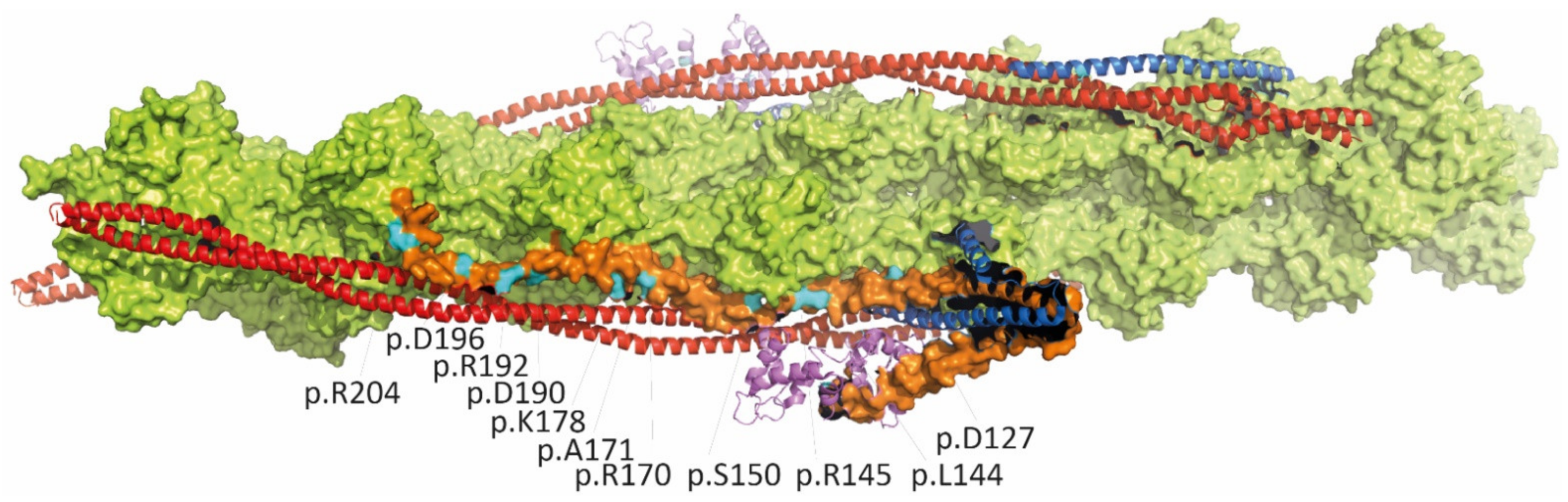
3.1.1. Cardiac Troponins (TNNI3, TNNT2, TNNC1) and Alpha-Tropomyosin (TPM1)
| Mutation | Age of Onset and Clinical Features | Family History | MAF 1 | Comments | References |
|---|---|---|---|---|---|
| TNNI3 | |||||
| p.D127Y | infant, HF, VAD | de novo | - | contractile dysfunctions and effects on thin filament structure | [53] |
| p.L144Q | adult, HF | unknown | - | [27] | |
| p.L144H | young adults, HF | familial | - | [54] | |
| p.R145W | children and adults, HF | familial, autosomal dominant | 3/280226 | variant also associated with HCM; Dutch founder mutation; segregation in several families | [27,39,55] |
| p.R145Q | children | familial, far relative HCM | - | associated with HCM | [55] |
| p.S150P | child, SCD | familial | - | one Chinese family with several affected members | [56] |
| c.549+2delT | infant, died at age 2 | de novo | - | predicts splicing defect and truncation | [55] |
| p.D168fsX176 | child, HF, died at age 28y | de novo | - | protein reduction | [57] |
| p.R170G | child, HF | de novo | - | [47] | |
| p.R170W | infant | de novo | - | variant also associated with HCM | [47,58] |
| p.R170Q | child, HF | de novo | - | variant also associated with HCM | [30,54] |
| p.A171T | adult, HF, AF | unknown | - | [27] | |
| p.E177fsX209 | child | de novo | - | [30] | |
| p.K178E | 6y, HF | de novo | - | [27] | |
| p.K178del | child | de novo | - | [55] | |
| p.D190H | mainly adults, HF, SCD | familial | - | named in ClinVar as p.D190G | [27] |
| p.R192C | child | familial | - | carries also mosaicism of p.R145Q; associated also with HCM in far relative | [55] |
| p.R192H | children, young adult, HF | de novo | - | independent reports of de novo mutations; variants also associated with HCM | [27,59,60] |
| p.K193E | adults, AF, SCD | familial | - | cousin developed HCM | [61] |
| p.I195fs | young adult, HF, HTx | de novo | - | dominant-negative effect | [62] |
| p.D196H | three adults, HF, HTx | familial, homozygous | - | heterozygous carrier asymptomatic | [63] |
| p.R204H | children, HF, HTx, VSD in one case | de novo | - | independent reports of de novo mutations | [59,64,65] |
| TNNT2 | |||||
| p.I89N | two adult cases within one family | familial | 0.00002 | mixed phenotype with HCM and DCM | [66] |
| p.R104C | children, young adult, HF | familial | - | mixed phenotype with HCM in the family | [67] |
| p.E69del | infant, HF, VAD | de novo | - | [28] | |
| p.E146K | child | familial | 0.00003 | variant also associated with other CMPs | [30] |
| TNNC1 | |||||
| p.A8V; p.D145E | two infants died | familial, compound heterozygous | 0.00001 0.0001 | HCM which evolved into RCM | [36] |
| TPM1 | |||||
| p.E62Q; p.M281T | child | familial, compound heterozygous | - 0.00001 | each single variant leads to a HCM like phenotype | [16] |
| ACTC1 | |||||
| p.D313H | child | familial | - | father was diagnosed with DCM | [30] |

3.1.2. Cardiac Actin (ACTC1)
3.1.3. Myosin Heavy and Light Chains (MYH7, MYL2 and MYL3)
| Mutation | Age of Onset and Clinical Features | Family History | MAF 1 | Comments | References |
|---|---|---|---|---|---|
| MYH7 | |||||
| p.Y386C | infant, coronary artery bridging | unknown | - | [91] | |
| p.R721K | adult, AF, | familial | - | in combination with ABCC9-p.R1186Q | [92] |
| p.G768R | adult, AF, | unknown | - | [39] | |
| death at age 42 | |||||
| infant, HTx | unknown | - | [93] | ||
| p.R783H | adult, AVB, | familial | 0.00002 | son has HCM | [39] |
| death at age 54 | |||||
| p.P838L | infant | de novo | - | [31] | |
| p.L840M | child | unknown | - | in combination with MYBPC3-p.P147L | [39] |
| p.R870C | two adults, AF | familial | 0.00002 | myofibrillar disarray, | [94] |
| cardiomyocyte necrosis, | |||||
| abnormal nuclei morphology | |||||
| p.I909M | adult, AVB, AF, death at age 56 | unknown | - | [39] | |
| p.T1188CfsX22 | adult, in combination with LVH | de novo | - | [95] | |
| MYL2 | |||||
| p.G57E | adult | absent | 0.000004 | in combination with MYL3-p.E143Khom | [32] |
| MYL3 | |||||
| MYL3-p.E143Khom | adult | absent | 0.00001 | in combination with MYL2-p.G57E | [32] |
3.1.4. Cardiac Myosin Binding Protein C (MYBPC3)
3.1.5. Titin (TTN)
3.2. Mutations in Genes Encoding Non-Saromere Proteins
3.2.1. Desmin (DES)
| Mutation | Age of Onset and Clinical Features | Family History | MAF 1 | Comments | References |
|---|---|---|---|---|---|
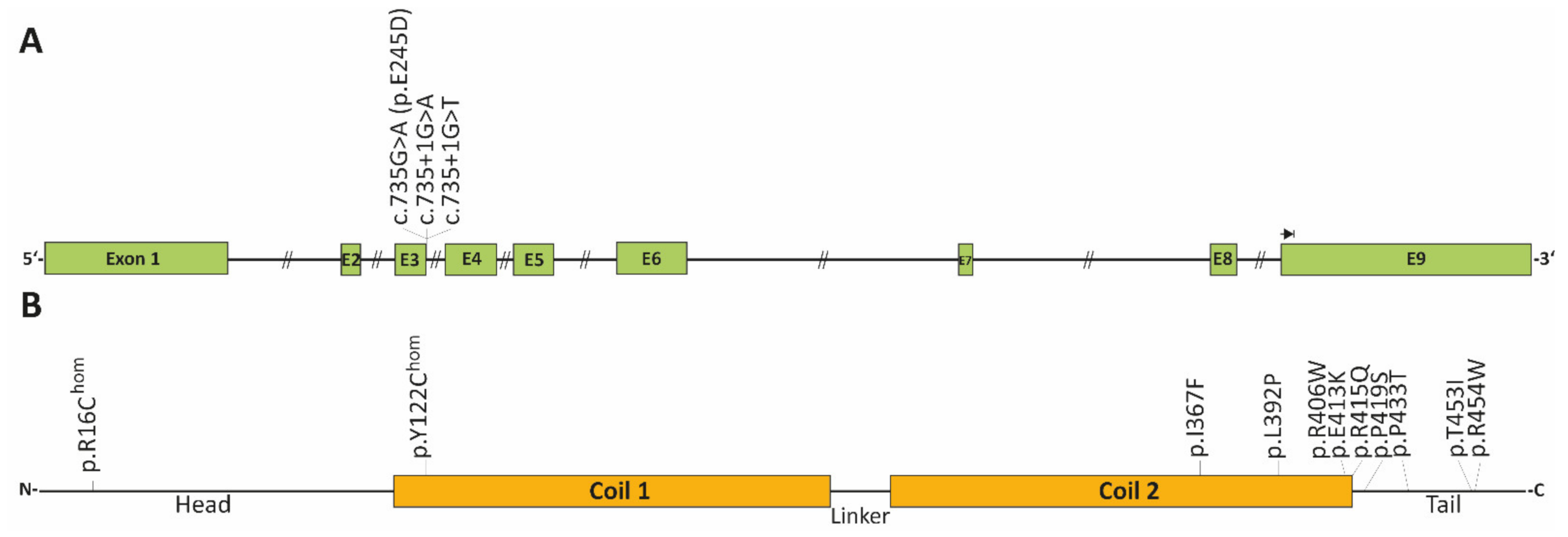
| c.735+1G>A | adult, SM | de novo | - | induces a splice defect, skipping of exon-3 | [133] |
| c.735+1G>T | adults, SM | two patients | - | induces a splice defect, skipping of exon-3 | [119] |
| p.R16C | adult, AVB, HTx | one patient | 0.000006570 | homozygous | [134] |
| p.Y122H | adult, AVB | one patient | - | homozygous | [14] |
| c.735G>C (p.E245D) | adults, AF | several family members, only index patient was genotyped | - | induces a splice defect, skipping of exon-3 | [116] |
| p.I367F | adults, AVB, SM | several family members | - | index patient diagnosed with HCM [135] | [15,135] |
| p.L392P | adult, AVB, SM | one patient | - | [135] | |
| p.R406W | adults, AVB | three affected members | - | a different index patient presented ACM in combination with SM [112] | [117,134] |
| p.E413K | adults, AVB, AF, SCD | four affected members | - | [136,137] | |
| p.R415Q | adult, AF | several family members | - | different phenotypes, unclear if a splice defect is caused (last bp of exon-6) | [15] |
| p.P419S | adults, AVB, SM | two patients | - | [135] | |
| p.P433T | adult, AVB, SM | one patient | - | [120] | |
| p.T453I | adult, AVB | de novo | - | [134] | |
| p.R454W | adults, AVB, SM | two patients | - | [112] |
3.2.2. Myopalladin (MYPN)
3.2.3. α-Actinin-2 (ACTN2)

3.2.4. Filamin-C (FLNC)
3.2.5. Lamin A/C (LMNA)
3.2.6. Transmembrane Protein 87B (TMEM87B)
3.2.7. αB-Crystallin (CRYAB)
3.2.8. Bcl2 Associated Athanogene 3 (BAG3)
3.2.9. Discoidin Cub and Lccl Domain Containing Protein-2 (DCBLD2)
4. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, epidemiology, and global burden of cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, P.; Myers, M. The classification of cardiac diagnosis. JAMA 1921, 77, 1414–1415. [Google Scholar]
- Gerull, B.; Klaassen, S.; Brodehl, A. The genetic landscape of cardiomyopathies. In Genetic Causes of Cardiac Disease; Erdmann, J., Moretti, A., Eds.; Springer: Cham, Switzerland, 2019; pp. 45–91. [Google Scholar]
- Butzner, M.; Leslie, D.L.; Cuffee, Y.; Hollenbeak, C.S.; Sciamanna, C.; Abraham, T. Stable rates of obstructive hypertrophic cardiomyopathy in a contemporary era. Front. Cardiovasc. Med. 2021, 8, 765876. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuddy, S.A.M.; Falk, R.H. Amyloidosis as a systemic disease in context. Can. J. Cardiol. 2020, 36, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef] [PubMed]
- Paisey, R.B.; Steeds, R.; Barrett, T.; Williams, D.; Geberhiwot, T.; Gunay-Aygun, M. Alstrom syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 1993. [Google Scholar]
- Starr, L.J.; Lindor, N.M.; Lin, A.E. Myhre syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 1993. [Google Scholar]
- Seferovic, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Gal, T.B.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the heart failure association of the european society of cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Gimeno, J.R.; Bahl, A.; Steffensen, U.; Steffensen, M.; Osman, E.; Thaman, R.; Mogensen, J.; Elliott, P.M.; Doi, Y.; et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J. Am. Coll. Cardiol. 2007, 49, 2419–2426. [Google Scholar] [CrossRef] [Green Version]
- DePasquale, E.C.; Nasir, K.; Jacoby, D.L. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J. Heart Lung Transplant. 2012, 31, 1269–1275. [Google Scholar] [CrossRef]
- Muchtar, E.; Blauwet, L.A.; Gertz, M.A. Restrictive cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 2017, 121, 819–837. [Google Scholar] [CrossRef]
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive cardiomyopathy is caused by a novel homozygous desmin (DES) mutation p.Y122H leading to a severe filament assembly defect. Genes 2019, 10, 918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripoll-Vera, T.; Zorio, E.; Gamez, J.M.; Molina, P.; Govea, N.; Cremer, D. Phenotypic patterns of cardiomyopathy caused by mutations in the desmin gene. A clinical and genetic study in two inherited heart disease units. Rev. Esp. Cardiol. 2015, 68, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Dorsch, L.M.; Kuster, D.W.D.; Jongbloed, J.D.H.; Boven, L.G.; van Spaendonck-Zwarts, K.Y.; Suurmeijer, A.J.H.; Vink, A.; du Marchie Sarvaas, G.J.; van den Berg, M.P.; van der Velden, J.; et al. The effect of tropomyosin variants on cardiomyocyte function and structure that underlie different clinical cardiomyopathy phenotypes. Int. J. Cardiol. 2021, 323, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Fichna, J.P.; Maruszak, A.; Zekanowski, C. Myofibrillar myopathy in the genomic context. J. Appl. Genet. 2018, 59, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Marmol, A.M.; Strasser, G.; Isamat, M.; Coulombe, P.A.; Yang, Y.; Roca, X.; Vela, E.; Mate, J.L.; Coll, J.; Fernandez-Figueras, M.T.; et al. A dysfunctional desmin mutation in a patient with severe generalized myopathy. Proc. Natl. Acad. Sci. USA 1998, 95, 11312–11317. [Google Scholar] [CrossRef] [Green Version]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prevost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tome, F.; Dupret, J.M.; et al. A missense mutation in the αB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef]
- Kley, R.A.; Hellenbroich, Y.; van der Ven, P.F.; Furst, D.O.; Huebner, A.; Bruchertseifer, V.; Peters, S.A.; Heyer, C.M.; Kirschner, J.; Schroder, R.; et al. Clinical and morphological phenotype of the filamin myopathy: A study of 31 German patients. Brain 2007, 130 Pt 12, 3250–3264. [Google Scholar] [CrossRef]
- Dhawan, P.S.; Liewluck, T.; Knapik, J.; Milone, M. Myofibrillar myopathy due to dominant LMNA mutations: A report of 2 cases. Muscle Nerve 2018, 57, E124–E126. [Google Scholar] [CrossRef]
- Odgerel, Z.; Sarkozy, A.; Lee, H.S.; McKenna, C.; Rankin, J.; Straub, V.; Lochmuller, H.; Paola, F.; D’Amico, A.; Bertini, E.; et al. Inheritance patterns and phenotypic features of myofibrillar myopathy associated with a BAG3 mutation. Neuromuscul. Disord. 2010, 20, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, G.; Barresi, R.; Wilson, I.J.; Hardy, S.A.; Griffin, H.; Hudson, J.; Elliott, H.R.; Ramesh, A.V.; Radunovic, A.; Winer, J.B.; et al. Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. J. Neurol. Neurosurg. Psychiatry 2014, 85, 331–338. [Google Scholar] [CrossRef]
- Izumi, R.; Niihori, T.; Aoki, Y.; Suzuki, N.; Kato, M.; Warita, H.; Takahashi, T.; Tateyama, M.; Nagashima, T.; Funayama, R.; et al. Exome sequencing identifies a novel TTN mutation in a family with hereditary myopathy with early respiratory failure. J. Hum. Genet. 2013, 58, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Weterman, M.A.; Barth, P.G.; van Spaendonck-Zwarts, K.Y.; Aronica, E.; Poll-The, B.T.; Brouwer, O.F.; van Tintelen, J.P.; Qahar, Z.; Bradley, E.J.; de Wissel, M.; et al. Recessive MYL2 mutations cause infantile type I muscle fibre disease and cardiomyopathy. Brain 2013, 136 Pt 1, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Cimiotti, D.; Budde, H.; Hassoun, R.; Jaquet, K. Genetic restrictive cardiomyopathy: Causes and consequences—An integrative approach. Int. J. Mol. Sci. 2021, 22, 558. [Google Scholar] [CrossRef]
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Investig. 2003, 111, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Peddy, S.B.; Vricella, L.A.; Crosson, J.E.; Oswald, G.L.; Cohn, R.D.; Cameron, D.E.; Valle, D.; Loeys, B.L. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics 2006, 117, 1830–1833. [Google Scholar] [CrossRef] [PubMed]
- Hager, S.; Mahrholdt, H.; Goldfarb, L.G.; Goebel, H.H.; Sechtem, U. Images in cardiovascular medicine. Giant right atrium in the setting of desmin-related restrictive cardiomyopathy. Circulation 2006, 113, e53–e55. [Google Scholar] [CrossRef] [PubMed]
- Kaski, J.P.; Syrris, P.; Burch, M.; Tome-Esteban, M.T.; Fenton, M.; Christiansen, M.; Andersen, P.S.; Sebire, N.; Ashworth, M.; Deanfield, J.E.; et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008, 94, 1478–1484. [Google Scholar] [CrossRef]
- Karam, S.; Raboisson, M.J.; Ducreux, C.; Chalabreysse, L.; Millat, G.; Bozio, A.; Bouvagnet, P. A de novo mutation of the beta cardiac myosin heavy chain gene in an infantile restrictive cardiomyopathy. Congenit. Heart Dis. 2008, 3, 138–143. [Google Scholar] [CrossRef]
- Caleshu, C.; Sakhuja, R.; Nussbaum, R.L.; Schiller, N.B.; Ursell, P.C.; Eng, C.; De Marco, T.; McGlothlin, D.; Burchard, E.G.; Rame, J.E. Furthering the link between the sarcomere and primary cardiomyopathies: Restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am. J. Med. Genet. Part A 2011, 155, 2229–2235. [Google Scholar] [CrossRef] [Green Version]
- Purevjav, E.; Arimura, T.; Augustin, S.; Huby, A.C.; Takagi, K.; Nunoda, S.; Kearney, D.L.; Taylor, M.D.; Terasaki, F.; Bos, J.M.; et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum. Mol. Genet. 2012, 21, 2039–2053. [Google Scholar] [CrossRef] [Green Version]
- Peled, Y.; Gramlich, M.; Yoskovitz, G.; Feinberg, M.S.; Afek, A.; Polak-Charcon, S.; Pras, E.; Sela, B.A.; Konen, E.; Weissbrod, O.; et al. Titin mutation in familial restrictive cardiomyopathy. Int. J. Cardiol. 2014, 171, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Lu, C.X.; Wang, Y.N.; Liu, F.; Chen, W.; Liu, Y.T.; Han, Y.C.; Cao, J.; Zhang, S.Y.; Zhang, X. Novel phenotype-genotype correlations of restrictive cardiomyopathy with myosin-binding protein C (MYBPC3) gene mutations tested by next-generation sequencing. J. Am. Heart Assoc. 2015, 4, e001879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploski, R.; Rydzanicz, M.; Ksiazczyk, T.M.; Franaszczyk, M.; Pollak, A.; Kosinska, J.; Michalak, E.; Stawinski, P.; Ziolkowska, L.; Bilinska, Z.T.; et al. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am. J. Med. Genet. Part A 2016, 170, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef]
- Yu, H.C.; Coughlin, C.R.; Geiger, E.A.; Salvador, B.J.; Elias, E.R.; Cavanaugh, J.L.; Chatfield, K.C.; Miyamoto, S.D.; Shaikh, T.H. Discovery of a potentially deleterious variant in TMEM87B in a patient with a hemizygous 2q13 microdeletion suggests a recessive condition characterized by congenital heart disease and restrictive cardiomyopathy. Mol. Case Stud. 2016, 2, a000844. [Google Scholar] [CrossRef] [Green Version]
- Kostareva, A.; Kiselev, A.; Gudkova, A.; Frishman, G.; Ruepp, A.; Frishman, D.; Smolina, N.; Tarnovskaya, S.; Nilsson, D.; Zlotina, A.; et al. Genetic spectrum of idiopathic restrictive cardiomyopathy uncovered by next-generation sequencing. PLoS ONE 2016, 11, e0163362. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschroder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef]
- Paller, M.S.; Martin, C.M.; Pierpont, M.E. Restrictive cardiomyopathy: An unusual phenotype of a lamin A variant. ESC Heart Fail. 2018, 5, 724–726. [Google Scholar] [CrossRef]
- Schanzer, A.; Rupp, S.; Graf, S.; Zengeler, D.; Jux, C.; Akinturk, H.; Gulatz, L.; Mazhari, N.; Acker, T.; Van Coster, R.; et al. Dysregulated autophagy in restrictive cardiomyopathy due to Pro209Leu mutation in BAG3. Mol. Genet. Metab. 2018, 123, 388–399. [Google Scholar] [CrossRef]
- Alhamoudi, K.M.; Barhoumi, T.; Al-Eidi, H.; Asiri, A.; Nashabat, M.; Alaamery, M.; Alharbi, M.; Alhaidan, Y.; Tabarki, B.; Umair, M.; et al. A homozygous nonsense mutation in DCBLD2 is a candidate cause of developmental delay, dysmorphic features and restrictive cardiomyopathy. Sci. Rep. 2021, 11, 12861. [Google Scholar] [CrossRef]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef] [PubMed]
- Thierfelder, L.; Watkins, H.; MacRae, C.; Lamas, R.; McKenna, W.; Vosberg, H.P.; Seidman, J.G.; Seidman, C.E. α-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 1994, 77, 701–712. [Google Scholar] [CrossRef]
- Gomes, A.V.; Liang, J.; Potter, J.D. Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J. Biol. Chem. 2005, 280, 30909–30915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimiotti, D.; Fujita-Becker, S.; Mohner, D.; Smolina, N.; Budde, H.; Wies, A.; Morgenstern, L.; Gudkova, A.; Sejersen, T.; Sjoberg, G.; et al. Infantile restrictive cardiomyopathy: cTnI-R170G/W impair the interplay of sarcomeric proteins and the integrity of thin filaments. PLoS ONE 2020, 15, e0229227. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Wen, H.; Edwards, T.; Metzger, J.M. Allele and species dependent contractile defects by restrictive and hypertrophic cardiomyopathy-linked troponin I mutants. J. Mol. Cell. Cardiol. 2008, 44, 891–904. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Charles, P.Y.; Nan, C.; Pinto, J.R.; Wang, Y.; Liang, J.; Wu, G.; Tian, J.; Feng, H.Z.; Potter, J.D.; et al. Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 49, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, L.; Jean-Charles, P.Y.; Nan, C.; Chen, G.; Tian, J.; Jin, J.P.; Gelb, I.J.; Huang, X. Dose-dependent diastolic dysfunction and early death in a mouse model with cardiac troponin mutations. J. Mol. Cell. Cardiol. 2013, 62, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Kawai, M.; Johnston, J.R.; Karam, T.; Wang, L.; Singh, R.K.; Pinto, J.R. Myosin rod hypophosphorylation and CB kinetics in papillary muscles from a TnC-A8V KI mouse model. Biophys. J. 2017, 112, 1726–1736. [Google Scholar] [CrossRef] [Green Version]
- Redwood, C.; Robinson, P. Alpha-tropomyosin mutations in inherited cardiomyopathies. J. Muscle Res. Cell Motil. 2013, 34, 285–294. [Google Scholar] [CrossRef]
- Hassoun, R.; Budde, H.; Mannherz, H.G.; Lódi, M.; Fujita-Becker, S.; Laser, K.T.; Gärtner, A.; Klingel, K.; Möhner, D.; Stehle, R.; et al. De novo missense mutations in TNNC1 and TNNI3 causing severe infantile cardiomyopathy affect myofilament structure and function and are modulated by troponin targeting agents. Int. J. Mol. Sci. 2021, 22, 9625. [Google Scholar] [CrossRef]
- Mouton, J.M.; Pellizzon, A.S.; Goosen, A.; Kinnear, C.J.; Herbst, P.G.; Brink, P.A.; Moolman-Smook, J.C. Diagnostic disparity and identification of two TNNI3 gene mutations, one novel and one arising de novo, in South African patients with restrictive cardiomyopathy and focal ventricular hypertrophy. Cardiovasc. J. Afr. 2015, 26, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Wijngaard, A.; Volders, P.; Van Tintelen, J.P.; Jongbloed, J.D.; van den Berg, M.P.; Lekanne Deprez, R.H.; Mannens, M.M.; Hofmann, N.; Slegtenhorst, M.; Dooijes, D.; et al. Recurrent and founder mutations in the Netherlands: Cardiac Troponin I (TNNI3) gene mutations as a cause of severe forms of hypertrophic and restrictive cardiomyopathy. Neth. Heart J. 2011, 19, 344–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Y.P.; Lu, C.X.; Zhao, X.Y.; Liang, R.J.; Lian, H.; Routledge, M.; Wu, W.; Zhang, X.; Fan, Z.J. Restrictive cardiomyopathy resulting from a troponin I type 3 mutation in a Chinese family. Chin. Med. Sci. J. 2016, 31, 1–7. [Google Scholar] [CrossRef]
- Kostareva, A.; Gudkova, A.; Sjöberg, G.; Mörner, S.; Semernin, E.; Krutikov, A.; Shlyakhto, E.; Sejersen, T. Deletion in TNNI3 gene is associated with restrictive cardiomyopathy. Int. J. Cardiol. 2009, 131, 410–412. [Google Scholar] [CrossRef]
- Mogensen, J.; Hey, T.; Lambrecht, S. A Systematic Review of Phenotypic Features Associated With Cardiac Troponin I Mutations in Hereditary Cardiomyopathies. Can. J. Cardiol. 2015, 31, 1377–1385. [Google Scholar] [CrossRef]
- Ding, W.H.; Han, L.; Xiao, Y.Y.; Mo, Y.; Yang, J.; Wang, X.F.; Jin, M. Role of Whole-exome sequencing in phenotype classification and clinical treatment of pediatric restrictive cardiomyopathy. Chin. Med. J. 2017, 130, 2823–2828. [Google Scholar] [CrossRef]
- Rai, T.S.; Ahmad, S.; Ahluwalia, T.S.; Ahuja, M.; Bahl, A.; Saikia, U.N.; Singh, B.; Talwar, K.K.; Khullar, M. Genetic and clinical profile of Indian patients of idiopathic restrictive cardiomyopathy with and without hypertrophy. Mol. Cell. Biochem. 2009, 331, 187–192. [Google Scholar] [CrossRef]
- Gerhardt, T.; Monserrat, L.; Landmesser, U.; Poller, W. A novel Troponin I mutation associated with severe restrictive cardiomyopathy-a case report of a 27-year-old woman with fatigue. Eur. Heart J. Case Rep. 2022, 6, ytac053. [Google Scholar] [CrossRef]
- Shah, S.; Yogasundaram, H.; Basu, R.; Wang, F.; Paterson, D.I.; Alastalo, T.P.; Oudit, G.Y. Novel dominant-negative mutation in cardiac troponin I causes severe restrictive cardiomyopathy. Circ. Heart Fail. 2017, 10, e003820. [Google Scholar] [CrossRef]
- Pantou, M.P.; Gourzi, P.; Gkouziouta, A.; Armenis, I.; Kaklamanis, L.; Zygouri, C.; Constantoulakis, P.; Adamopoulos, S.; Degiannis, D. A case report of recessive restrictive cardiomyopathy caused by a novel mutation in cardiac troponin I (TNNI3). BMC Med. Genet. 2019, 20, 61. [Google Scholar] [CrossRef]
- Yang, S.W.; Hitz, M.P.; Andelfinger, G. Ventricular septal defect and restrictive cardiomyopathy in a paediatric TNNI3 mutation carrier. Cardiol. Young 2010, 20, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Gambarin, F.I.; Tagliani, M.; Arbustini, E. Pure restrictive cardiomyopathy associated with cardiac troponin I gene mutation: Mismatch between the lack of hypertrophy and the presence of disarray. Heart 2008, 94, 1257. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.C.; Michels, V.V.; Pellikka, P.A.; Ballew, J.D.; Karst, M.L.; Herron, K.J.; Nelson, S.M.; Rodeheffer, R.J.; Olson, T.M. Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin. Genet. 2008, 74, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezekian, J.E.; Clippinger, S.R.; Garcia, J.M.; Yang, Q.; Denfield, S.; Jeewa, A.; Dreyer, W.J.; Zou, W.; Fan, Y.; Allen, H.D.; et al. Variant R94C in TNNT2-encoded troponin t predisposes to pediatric restrictive cardiomyopathy and sudden death through impaired thin filament relaxation resulting in myocardial diastolic dysfunction. J. Am. Heart Assoc. 2020, 9, e015111. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, H.; Inazawa, J.; Ariyama, T.; Nishino, H.; Ochiai, Y.; Ohkubo, I.; Miwa, T. Reexamination of chromosomal loci of human muscle actin genes by fluorescence in situ hybridization. Jpn. J. Hum. Genet. 1995, 40, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Gunning, P.; Ponte, P.; Kedes, L.; Eddy, R.; Shows, T. Chromosomal location of the co-expressed human skeletal and cardiac actin genes. Proc. Natl. Acad. Sci. USA 1984, 81, 1813–1817. [Google Scholar] [CrossRef] [Green Version]
- Squire, J. Special issue: The actin-myosin interaction in muscle: Background and overview. Int. J. Mol. Sci. 2019, 20, 5715. [Google Scholar] [CrossRef] [Green Version]
- Olson, T.M.; Michels, V.V.; Thibodeau, S.N.; Tai, Y.S.; Keating, M.T. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 1998, 280, 750–752. [Google Scholar] [CrossRef] [Green Version]
- Olson, T.M.; Doan, T.P.; Kishimoto, N.Y.; Whitby, F.G.; Ackerman, M.J.; Fananapazir, L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2000, 32, 1687–1694. [Google Scholar] [CrossRef]
- Klaassen, S.; Probst, S.; Oechslin, E.; Gerull, B.; Krings, G.; Schuler, P.; Greutmann, M.; Hurlimann, D.; Yegitbasi, M.; Pons, L.; et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 2008, 117, 2893–2901. [Google Scholar] [CrossRef] [PubMed]
- Greenway, S.C.; McLeod, R.; Hume, S.; Roslin, N.M.; Alvarez, N.; Giuffre, M.; Zhan, S.H.; Shen, Y.; Preuss, C.; Andelfinger, G.; et al. Exome sequencing identifies a novel variant in ACTC1 associated with familial atrial septal defect. Can. J. Cardiol. 2014, 30, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, F.; Lyon, R.C.; Chen, J. Getting the skinny on thick filament regulation in cardiac muscle biology and disease. Trends Cardiovasc. Med. 2014, 24, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warrick, H.M.; Spudich, J.A. Myosin structure and function in cell motility. Annu. Rev. Cell Biol. 1987, 3, 379–421. [Google Scholar] [CrossRef]
- Alamo, L.; Ware, J.S.; Pinto, A.; Gillilan, R.E.; Seidman, J.G.; Seidman, C.E.; Padron, R. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. eLife 2017, 6, e24634. [Google Scholar] [CrossRef]
- Colegrave, M.; Peckham, M. Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat. Rec. 2014, 297, 1670–1680. [Google Scholar] [CrossRef]
- Vale, R.D.; Milligan, R.A. The way things move: Looking under the hood of molecular motor proteins. Science 2000, 288, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Trybus, K.M. Role of myosin light chains. J. Muscle Res. Cell. Motil. 1994, 15, 587–594. [Google Scholar] [CrossRef]
- Wolny, M.; Colegrave, M.; Colman, L.; White, E.; Knight, P.J.; Peckham, M. Cardiomyopathy mutations in the tail of beta-cardiac myosin modify the coiled-coil structure and affect integration into thick filaments in muscle sarcomeres in adult cardiomyocytes. J. Biol. Chem. 2013, 288, 31952–31962. [Google Scholar] [CrossRef] [Green Version]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Moller, D.V.; Andersen, P.S.; Hedley, P.; Ersboll, M.K.; Bundgaard, H.; Moolman-Smook, J.; Christiansen, M.; Kober, L. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur. J. Hum. Genet. 2009, 17, 1241–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferradini, V.; Parca, L.; Martino, A.; Lanzillo, C.; Silvetti, E.; Calo, L.; Caselli, S.; Novelli, G.; Helmer-Citterich, M.; Sangiuolo, F.C.; et al. Variants in MHY7 gene cause arrhythmogenic cardiomyopathy. Genes 2021, 12, 793. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Karst, M.L.; Whitby, F.G.; Driscoll, D.J. Myosin light chain mutation causes autosomal recessive cardiomyopathy with mid-cavitary hypertrophy and restrictive physiology. Circulation 2002, 105, 2337–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, C.C.; Kazmierczak, K.; Liang, J.; Kanashiro-Takeuchi, R.; Irving, T.C.; Gomes, A.V.; Wang, Y.; Burghardt, T.P.; Szczesna-Cordary, D. Hypercontractile mutant of ventricular myosin essential light chain leads to disruption of sarcomeric structure and function and results in restrictive cardiomyopathy in mice. Cardiovasc. Res. 2017, 113, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Poetter, K.; Jiang, H.; Hassanzadeh, S.; Master, S.R.; Chang, A.; Dalakas, M.C.; Rayment, I.; Sellers, J.R.; Fananapazir, L.; Epstein, N.D. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat. Genet. 1996, 13, 63–69. [Google Scholar] [CrossRef]
- Flavigny, J.; Richard, P.; Isnard, R.; Carrier, L.; Charron, P.; Bonne, G.; Forissier, J.F.; Desnos, M.; Dubourg, O.; Komajda, M.; et al. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J. Mol. Med. 1998, 76, 208–214. [Google Scholar] [CrossRef]
- Osborn, D.P.S.; Emrahi, L.; Clayton, J.; Tabrizi, M.T.; Wan, A.Y.B.; Maroofian, R.; Yazdchi, M.; Garcia, M.L.E.; Galehdari, H.; Hesse, C.; et al. Autosomal recessive cardiomyopathy and sudden cardiac death associated with variants in MYL3. Genet. Med. 2021, 23, 787–792. [Google Scholar] [CrossRef]
- Greenway, S.C.; Wilson, G.J.; Wilson, J.; George, K.; Kantor, P.F. Sudden death in an infant with angina, restrictive cardiomyopathy, and coronary artery bridging: An unusual phenotype for a beta-myosin heavy chain (MYH7) sarcomeric protein mutation. Circ. Heart Fail. 2012, 5, e92–e93. [Google Scholar] [CrossRef] [Green Version]
- Neagoe, O.; Ciobanu, A.; Diaconu, R.; Mirea, O.; Donoiu, I.; Militaru, C. A rare case of familial restrictive cardiomyopathy, with mutations in MYH7 and ABCC9 genes. Discoveries 2019, 7, e99. [Google Scholar] [CrossRef]
- Ware, S.M.; Quinn, M.E.; Ballard, E.T.; Miller, E.; Uzark, K.; Spicer, R.L. Pediatric restrictive cardiomyopathy associated with a mutation in beta-myosin heavy chain. Clin. Genet. 2008, 73, 165–170. [Google Scholar] [CrossRef]
- Kawano, H.; Kawamura, K.; Kanda, M.; Ishijima, M.; Abe, K.; Hayashi, T.; Matsumoto, Y.; Kimura, A.; Maemura, K. Histopathological changes of myocytes in restrictive cardiomyopathy. Med. Mol. Morphol. 2021, 54, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.L.; Guo, S.; Jin, J.Y.; He, Z.J.; Zhao, S.P.; Xiang, R.; Zhao, W. Whole exome sequencing identified a 13 base pair MYH7 deletion-mutation in a patient with restrictive cardiomyopathy and left ventricle hypertrophy. Ann. Clin. Lab. Sci. 2019, 49, 838–840. [Google Scholar] [PubMed]
- Gerull, B. The rapidly evolving role of titin in cardiac physiology and cardiomyopathy. Can. J. Cardiol. 2015, 31, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitas, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Ware, J.S.; Cook, S.A. Role of titin in cardiomyopathy: From DNA variants to patient stratification. Nat. Rev. Cardiol. 2018, 15, 241–252. [Google Scholar] [CrossRef]
- McAfee, Q.; Chen, C.Y.; Yang, Y.; Caporizzo, M.A.; Morley, M.; Babu, A.; Jeong, S.; Brandimarto, J.; Bedi, K.C., Jr.; Flam, E.; et al. Truncated titin proteins in dilated cardiomyopathy. Sci. Transl. Med. 2021, 13, eabd7287. [Google Scholar] [CrossRef]
- Akinrinade, O.; Helio, T.; Lekanne Deprez, R.H.; Jongbloed, J.D.H.; Boven, L.G.; van den Berg, M.P.; Pinto, Y.M.; Alastalo, T.P.; Myllykangas, S.; Spaendonck-Zwarts, K.V.; et al. Relevance of titin missense and non-frameshifting insertions/deletions variants in dilated cardiomyopathy. Sci. Rep. 2019, 9, 4093. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Dayal, A.A.; Medvedeva, N.V.; Nekrasova, T.M.; Duhalin, S.D.; Surin, A.K.; Minin, A.A. Desmin interacts directly with mitochondria. Int. J. Mol. Sci. 2020, 21, 8122. [Google Scholar] [CrossRef]
- Patel, D.M.; Green, K.J. Desmosomes in the heart: A review of clinical and mechanistic analyses. Cell Commun. Adhes. 2014, 21, 109–128. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.; Cowin, P. Deconstructing desmoplakin. Nat. Cell Biol. 2001, 3, E270–E272. [Google Scholar] [CrossRef] [PubMed]
- Gorza, L.; Sorge, M.; Secli, L.; Brancaccio, M. Master Regulators of muscle atrophy: Role of costamere components. Cells 2021, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Wiche, G. Plectin-mediated intermediate filament functions: Why isoforms matter. Cells 2021, 10, 2154. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Colucci-Guyon, E.; Pincon-Raymond, M.; Mericskay, M.; Pournin, S.; Paulin, D.; Babinet, C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev. Biol. 1996, 175, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Capetanaki, Y.; Milner, D.J.; Weitzer, G. Desmin in muscle formation and maintenance: Knockouts and consequences. Cell Struct. Funct. 1997, 22, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, I.; Dieding, M.; Klauke, B.; Brodehl, A.; Gaertner-Rommel, A.; Walhorn, V.; Gummert, J.; Schulz, U.; Paluszkiewicz, L.; Anselmetti, D.; et al. A novel desmin (DES) indel mutation causes severe atypical cardiomyopathy in combination with atrioventricular block and skeletal myopathy. Mol. Genet. Genom. Med. 2018, 6, 288–293. [Google Scholar] [CrossRef] [Green Version]
- Marakhonov, A.V.; Brodehl, A.; Myasnikov, R.P.; Sparber, P.A.; Kiseleva, A.V.; Kulikova, O.V.; Meshkov, A.N.; Zharikova, A.A.; Koretsky, S.N.; Kharlap, M.S.; et al. Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum. Mutat. 2019, 40, 734–741. [Google Scholar] [CrossRef]
- Kubanek, M.; Schimerova, T.; Piherova, L.; Brodehl, A.; Krebsova, A.; Ratnavadivel, S.; Stanasiuk, C.; Hansikova, H.; Zeman, J.; Palecek, T.; et al. Desminopathy: Novel desmin variants, a new cardiac phenotype, and further evidence for secondary mitochondrial dysfunction. J. Clin. Med. 2020, 9, 937. [Google Scholar] [CrossRef] [Green Version]
- Protonotarios, A.; Brodehl, A.; Asimaki, A.; Jager, J.; Quinn, E.; Stanasiuk, C.; Ratnavadivel, S.; Futema, M.; Akhtar, M.M.; Gossios, T.D.; et al. The novel desmin variant p.Leu115Ile is associated with a unique form of biventricular arrhythmogenic cardiomyopathy. Can. J. Cardiol. 2021, 37, 857–866. [Google Scholar] [CrossRef]
- Fischer, B.; Dittmann, S.; Brodehl, A.; Unger, A.; Stallmeyer, B.; Paul, M.; Seebohm, G.; Kayser, A.; Peischard, S.; Linke, W.A.; et al. Functional characterization of novel alpha-helical rod domain desmin (DES) pathogenic variants associated with dilated cardiomyopathy, atrioventricular block and a risk for sudden cardiac death. Int. J. Cardiol. 2021, 329, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Clemen, C.S.; Fischer, D.; Reimann, J.; Eichinger, L.; Muller, C.R.; Muller, H.D.; Goebel, H.H.; Schroder, R. How much mutant protein is needed to cause a protein aggregate myopathy in vivo? Lessons from an exceptional desminopathy. Hum. Mutat. 2009, 30, E490–E499. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Hain, C.; Flottmann, F.; Ratnavadivel, S.; Gaertner, A.; Klauke, B.; Kalinowski, J.; Körperich, H.; Gummert, J.; Paluszkiewicz, L.; et al. The desmin mutation DES-c.735G>C causes severe restrictive cardiomyopathy by inducing in-frame skipping of exon-3. Biomedicines 2021, 9, 1400. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, R.; Wang, Y.; Cao, L.; Lin, C.; Liu, F.; Hu, R.; Nan, J.; Zhuang, X.; Lu, X.; et al. Features of myocardial injury detected by cardiac magnetic resonance in a patient with desmin-related restrictive cardiomyopathy. ESC Heart Fail. 2021, 8, 5560–5564. [Google Scholar] [CrossRef]
- Herrmann, H.; Cabet, E.; Chevalier, N.R.; Moosmann, J.; Schultheis, D.; Haas, J.; Schowalter, M.; Berwanger, C.; Weyerer, V.; Agaimy, A.; et al. Dual Functional states of R406W-desmin assembly complexes cause cardiomyopathy with severe intercalated disc derangement in humans and in knock-in mice. Circulation 2020, 142, 2155–2171. [Google Scholar] [CrossRef]
- Ojrzynska, N.; Bilinska, Z.T.; Franaszczyk, M.; Ploski, R.; Grzybowski, J. Restrictive cardiomyopathy due to novel desmin gene mutation. Kardiol. Pol. 2017, 75, 723. [Google Scholar] [CrossRef] [Green Version]
- Jurcu, T.R.; Bastian, A.E.; Militaru, S.; Popa, A.; Manole, E.; Popescu, B.A.; Tallila, J.; Popescu, B.O.; Ginghina, C.D. Discovery of a new mutation in the desmin gene in a young patient with cardiomyopathy and muscular weakness. Rom. J. Morphol. Embryol. 2017, 58, 225–230. [Google Scholar]
- Sharma, S.; Juneja, R.; Sharma, G.; Arava, S.; Ray, R. Desmin-related restrictive cardiomyopathy in a pediatric patient: A case report. Indian J. Pathol. Microbiol. 2013, 56, 402–404. [Google Scholar]
- Pinol-Ripoll, G.; Shatunov, A.; Cabello, A.; Larrode, P.; de la Puerta, I.; Pelegrin, J.; Ramos, F.J.; Olive, M.; Goldfarb, L.G. Severe infantile-onset cardiomyopathy associated with a homozygous deletion in desmin. Neuromuscul. Disord. 2009, 19, 418–422. [Google Scholar] [CrossRef] [Green Version]
- Bar, H.; Mucke, N.; Kostareva, A.; Sjoberg, G.; Aebi, U.; Herrmann, H. Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc. Natl. Acad. Sci. USA 2005, 102, 15099–15104. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Hedde, P.N.; Dieding, M.; Fatima, A.; Walhorn, V.; Gayda, S.; Saric, T.; Klauke, B.; Gummert, J.; Anselmetti, D.; et al. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J. Biol. Chem. 2012, 287, 16047–16057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, H.; Aebi, U. Intermediate filaments: Structure and assembly. Cold Spring Harb. Perspect. Biol. 2016, 8, a018242. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, R.A.; Hatzfeld, M.; Franke, W.W.; Lustig, A.; Schulthess, T.; Engel, J. Characterization of dimer subunits of intermediate filament proteins. J. Mol. Biol. 1986, 192, 337–349. [Google Scholar] [CrossRef]
- Parry, D.A.; Strelkov, S.V.; Burkhard, P.; Aebi, U.; Herrmann, H. Towards a molecular description of intermediate filament structure and assembly. Exp. Cell Res. 2007, 313, 2204–2216. [Google Scholar] [CrossRef]
- Herrmann, H.; Haner, M.; Brettel, M.; Ku, N.O.; Aebi, U. Characterization of distinct early assembly units of different intermediate filament proteins. J. Mol. Biol. 1999, 286, 1403–1420. [Google Scholar] [CrossRef]
- Herrmann, H.; Aebi, U. Intermediate filaments: Molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu. Rev. Biochem. 2004, 73, 749–789. [Google Scholar] [CrossRef]
- Colakoglu, G.; Brown, A. Intermediate filaments exchange subunits along their length and elongate by end-to-end annealing. J. Cell Biol. 2009, 185, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Winheim, S.; Hieb, A.R.; Silbermann, M.; Surmann, E.M.; Wedig, T.; Herrmann, H.; Langowski, J.; Mucke, N. Deconstructing the late phase of vimentin assembly by total internal reflection fluorescence microscopy (TIRFM). PLoS ONE 2011, 6, e19202. [Google Scholar] [CrossRef] [Green Version]
- Noding, B.; Herrmann, H.; Koster, S. Direct observation of subunit exchange along mature vimentin intermediate filaments. Biophys. J. 2014, 107, 2923–2931. [Google Scholar] [CrossRef] [Green Version]
- Park, K.Y.; Dalakas, M.C.; Goebel, H.H.; Ferrans, V.J.; Semino-Mora, C.; Litvak, S.; Takeda, K.; Goldfarb, L.G. Desmin splice variants causing cardiac and skeletal myopathy. J. Med. Genet. 2000, 37, 851–857. [Google Scholar] [CrossRef]
- Arbustini, E.; Pasotti, M.; Pilotto, A.; Pellegrini, C.; Grasso, M.; Previtali, S.; Repetto, A.; Bellini, O.; Azan, G.; Scaffino, M.; et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur. J. Heart Fail. 2006, 8, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Armstrong, J.; Miralles, F.; Pou, A.; Fardeau, M.; Gonzalez, L.; Martinez, F.; Fischer, D.; Martinez Matos, J.A.; Shatunov, A.; et al. Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul. Disord. 2007, 17, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruszczyk, P.; Kostera-Pruszczyk, A.; Shatunov, A.; Goudeau, B.; Draminska, A.; Takeda, K.; Sambuughin, N.; Vicart, P.; Strelkov, S.V.; Goldfarb, L.G.; et al. Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. Int. J. Cardiol. 2007, 117, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Bar, H.; Goudeau, B.; Walde, S.; Casteras-Simon, M.; Mucke, N.; Shatunov, A.; Goldberg, Y.P.; Clarke, C.; Holton, J.L.; Eymard, B.; et al. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum. Mutat. 2007, 28, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Bang, M.L.; Mudry, R.E.; McElhinny, A.S.; Trombitas, K.; Geach, A.J.; Yamasaki, R.; Sorimachi, H.; Granzier, H.; Gregorio, C.C.; Labeit, S. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J. Cell Biol. 2001, 153, 413–427. [Google Scholar] [CrossRef] [Green Version]
- Filomena, M.C.; Yamamoto, D.L.; Carullo, P.; Medvedev, R.; Ghisleni, A.; Piroddi, N.; Scellini, B.; Crispino, R.; D’Autilia, F.; Zhang, J.; et al. Myopalladin knockout mice develop cardiac dilation and show a maladaptive response to mechanical pressure overload. eLife 2021, 10, e58313. [Google Scholar] [CrossRef]
- Duboscq-Bidot, L.; Xu, P.; Charron, P.; Neyroud, N.; Dilanian, G.; Millaire, A.; Bors, V.; Komajda, M.; Villard, E. Mutations in the Z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc. Res. 2008, 77, 118–125. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Yeates, L.; Semsarian, C. Analysis of the Z-disc genes PDLIM3 and MYPN in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2010, 145, 601–602. [Google Scholar] [CrossRef]
- Miyatake, S.; Mitsuhashi, S.; Hayashi, Y.K.; Purevjav, E.; Nishikawa, A.; Koshimizu, E.; Suzuki, M.; Yatabe, K.; Tanaka, Y.; Ogata, K.; et al. Biallelic mutations in MYPN, encoding myopalladin, are associated with childhood-onset, slowly progressive nemaline myopathy. Am. J. Hum. Genet. 2017, 100, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Beggs, A.H.; Byers, T.J.; Knoll, J.H.; Boyce, F.M.; Bruns, G.A.; Kunkel, L.M. Cloning and characterization of two human skeletal muscle alpha-actinin genes located on chromosomes 1 and 11. J. Biol. Chem. 1992, 267, 9281–9288. [Google Scholar] [CrossRef]
- Tiso, N.; Majetti, M.; Stanchi, F.; Rampazzo, A.; Zimbello, R.; Nava, A.; Danieli, G.A. Fine mapping and genomic structure of ACTN2, the human gene coding for the sarcomeric isoform of α-actinin-2, expressed in skeletal and cardiac muscle. Biochem. Biophys. Res. Commun. 1999, 265, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Wadmore, K.; Azad, A.J.; Gehmlich, K. The role of Z-disc proteins in myopathy and cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 3058. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Delalande, O.; Hubert, J.F.; Le Rumeur, E. The spectrin family of proteins: A unique coiled-coil fold for various molecular surface properties. J. Struct. Biol. 2014, 186, 392–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davison, M.D.; Critchley, D.R. alpha-Actinins and the DMD protein contain spectrin-like repeats. Cell 1988, 52, 159–160. [Google Scholar] [CrossRef]
- Djinovic-Carugo, K.; Gautel, M.; Ylanne, J.; Young, P. The spectrin repeat: A structural platform for cytoskeletal protein assemblies. FEBS Lett. 2002, 513, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro Ede, A., Jr.; Pinotsis, N.; Ghisleni, A.; Salmazo, A.; Konarev, P.V.; Kostan, J.; Sjoblom, B.; Schreiner, C.; Polyansky, A.A.; Gkougkoulia, E.A.; et al. The structure and regulation of human muscle α-actinin. Cell 2014, 159, 1447–1460. [Google Scholar] [CrossRef] [Green Version]
- Mohapatra, B.; Jimenez, S.; Lin, J.H.; Bowles, K.R.; Coveler, K.J.; Marx, J.G.; Chrisco, M.A.; Murphy, R.T.; Lurie, P.R.; Schwartz, R.J.; et al. Mutations in the muscle LIM protein and α-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol. Genet. Metab. 2003, 80, 207–215. [Google Scholar] [CrossRef]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horvath, A.; Di Mauro, V.; Koivumaki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Kramer, E.; et al. Disease modeling of a mutation in α-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, 11, e11115. [Google Scholar] [CrossRef]
- Park, J.; Cho, Y.G.; Park, H.W.; Cho, J.S. Case report: Novel likely pathogenic ACTN2 variant causing heterogeneous phenotype in a korean family with left ventricular non-compaction. Front. Pediatr. 2021, 9, 609389. [Google Scholar] [CrossRef]
- Good, J.M.; Fellmann, F.; Bhuiyan, Z.A.; Rotman, S.; Pruvot, E.; Schlapfer, J. ACTN2 variant associated with a cardiac phenotype suggestive of left-dominant arrhythmogenic cardiomyopathy. HeartRhythm Case Rep. 2020, 6, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Noguchi, S.; Sonehara, K.; Nakamura-Shindo, K.; Taniguchi, A.; Kajikawa, H.; Nakamura, H.; Ishikawa, K.; Ogawa, M.; Hayashi, S.; et al. A recurrent homozygous ACTN2 variant associated with core myopathy. Acta Neuropathol. 2021, 142, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Vorgerd, M.; van der Ven, P.F.; Bruchertseifer, V.; Lowe, T.; Kley, R.A.; Schroder, R.; Lochmuller, H.; Himmel, M.; Koehler, K.; Furst, D.O.; et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am. J. Hum. Genet. 2005, 77, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Duff, R.M.; Tay, V.; Hackman, P.; Ravenscroft, G.; McLean, C.; Kennedy, P.; Steinbach, A.; Schoffler, W.; van der Ven, P.F.M.; Furst, D.O.; et al. Mutations in the N-terminal actin-binding domain of filamin C cause a distal myopathy. Am. J. Hum. Genet. 2011, 88, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Chakarova, C.; Wehnert, M.S.; Uhl, K.; Sakthivel, S.; Vosberg, H.P.; van der Ven, P.F.; Furst, D.O. Genomic structure and fine mapping of the two human filamin gene paralogues FLNB and FLNC and comparative analysis of the filamin gene family. Hum. Genet. 2000, 107, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Nakamura, F. Structure and function of filamin c in the muscle Z-disc. Int. J. Mol. Sci. 2020, 21, 2696. [Google Scholar] [CrossRef] [Green Version]
- Pudas, R.; Kiema, T.R.; Butler, P.J.; Stewart, M.; Ylanne, J. Structural basis for vertebrate filamin dimerization. Structure 2005, 13, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Ven, P.F.; Obermann, W.M.; Lemke, B.; Gautel, M.; Weber, K.; Furst, D.O. Characterization of muscle filamin isoforms suggests a possible role of gamma-filamin/ABP-L in sarcomeric Z-disc formation. Cell Motil. Cytoskelet. 2000, 45, 149–162. [Google Scholar] [CrossRef]
- Labeit, S.; Lahmers, S.; Burkart, C.; Fong, C.; McNabb, M.; Witt, S.; Witt, C.; Labeit, D.; Granzier, H. Expression of distinct classes of titin isoforms in striated and smooth muscles by alternative splicing, and their conserved interaction with filamins. J. Mol. Biol. 2006, 362, 664–681. [Google Scholar] [CrossRef]
- Gonzalez-Morales, N.; Holenka, T.K.; Schock, F. Filamin actin-binding and titin-binding fulfill distinct functions in Z-disc cohesion. PLoS Genet. 2017, 13, e1006880. [Google Scholar] [CrossRef]
- Gontier, Y.; Taivainen, A.; Fontao, L.; Sonnenberg, A.; van der Flier, A.; Carpen, O.; Faulkner, G.; Borradori, L. The Z-disc proteins myotilin and FATZ-1 interact with each other and are connected to the sarcolemma via muscle-specific filamins. J. Cell Sci. 2005, 118 Pt 16, 3739–3749. [Google Scholar] [CrossRef] [Green Version]
- Thompson, T.G.; Chan, Y.M.; Hack, A.A.; Brosius, M.; Rajala, M.; Lidov, H.G.; McNally, E.M.; Watkins, S.; Kunkel, L.M. Filamin 2 (FLN2): A muscle-specific sarcoglycan interacting protein. J. Cell Biol. 2000, 148, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Mas, R.; Gutierrez-Fernandez, A.; Gomez, J.; Coto, E.; Astudillo, A.; Puente, D.A.; Reguero, J.R.; Alvarez, V.; Moris, C.; Leon, D.; et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat. Commun. 2014, 5, 5326. [Google Scholar] [CrossRef] [PubMed]
- Begay, R.L.; Tharp, C.A.; Martin, A.; Graw, S.L.; Sinagra, G.; Miani, D.; Sweet, M.E.; Slavov, D.B.; Stafford, N.; Zeller, M.J.; et al. FLNC gene splice mutations cause dilated cardiomyopathy. JACC Basic Transl. Sci. 2016, 1, 344–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augusto, J.B.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; Treibel, T.A.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: A comprehensive genotype-imaging phenotype study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef]
- Van Waning, J.I.; Hoedemaekers, Y.M.; te Rijdt, W.P.; Jpma, A.I.; Heijsman, D.; Caliskan, K.; Hoendermis, E.S.; Willems, T.P.; van den Wijngaard, A.; Suurmeijer, A. FLNC missense variants in familial noncompaction cardiomyopathy. Cardiogenetics 2019, 9, 9–13. [Google Scholar] [CrossRef] [Green Version]
- Reinstein, E.; Gutierrez-Fernandez, A.; Tzur, S.; Bormans, C.; Marcu, S.; Tayeb-Fligelman, E.; Vinkler, C.; Raas-Rothschild, A.; Irge, D.; Landau, M.; et al. Congenital dilated cardiomyopathy caused by biallelic mutations in Filamin C. Eur. J. Hum. Genet. 2016, 24, 1792–1796. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wu, J.; Han, C.; Li, Y.; Guo, Y.; Tong, X. A mutation in the filamin c gene causes myofibrillar myopathy with lower motor neuron syndrome: A case report. BMC Neurol. 2019, 19, 198. [Google Scholar] [CrossRef] [Green Version]
- Dalkilic, I.; Schienda, J.; Thompson, T.G.; Kunkel, L.M. Loss of FilaminC (FLNc) results in severe defects in myogenesis and myotube structure. Mol. Cell. Biol. 2006, 26, 6522–6534. [Google Scholar] [CrossRef] [Green Version]
- Ruparelia, A.A.; Zhao, M.; Currie, P.D.; Bryson-Richardson, R.J. Characterization and investigation of zebrafish models of filamin-related myofibrillar myopathy. Hum. Mol. Genet. 2012, 21, 4073–4083. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Mitsuhashi, H.; Isogai, S.; Nakata, T.; Kawakami, A.; Nonaka, I.; Noguchi, S.; Hayashi, Y.K.; Nishino, I.; Kudo, A. Filamin C plays an essential role in the maintenance of the structural integrity of cardiac and skeletal muscles, revealed by the medaka mutant zacro. Dev. Biol. 2012, 361, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Deo, R.C.; Musso, G.; Tasan, M.; Tang, P.; Poon, A.; Yuan, C.; Felix, J.F.; Vasan, R.S.; Beroukhim, R.; De Marco, T.; et al. Prioritizing causal disease genes using unbiased genomic features. Genome Biol. 2014, 15, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevessier, F.; Schuld, J.; Orfanos, Z.; Plank, A.C.; Wolf, L.; Maerkens, A.; Unger, A.; Schlotzer-Schrehardt, U.; Kley, R.A.; Von Horsten, S.; et al. Myofibrillar instability exacerbated by acute exercise in filaminopathy. Hum. Mol. Genet. 2015, 24, 7207–7220. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Khudiakov, A.; Tarnovskaya, S.; Liu, J.; Sergushichev, A.; Kazakov, S.; Frishman, D.; Smolina, N.; et al. De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum. Mutat. 2018, 39, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, A.A.; Oorschot, V.; Ramm, G.; Bryson-Richardson, R.J. FLNC myofibrillar myopathy results from impaired autophagy and protein insufficiency. Hum. Mol. Genet. 2016, 25, 2131–2142. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Z.; Zhang, L.; Zhu, M.; Tan, C.; Zhou, X.; Evans, S.M.; Fang, X.; Feng, W.; Chen, J. Loss of Filamin C is catastrophic for heart function. Circulation 2020, 141, 869–871. [Google Scholar] [CrossRef]
- Rodina, N.; Khudiakov, A.; Perepelina, K.; Muravyev, A.; Boytsov, A.; Zlotina, A.; Sokolnikova, P.; Kostareva, A. Generation of iPSC line (FAMRCi009-A) from patient with familial progressive cardiac conduction disorder carrying genetic variant FLNC p.Val2264Met. Stem Cell Res. 2021, 59, 102640. [Google Scholar] [CrossRef]
- Perepelina, K.; Khudiakov, A.; Rodina, N.; Boytsov, A.; Vavilova, T.; Zlotina, A.; Sokolnikova, P.; Kostareva, A. Generation of iPSC line FAMRCi010-A from patient with restrictive cardiomyopathy carrying genetic variant FLNC p.Gly2011Arg. Stem Cell Res. 2021, 59, 102639. [Google Scholar] [CrossRef]
- Tucker, N.R.; McLellan, M.A.; Hu, D.; Ye, J.; Parsons, V.A.; Mills, R.W.; Clauss, S.; Dolmatova, E.; Shea, M.A.; Milan, D.J.; et al. Novel mutation in FLNC (Filamin C) causes familial restrictive cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001780. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Wei, Q.; Wu, B.; Liu, X.; Mading, A.; Yang, L.; Li, Y.; Liu, F.; Pan, X.; Wang, H. Clinical exome sequencing revealed that FLNC variants contribute to the early diagnosis of cardiomyopathies in infant patients. Transl. Pediatr. 2020, 9, 21–33. [Google Scholar] [CrossRef]
- Roldan-Sevilla, A.; Palomino-Doza, J.; de Juan, J.; Sanchez, V.; Dominguez-Gonzalez, C.; Salguero-Bodes, R.; Arribas-Ynsaurriaga, F. Missense mutations in the FLNC gene causing familial restrictive cardiomyopathy. Circ. Genom. Precis. Med. 2019, 12, e002388. [Google Scholar] [CrossRef]
- Schubert, J.; Tariq, M.; Geddes, G.; Kindel, S.; Miller, E.M.; Ware, S.M. Novel pathogenic variants in Filamin C identified in pediatric restrictive cardiomyopathy. Hum. Mutat. 2018, 39, 2083–2096. [Google Scholar] [CrossRef] [PubMed]
- Aebi, U.; Cohn, J.; Buhle, L.; Gerace, L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature 1986, 323, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Dobrzynska, A.; Gonzalo, S.; Shanahan, C.; Askjaer, P. The nuclear lamina in health and disease. Nucleus 2016, 7, 233–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [Green Version]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Shan, H.; Huang, J.; Li, N.; Hou, C.; Pu, J. A novel lamin A/C gene missense mutation (445 V > E) in immunoglobulin-like fold associated with left ventricular non-compaction. Europace 2016, 18, 617–622. [Google Scholar] [CrossRef]
- Raffaele Di Barletta, M.; Ricci, E.; Galluzzi, G.; Tonali, P.; Mora, M.; Morandi, L.; Romorini, A.; Voit, T.; Orstavik, K.H.; Merlini, L.; et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2000, 66, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Shackleton, S.; Lloyd, D.J.; Jackson, S.N.; Evans, R.; Niermeijer, M.F.; Singh, B.M.; Schmidt, H.; Brabant, G.; Kumar, S.; Durrington, P.N.; et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat. Genet. 2000, 24, 153–156. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Hirata, T.; Fujita, M.; Nakamura, S.; Gotoh, K.; Motooka, D.; Murakami, Y.; Maeda, Y.; Kinoshita, T. Post-Golgi anterograde transport requires GARP-dependent endosome-to-TGN retrograde transport. Mol. Biol. Cell 2015, 26, 3071–3084. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.W.; Raeker, M.O.; Geisler, S.B.; Thomas, P.E.; Simmons, T.A.; Bernat, J.A.; Thorsson, T.; Innis, J.W. Functional analysis of candidate genes in 2q13 deletion syndrome implicates FBLN7 and TMEM87B deficiency in congenital heart defects and FBLN7 in craniofacial malformations. Hum. Mol. Genet. 2014, 23, 4272–4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido, C.; Paul, C.; Seigneuric, R.; Kampinga, H.H. The small heat shock proteins family: The long forgotten chaperones. Int. J. Biochem. Cell Biol. 2012, 44, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Dubin, R.A.; Ally, A.H.; Chung, S.; Piatigorsky, J. Human αB-crystallin gene and preferential promoter function in lens. Genomics 1990, 7, 594–601. [Google Scholar] [CrossRef]
- Dimauro, I.; Antonioni, A.; Mercatelli, N.; Caporossi, D. The role of αB-crystallin in skeletal and cardiac muscle tissues. Cell Stress Chaperones 2018, 23, 491–505. [Google Scholar] [CrossRef]
- Chepelinsky, A.B.; Piatigorsky, J.; Pisano, M.M.; Dubin, R.A.; Wistow, G.; Limjoco, T.I.; Klement, J.F.; Jaworski, C.J. Lens protein gene expression: Alpha-crystallins and MIP. Lens Eye Toxic Res. 1991, 8, 319–344. [Google Scholar]
- Sacconi, S.; Feasson, L.; Antoine, J.C.; Pecheux, C.; Bernard, R.; Cobo, A.M.; Casarin, A.; Salviati, L.; Desnuelle, C.; Urtizberea, A. A novel CRYAB mutation resulting in multisystemic disease. Neuromuscul. Disord. 2012, 22, 66–72. [Google Scholar] [CrossRef]
- Safieh, L.A.; Khan, A.O.; Alkuraya, F.S. Identification of a novel CRYAB mutation associated with autosomal recessive juvenile cataract in a Saudi family. Mol. Vis. 2009, 15, 980–984. [Google Scholar]
- Inagaki, N.; Hayashi, T.; Arimura, T.; Koga, Y.; Takahashi, M.; Shibata, H.; Teraoka, K.; Chikamori, T.; Yamashina, A.; Kimura, A. αB-crystallin mutation in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 342, 379–386. [Google Scholar] [CrossRef]
- Peschek, J.; Braun, N.; Franzmann, T.M.; Georgalis, Y.; Haslbeck, M.; Weinkauf, S.; Buchner, J. The eye lens chaperone α-crystallin forms defined globular assemblies. Proc. Natl. Acad. Sci. USA 2009, 106, 13272–13277. [Google Scholar] [CrossRef] [Green Version]
- Ganea, E. Chaperone-like activity of alpha-crystallin and other small heat shock proteins. Curr. Protein Pept. Sci. 2001, 2, 205–225. [Google Scholar] [CrossRef] [PubMed]
- Bullard, B.; Ferguson, C.; Minajeva, A.; Leake, M.C.; Gautel, M.; Labeit, D.; Ding, L.; Labeit, S.; Horwitz, J.; Leonard, K.R.; et al. Association of the chaperone αB-crystallin with titin in heart muscle. J. Biol. Chem. 2004, 279, 7917–7924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jehle, S.; Rajagopal, P.; Bardiaux, B.; Markovic, S.; Kuhne, R.; Stout, J.R.; Higman, V.A.; Klevit, R.E.; van Rossum, B.J.; Oschkinat, H. Solid-state NMR and SAXS studies provide a structural basis for the activation of αB-crystallin oligomers. Nat. Struct. Mol. Biol. 2010, 17, 1037–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasekaran, N.S.; Connell, P.; Christians, E.S.; Yan, L.J.; Taylor, R.P.; Orosz, A.; Zhang, X.Q.; Stevenson, T.J.; Peshock, R.M.; Leopold, J.A.; et al. Human αB-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 2007, 130, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturner, E.; Behl, C. The role of the multifunctional BAG3 protein in cellular protein quality control and in disease. Front. Mol. Neurosci. 2017, 10, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, S.; Xie, Z.; Reed, J.C. An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J. Biol. Chem. 1999, 274, 781–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Koren, S.A.; Cvetojevic, G.; Girardi, P.; Johnson, G.V.W. The role of BAG3 in health and disease: A “Magic BAG of Tricks”. J. Cell. Biochem. 2022, 123, 4–21. [Google Scholar] [CrossRef]
- Kogel, D.; Linder, B.; Brunschweiger, A.; Chines, S.; Behl, C. At the crossroads of apoptosis and autophagy: Multiple roles of the Co-chaperone bag3 in stress and therapy resistance of cancer. Cells 2020, 9, 574. [Google Scholar] [CrossRef] [Green Version]
- Takayama, S.; Reed, J.C. Molecular chaperone targeting and regulation by BAG family proteins. Nat. Cell Biol. 2001, 3, E237–E241. [Google Scholar] [CrossRef]
- Sondermann, H.; Scheufler, C.; Schneider, C.; Hohfeld, J.; Hartl, F.U.; Moarefi, I. Structure of a Bag/Hsc70 complex: Convergent functional evolution of Hsp70 nucleotide exchange factors. Science 2001, 291, 1553–1557. [Google Scholar] [CrossRef]
- Shemetov, A.A.; Gusev, N.B. Biochemical characterization of small heat shock protein HspB8 (Hsp22)-Bag3 interaction. Arch. Biochem. Biophys. 2011, 513, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Morelli, F.F.; Mediani, L.; Heldens, L.; Bertacchini, J.; Bigi, I.; Carra, A.D.; Vinet, J.; Carra, S. An interaction study in mammalian cells demonstrates weak binding of HSPB2 to BAG3, which is regulated by HSPB3 and abrogated by HSPB8. Cell Stress Chaperones 2017, 22, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hishiya, A.; Salman, M.N.; Carra, S.; Kampinga, H.H.; Takayama, S. BAG3 directly interacts with mutated αB-crystallin to suppress its aggregation and toxicity. PLoS ONE 2011, 6, e16828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, J.N.; Tse, E.; Freilich, R.; Mok, S.A.; Makley, L.N.; Southworth, D.R.; Gestwicki, J.E. BAG3 is a modular, scaffolding protein that physically links heat shock protein 70 (Hsp70) to the small heat shock proteins. J. Mol. Biol. 2017, 429, 128–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, M.; Poirier, D.J.; Seguin, S.J.; Lambert, H.; Carra, S.; Charette, S.J.; Landry, J. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem. J. 2009, 425, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, W.; Sadoshima, J. BAG3 plays a central role in proteostasis in the heart. J. Clin. Investig. 2017, 127, 2900–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Zuchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Semmler, A.L.; Sacconi, S.; Bach, J.E.; Liebe, C.; Burmann, J.; Kley, R.A.; Ferbert, A.; Anderheiden, R.; Van den Bergh, P.; Martin, J.J.; et al. Unusual multisystemic involvement and a novel BAG3 mutation revealed by NGS screening in a large cohort of myofibrillar myopathies. Orphanet J. Rare Dis. 2014, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Ooms, A.; Graf-Riesen, K.; Kuppusamy, M.; Unger, A.; Schuld, J.; Daerr, J.; Lother, A.; Geisen, C.; Hein, L.; et al. Overexpression of human BAG3(P209L) in mice causes restrictive cardiomyopathy. Nat. Commun. 2021, 12, 3575. [Google Scholar] [CrossRef]
- Fang, X.; Bogomolovas, J.; Zhou, P.S.; Mu, Y.; Ma, X.; Chen, Z.; Zhang, L.; Zhu, M.; Veevers, J.; Ouyang, K.; et al. P209L mutation in Bag3 does not cause cardiomyopathy in mice. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H392–H399. [Google Scholar] [CrossRef]
- Kobuke, K.; Furukawa, Y.; Sugai, M.; Tanigaki, K.; Ohashi, N.; Matsumori, A.; Sasayama, S.; Honjo, T.; Tashiro, K. ESDN, a novel neuropilin-like membrane protein cloned from vascular cells with the longest secretory signal sequence among eukaryotes, is up-regulated after vascular injury. J. Biol. Chem. 2001, 276, 34105–34114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmoker, A.M.; Ebert, A.M.; Ballif, B.A. The DCBLD receptor family: Emerging signaling roles in development, homeostasis and disease. Biochem. J. 2019, 476, 931–950. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.M.; Esmailzadeh, L.; Zhang, J.; Guo, X.; Asadi, A.; Krassilnikova, S.; Fassaei, H.R.; Luo, G.; Al-Lamki, R.S.; Takahashi, T.; et al. ESDN is a marker of vascular remodeling and regulator of cell proliferation in graft arteriosclerosis. Am. J. Transplant. 2007, 7, 2098–2105. [Google Scholar] [CrossRef] [Green Version]
- Nie, L.; Guo, X.; Esmailzadeh, L.; Zhang, J.; Asadi, A.; Collinge, M.; Li, X.; Kim, J.D.; Woolls, M.; Jin, S.W.; et al. Transmembrane protein ESDN promotes endothelial VEGF signaling and regulates angiogenesis. J. Clin. Investig. 2013, 123, 5082–5097. [Google Scholar] [CrossRef]
- Xie, P.; Yuan, F.Q.; Huang, M.S.; Zhang, W.; Zhou, H.H.; Li, X.; Liu, Z.Q. DCBLD2 affects the development of colorectal cancer via emt and angiogenesis and modulates 5-FU drug resistance. Front. Cell Dev. Biol. 2021, 9, 669285. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Adams, K.F.; Anand, I.; Arias-Mendoza, A.; Biering-Sorensen, T.; et al. Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N. Engl. J. Med. 2021, 384, 105–116. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Ozcan, A.; Krajeski, R.; Ioannidi, E.; Lee, B.; Gardner, A.; Makarova, K.S.; Koonin, E.V.; Abudayyeh, O.O.; Gootenberg, J.S. Programmable RNA targeting with the single-protein CRISPR effector Cas7-11. Nature 2021, 597, 720–725. [Google Scholar] [CrossRef]
- Jungmann, A.; Leuchs, B.; Rommelaere, J.; Katus, H.A.; Muller, O.J. Protocol for efficient generation and characterization of adeno-associated viral vectors. Hum. Gene Ther. Methods 2017, 28, 235–246. [Google Scholar] [CrossRef]
- Weinmann, J.; Weis, S.; Sippel, J.; Tulalamba, W.; Remes, A.; El Andari, J.; Herrmann, A.K.; Pham, Q.H.; Borowski, C.; Hille, S.; et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020, 11, 5432. [Google Scholar] [CrossRef] [PubMed]
- Fomin, A.; Gartner, A.; Cyganek, L.; Tiburcy, M.; Tuleta, I.; Wellers, L.; Folsche, L.; Hobbach, A.J.; von Frieling-Salewsky, M.; Unger, A.; et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci. Transl. Med. 2021, 13, eabd3079. [Google Scholar] [CrossRef] [PubMed]






| Mutation | Clinical Features | Family History | MAF 1 | Comments | References |
|---|---|---|---|---|---|
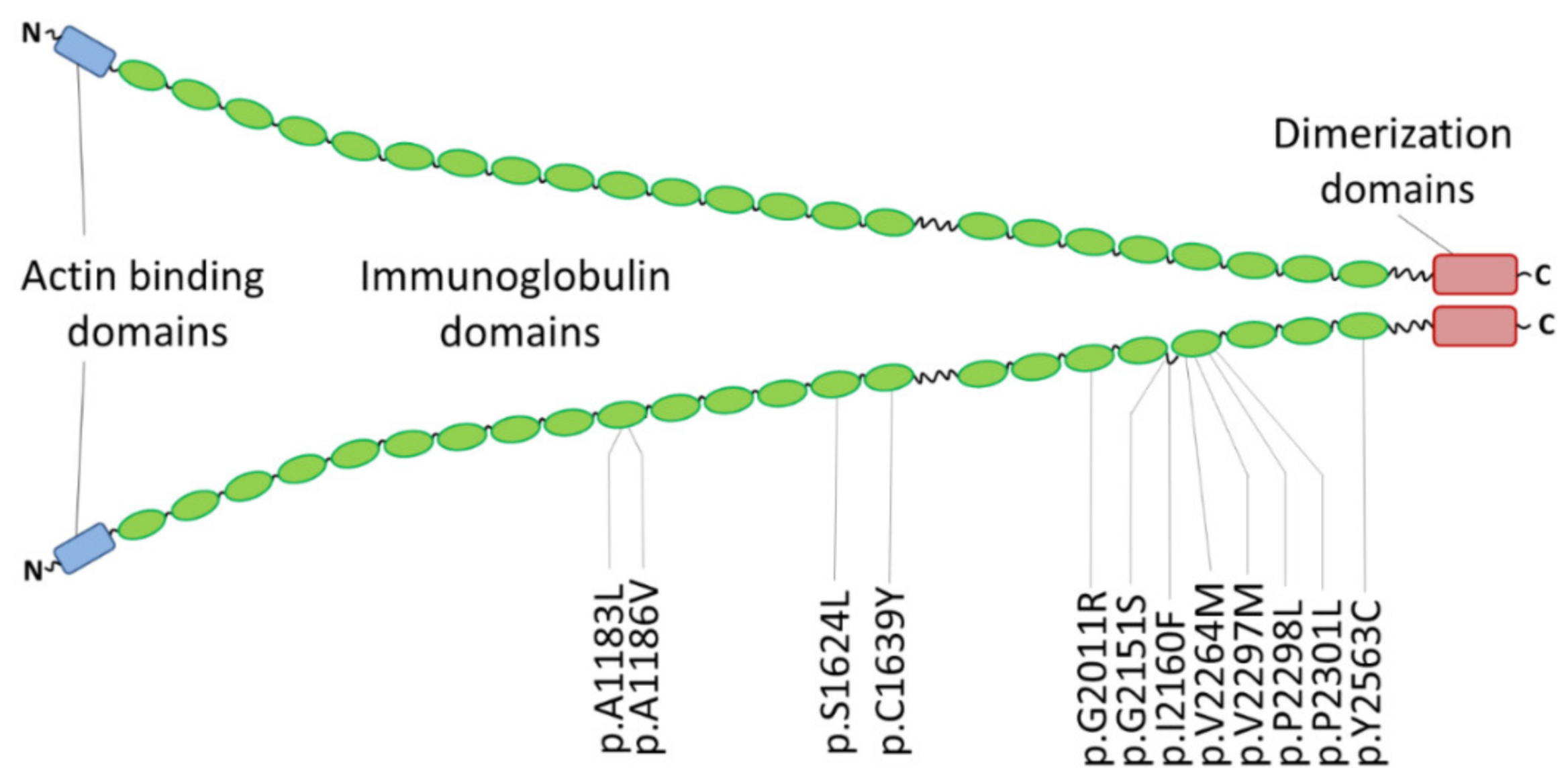
| p.A1183L | RCM and congenital myopathy | one patient | - | [176] | |
| p.A1186V | RCM and congenital myopathy | three unrelated index patients | - | de novo | [176] |
| RCM | one patient | - | de novo, early onset | [182] | |
| p.S1624L | RCM | four affected family members | 0.00003 | [37] | |
| p.C1639Y | RCM | one patient | - | de novo, early onset | [182] |
| p.G2011R | RCM | one patient | - | iPSC model | [180] |
| p.G2151S | RCM | two patients | - | in addition PTPN11-p.Q510R | [183] |
| p.I2160F | RCM | three affected family members | - | [37] | |
| p.V2264M | RCM, SM | one patient | - | iPSC model | [179] |
| p.V2297M | RCM, AF | five affected family members | 0.000004 | [181] | |
| p.P2298L | RCM | eight patients (four genotyped) | - | [184] | |
| p.P2301L | RCM, AF, muscular weakness | one patient | - | de novo | [183] |
| p.Y2563C | RCM | two monozygotic twins | - | de novo | [184] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brodehl, A.; Gerull, B. Genetic Insights into Primary Restrictive Cardiomyopathy. J. Clin. Med. 2022, 11, 2094. https://doi.org/10.3390/jcm11082094
Brodehl A, Gerull B. Genetic Insights into Primary Restrictive Cardiomyopathy. Journal of Clinical Medicine. 2022; 11(8):2094. https://doi.org/10.3390/jcm11082094
Chicago/Turabian StyleBrodehl, Andreas, and Brenda Gerull. 2022. "Genetic Insights into Primary Restrictive Cardiomyopathy" Journal of Clinical Medicine 11, no. 8: 2094. https://doi.org/10.3390/jcm11082094
APA StyleBrodehl, A., & Gerull, B. (2022). Genetic Insights into Primary Restrictive Cardiomyopathy. Journal of Clinical Medicine, 11(8), 2094. https://doi.org/10.3390/jcm11082094