
Treatment of Transthyretin Amyloid Cardiomyopathy: The Current Options, the Future, and the Challenges


Abstract
:1. Introduction
2. Symptomatic Treatments
3. Supportive Therapy with Epigallocatechin-3-Gallate (Green Tea)
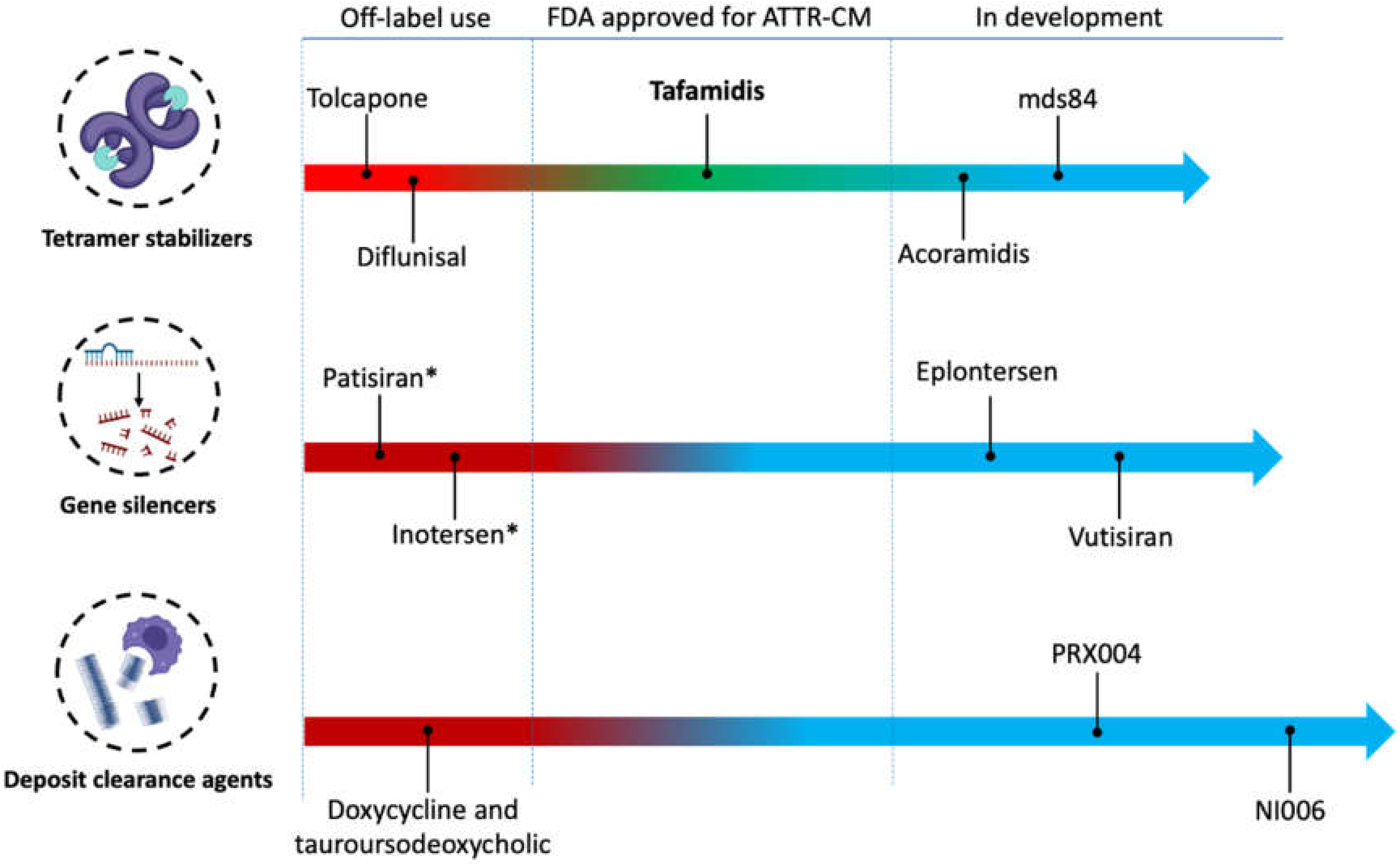
4. Specific Treatments
4.1. Transthyretin Tetramer Stabilization
4.1.1. Tolcapone
4.1.2. Diflunisal
4.1.3. Tafamidis
4.1.4. Acoramidis
4.1.5. Bivalent TTR Stabilizers
4.2. Transthyretin Gene Silencers
4.2.1. Patisiran
4.2.2. Inotersen
4.2.3. Revusiran
4.2.4. Second Generation TTR Gene Silencers
4.3. Gene Editing
4.4. Enhancing Amyloid Clearance
4.4.1. Doxycycline and Tauroursodeoxycholic Acid
4.4.2. Human Monoclonal Antibodies
5. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gillmore, J.D.; Hawkins, P.N. Pathophysiology and treatment of systemic amyloidosis. Nat. Rev. Nephrol. 2013, 9, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, J.; Hanna, M. Cardiac amyloidosis: An update on diagnosis and treatment. Cleve Clin. J. Med. 2017, 84 (Suppl. S3), 12–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kholova, I.; Niessen, H.W. Amyloid in the cardiovascular system: A review. J. Clin. Pathol. 2005, 58, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Muchtar, E.; Buadi, F.K.; Dispenzieri, A.; Gertz, M.A. Immunoglobulin Light-Chain Amyloidosis: From Basics to New Developments in Diagnosis, Prognosis and Therapy. Acta Haematol. 2016, 135, 172–190. [Google Scholar] [CrossRef] [PubMed]
- Liz, M.A.; Mar, F.M.; Franquinho, F.; Sousa, M.M. Aboard transthyretin: From transport to cleavage. IUBMB Life 2010, 62, 429–435. [Google Scholar] [CrossRef]
- Maleszewski, J.J. Cardiac amyloidosis: Pathology, nomenclature, and typing. Cardiovasc. Pathol. 2015, 24, 343–350. [Google Scholar] [CrossRef]
- Kelly, J.W.; Colon, W.; Lai, Z.; Lashuel, H.A.; Mcculloch, J.; Mccutchen, S.L.; Miroy, G.J.; Peterson, S.A. Transthyretin quaternary and tertiary structural changes facilitate misassembly into amyloid. Adv. Protein Chem. 1997, 50, 161–181. [Google Scholar]
- Herbert, J.; Wilcox, J.N.; Pham, K.-T.C.; Fremeau, R.T.; Zeviani, M.; Dwork, A.; Soprano, D.R.; Makover, A.; Goodman, D.S.; Zimmerman, E.A.; et al. Transthyretin: A choroid plexus-specific transport protein in human brain. The 1986 S. Weir Mitchell award. Neurology 1986, 36, 900–911. [Google Scholar] [CrossRef]
- Merlini, G.; Bellotti, V. Molecular mechanisms of amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Nativi-Nicolau, J.N.; Karam, C.; Khella, S.; Maurer, M.S. Screening for ATTR amyloidosis in the clinic: Overlapping disorders, misdiagnosis, and multiorgan awareness. Heart Fail. Rev. 2021, 1–9. [Google Scholar] [CrossRef]
- Rapezzi, C.; Merlini, G.; Quarta, C.C.; Riva, L.; Longhi, S.; Leone, O.; Salvi, F.; Ciliberti, P.; Pastorelli, F.; Biagini, E.; et al. Systemic cardiac amyloidoses: Disease profiles and clinical courses of the 3 main types. Circulation 2009, 120, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperry, B.W.; Reyes, B.A.; Ikram, A.; Donnelly, J.; Phelan, D.; Jaber, W.A.; Shapiro, D.; Evans, P.J.; Maschke, S.; Kilpatrick, S.E.; et al. Tenosynovial and Cardiac Amyloidosis in Patients Undergoing Carpal Tunnel Release. J. Am. Coll. Cardiol. 2018, 72, 2040–2050. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.-G.; Ikeda, S.-i.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, M.S.; Hanna, M.; Grogan, M.; Dispenzieri, A.; Witteles, R.; Drachman, B.; Judge, D.P.; Lenihan, D.J.; Gottlieb, S.S.; Shah, S.J.; et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J. Am. Coll. Cardiol. 2016, 68, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.N.; Ruberg, F.L. Transthyretin V122I (pV142I)* cardiac amyloidosis: An age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet. Med. 2017, 19, 733–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruberg, F.L.; Berk, J.L. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012, 126, 1286–1300. [Google Scholar] [CrossRef] [Green Version]
- Westermark, P.; Sletten, K.; Johansson, B.; Cornwell, G.G., 3rd. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 1990, 87, 2843–2845. [Google Scholar] [CrossRef] [Green Version]
- Kittleson, M.M.; Maurer, M.S.; Ambardekar, A.V.; Bullock-Palmer, R.P.; Chang, P.P.; Eisen, H.J.; Nair, A.P.; Nativi-Nicolau, J.; Ruberg, F.L.; American Heart Association Heart Failure and Transplantation Committee of the Council on Clinical Cardiology. Cardiac. Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation 2020, 142, e7–e22. [Google Scholar] [CrossRef]
- Conceição, I.; González-Duarte, A.; Obici, L.; Schmidt, H.H.J.; Simoneau, D.; Ong, M.L.; Amass, L. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J. Peripher. Nerv. Syst. 2016, 21, 5–9. [Google Scholar] [CrossRef]
- Nativi-Nicolau, J.; Maurer, M.S. Amyloidosis cardiomyopathy: Update in the diagnosis and treatment of the most common types. Curr. Opin. Cardiol. 2018, 33, 571–579. [Google Scholar] [CrossRef]
- Witteles, R.M.; Bokhari, S.; Damy, T.; Elliott, P.; Falk, R.H.; Fine, N.M.; Gospodinova, M.; Obici, L.; Rapezzi, C.; Garcia-Pavia, P. Screening for Transthyretin Amyloid Cardiomyopathy in Everyday Practice. JACC Heart Fail. 2019, 7, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Rintell, D.; Heath, D.; Braga Mendendez, F.; Cross, E.; Cross, T.; Knobel, V.; Gagnon, B.; Turtle, C.; Cohen, A.; Kalmykov, E.; et al. Patient and family experience with transthyretin amyloid cardiomyopathy (ATTR-CM) and polyneuropathy (ATTR-PN) amyloidosis: Results of two focus groups. Orphanet J. Rare Dis. 2021, 16, 70. [Google Scholar] [CrossRef] [PubMed]
- Grande-Trillo, A.; Baliellas, C.; Lladó, L.; Casasnovas, C.; Franco-Baux, J.V.; Gracia-Sánchez, L.; Bravo, M.G.; González-Vilatarsana, E.; Caballero-Gullón, L.; Echeverri, E.; et al. Transthyretin amyloidosis with cardiomyopathy after domino liver transplantation: Results of a cross-sectional study. Am. J. Transplant. 2021, 21, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Falk, R.H.; Alexander, K.M.; Liao, R.; Dorbala, S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J. Am. Coll. Cardiol. 2016, 68, 1323–1341. [Google Scholar] [CrossRef]
- Castaño, A.; Drachman, B.M.; Judge, D.; Maurer, M.S. Natural history and therapy of TTR-cardiac amyloidosis: Emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail. Rev. 2015, 20, 163–178. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, J.T. Cardiac amyloidosis. Two cases with digitalis sensitivity. Ann. Intern. Med. 1961, 55, 989–994. [Google Scholar] [CrossRef]
- Palma, J.A.; Gonzalez-Duarte, A.; Kaufmann, H. Orthostatic hypotension in hereditary transthyretin amyloidosis: Epidemiology, diagnosis and management. Clin. Auton. Res. 2019, 29 (Suppl. S1), 33–44. [Google Scholar] [CrossRef] [Green Version]
- Aimo, A.; Rapezzi, C.; Vergaro, G.; Giannoni, A.; Spini, V.; Passino, C.; Emdin, M. Management of complications of cardiac amyloidosis: 10 questions and answers. Eur. J. Prev. Cardiol. 2021, 28, 1000–1005. [Google Scholar] [CrossRef]
- Mitrani, L.R.; Santos, J.D.L.; Driggin, E.; Kogan, R.; Helmke, S.; Goldsmith, J.; Biviano, A.B.; Maurer, M.S. Anticoagulation with warfarin compared to novel oral anticoagulants for atrial fibrillation in adults with transthyretin cardiac amyloidosis: Comparison of thromboembolic events and major bleeding. Amyloid 2021, 28, 30–34. [Google Scholar] [CrossRef]
- Givens, R.C.; Russo, C.; Green, P.; Maurer, M.S. Comparison of cardiac amyloidosis due to wild-type and V122I transthyretin in older adults referred to an academic medical center. Aging Health 2013, 9, 229–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinney, J.H.; Whelan, C.J.; Petrie, A.; Dungu, J.; Banypersad, S.M.; Sattianayagam, P.; Wechalekar, A.; Gibbs, S.D.J.; Venner, C.P.; Wassef, N.; et al. Senile systemic amyloidosis: Clinical features at presentation and outcome. J. Am. Heart Assoc. 2013, 2, e000098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varr, B.C.; Zarafshar, S.; Coakley, T.; Liedtke, M.; Lafayette, R.A.; Arai, S.; Schrier, S.L.; Witteles, R.M. Implantable cardioverter-defibrillator placement in patients with cardiac amyloidosis. Heart Rhythm. 2014, 11, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Ternacle, J.; Krapf, L.; Mohty, D.; Magne, J.; Nguyen, A.; Galat, A.; Gallet, R.; Teiger, E.; Côté, N.; Clavel, M.A.; et al. Aortic Stenosis and Cardiac Amyloidosis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 2638–2651. [Google Scholar] [CrossRef]
- Nagle, D.G.; Ferreira, D.; Zhou, Y.-D. Epigallocatechin-3-gallate (EGCG): Chemical and biomedical perspectives. Phytochemistry 2006, 67, 1849–1855. [Google Scholar] [CrossRef] [Green Version]
- Kristen, A.V.; Lehrke, S.; Buss, S.; Mereles, D.; Steen, H.; Ehlermann, P.; Hardt, S.; Giannitsis, E.; Schreiner, R.; Haberkorn, U.; et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin. Res. Cardiol. 2012, 101, 805–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappelli, F.; Martone, R.; Taborchi, G.; Morini, S.; Bartolini, S.; Angelotti, P.; Farsetti, S.; Di Mario, C.; Perfetto, F. Epigallocatechin-3-gallate tolerability and impact on survival in a cohort of patients with transthyretin-related cardiac amyloidosis. A single-center retrospective study. Intern. Emerg. Med. 2018, 13, 873–880. [Google Scholar] [CrossRef]
- Ericzon, B.G.; Wilczek, H.E.; Larsson, M.; Wijayatunga, P.; Stangou, A.; Pena, J.R.; Furtado, E.; Barroso, E.; Daniel, J.; Samuel, D.; et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation 2015, 99, 1847–1854. [Google Scholar] [CrossRef]
- Liepnieks, J.J.; Zhang, L.Q.; Benson, M.D. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology 2010, 75, 324–327. [Google Scholar] [CrossRef] [Green Version]
- Barreiros, A.-P.; Post, F.; Hoppe-Lotichius, M.; Linke, R.P.; Vahl, C.F.; Schäfers, H.-J.; Galle, P.R.; Otto, G. Liver transplantation and combined liver-heart transplantation in patients with familial amyloid polyneuropathy: A single-center experience. Liver Transpl. 2010, 16, 314–323. [Google Scholar] [CrossRef]
- González-López, E.; Sainz, A.L.; Garcia-Pavia, P. Diagnosis and Treatment of Transthyretin Cardiac Amyloidosis. Prog. Hope Rev. Esp. Cardiol. Engl. Ed. 2017, 70, 991–1004. [Google Scholar] [CrossRef]
- Almeida, M.; Gales, L.; Damas, A.M.; Cardoso, I.; Saraiva, M.J. Small transthyretin (TTR) ligands as possible therapeutic agents in TTR amyloidoses. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Mangione, P.P.; Verona, G.; Corazza, A.; Marcoux, J.; Canetti, D.; Giorgetti, S.; Raimondi, S.; Stoppini, M.; Esposito, M.; Relini, A.; et al. Plasminogen activation triggers transthyretin amyloidogenesis in vitro. J. Biol. Chem. 2018, 293, 14192–14199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, M.; Macedo, B.; Cardoso, I.; Alves, I.; Valencia, G.; Arsequell, G.; Planas, A.; Saraiva, M.J. Selective binding to transthyretin and tetramer stabilization in serum from patients with familial amyloidotic polyneuropathy by an iodinated diflunisal derivative. Biochem. J. 2004, 381 Pt 2, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Gamez, J.; Salvadó, M.; Reig, N.; Suñé, P.; Casasnovas, C.; Rojas-Garcia, R.; Insa, R. Transthyretin stabilization activity of the catechol-O-methyltransferase inhibitor tolcapone (SOM0226) in hereditary ATTR amyloidosis patients and asymptomatic carriers: Proof-of-concept study. Amyloid 2019, 26, 74–84. [Google Scholar] [CrossRef]
- Larsen, K.R.; Dajani, E.Z.; Dajani, N.E.; Dayton, M.T.; Moore, J.G. Effects of tolcapone, a catechol-O-methyltransferase inhibitor, and Sinemet on intestinal electrolyte and fluid transport in conscious dogs. Dig. Dis. Sci. 1998, 43, 1806–1813. [Google Scholar] [CrossRef]
- Kaakkola, S. Clinical pharmacology, therapeutic use and potential of COMT inhibitors in Parkinson’s disease. Drugs 2000, 59, 1233–1250. [Google Scholar] [CrossRef]
- Van Booven, D.; Marsh, S.; McLeod, H.; Whirl-Carrillo, M.; Sangkuhl, K.; Klein, T.E.; Altman, R.B. Cytochrome P450 2C9-CYP2C9. Pharm. Genom. 2010, 20, 277–281. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [Green Version]
- Wixner, J.; Westermark, P.; Ihse, E.; Pilebro, B.; Lundgren, H.-E.; Anan, I. The Swedish open-label diflunisal trial (DFNS01) on hereditary transthyretin amyloidosis and the impact of amyloid fibril composition. Amyloid 2019, 26 (Suppl. S1), 39–40. [Google Scholar] [CrossRef] [Green Version]
- Siddiqi, O.K.; Mints, Y.Y.; Berk, J.L.; Connors, L.; Doros, G.; Gopal, D.M.; Kataria, S.; Lohrmann, G.; Pipilas, A.R.; Ruberg, F.L. Diflunisal treatment is associated with improved survival for patients with early stage wild-type transthyretin (ATTR) amyloid cardiomyopathy: The Boston University Amyloidosis Center experience. Amyloid 2022, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lohrmann, G.; Pipilas, A.; Mussinelli, R.; Gopal, D.M.; Berk, J.L.; Connors, L.H.; Vellanki, N.; Hellawell, J.; Siddiqi, O.K.; Fox, J.; et al. Stabilization of Cardiac Function With Diflunisal in Transthyretin (ATTR) Cardiac Amyloidosis. J. Card. Fail. 2020, 26, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Razavi, H.; Palaninathan, S.K.; Powers, E.T.; Wiseman, R.L.; Purkey, H.E.; Mohamedmohaideen, N.N.; Deechongkit, S.; Chiang, K.P.; Dendle, M.T.A.; Sacchettini, J.C.; et al. Benzoxazoles as transthyretin amyloid fibril inhibitors: Synthesis, evaluation, and mechanism of action. Angew. Chem. Int. Ed. Engl. 2003, 42, 2758–2761. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Egolum, U.; Parker, S.; Andrews, E.; Ombengi, D.; Ling, H. Tafamidis: A First-in-Class Transthyretin Stabilizer for Transthyretin Amyloid Cardiomyopathy. Ann. Pharmacother. 2020, 54, 470–477. [Google Scholar] [CrossRef]
- Coelho, T.; Maia, L.; Da Silva, A.M.; Cruz, M.W.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.; Campistol, J.M.; Conceicao, I.; Schmidt, H.H.-J.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef]
- Lozeron, P.; Theaudin, M.; Mincheva, Z.; Ducot, B.; Lacroix, C.; Adams, D.; French Network for FAP (CORNAMYL). Effect on disability and safety of Tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. Eur. J. Neurol. 2013, 20, 1539–1545. [Google Scholar] [CrossRef]
- Merlini, G.; Planté-Bordeneuve, V.; Judge, D.; Schmidt, H.; Obici, L.; Perlini, S.; Packman, J.; Tripp, T.; Grogan, D.R. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J. Cardiovasc. Transl. Res. 2013, 6, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Maurer, M.S.; Grogan, D.R.; Judge, D.P.; Mundayat, R.; Packman, J.; Lombardo, I.; Quyyumi, A.A.; Aarts, J.; Falk, R.H. Tafamidis in transthyretin amyloid cardiomyopathy: Effects on transthyretin stabilization and clinical outcomes. Circ. Heart Fail. 2015, 8, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Bézard, M.; Kharoubi, M.; Galat, A.; Poullot, E.; Guendouz, S.; Fanen, P.; Funalot, B.; Moktefi, A.; Lefaucheur, J.; Abulizi, M.; et al. Natural history and impact of treatment with tafamidis on major cardiovascular outcome-free survival time in a cohort of patients with transthyretin amyloidosis. Eur. J. Heart Fail. 2021, 23, 264–274. [Google Scholar] [CrossRef]
- Cho, Y.; Baranczak, A.; Helmke, S.; Teruya, S.; Horn, E.M.; Maurer, M.S.; Kelly, J.W. Personalized medicine approach for optimizing the dose of tafamidis to potentially ameliorate wild-type transthyretin amyloidosis (cardiomyopathy). Amyloid 2015, 22, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Lockwood, P.A.; Le, V.H.; O’Gorman, M.T.; Patterson, T.A.; Sultan, M.B.; Tankisheva, E.; Wang, Q.; Riley, S. The Bioequivalence of Tafamidis 61-mg Free Acid Capsules and Tafamidis Meglumine 4 x 20-mg Capsules in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 9, 849–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damy, T.; Garcia-Pavia, P.; Hanna, M.; Judge, D.P.; Merlini, G.; Gundapaneni, B.; Patterson, T.A.; Riley, S.; Schwartz, J.H.; Sultan, M.B.; et al. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study. Eur. J. Heart Fail. 2021, 23, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Kristen, A.; Gundapaneni, B.; Sultan, M.; Hanna, M. Benefits of tafamidis in patients with advanced transthyretin amyloid cardiomyopathy. Eur. Heart J. 2020, 41 (Suppl. S2). [Google Scholar] [CrossRef]
- Li, H.; Rozenbaum, M.; Casey, M.; Sultan, M. Estimating treatment effect of tafamidis on hospitalisation in NYHA class III ATTR-CM patients in the presence of death using principal stratification. Eur. Heart J. 2021, 42 (Suppl. S1), ehab724.0829. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Judge, D.; Heitner, S.B.; Falk, R.H.; Maurer, M.S.; Shah, S.; Witteles, R.M.; Grogan, M.; Selby, V.N.; Jacoby, D.; Hanna, M.; et al. Transthyretin Stabilization by AG10 in Symptomatic Transthyretin Amyloid Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 285–295. [Google Scholar] [CrossRef]
- Nelson, L.T.; Paxman, R.J.; Xu, J.; Webb, B.; Powers, E.T.; Kelly, J.W. Blinded potency comparison of transthyretin kinetic stabilisers by subunit exchange in human plasma. Amyloid 2021, 28, 24–29. [Google Scholar] [CrossRef]
- Trial of Acoramidis for Transthyretin Amyloid Cardiomyopathy Misses Primary Endpoint. 2021. Available online: https://www.healio.com/news/cardiology/20211228/trial-of-acoramidis-for-transthyretin-amyloid-cardiomyopathy-misses-primary-endpoint (accessed on 21 March 2022).
- Corazza, A.; Verona, G.; Waudby, C.A.; Mangione, P.P.; Bingham, R.; Uings, I.; Canetti, D.; Nocerino, P.; Taylor, G.W.; Pepys, M.B.; et al. Binding of Monovalent and Bivalent Ligands by Transthyretin Causes Different Short- and Long-Distance Conformational Changes. J. Med. Chem. 2019, 62, 8274–8283. [Google Scholar] [CrossRef] [Green Version]
- Verona, G.; Mangione, P.P.; Raimondi, S.; Giorgetti, S.; Faravelli, G.; Porcari, R.; Corazza, A.; Gillmore, J.D.; Hawkins, P.N.; Pepys, M.B.; et al. Inhibition of the mechano-enzymatic amyloidogenesis of transthyretin: Role of ligand affinity, binding cooperativity and occupancy of the inner channel. Sci. Rep. 2017, 7, 182. [Google Scholar] [CrossRef] [Green Version]
- Franz, C.; Hoffmann, K.; Hinz, U.; Singer, R.; Hund, E.; Gotthardt, D.N.; Ganten, T.; Kristen, A.V.; Hegenbart, U.; Schönland, S.; et al. Modified body mass index and time interval between diagnosis and operation affect survival after liver transplantation for hereditary amyloidosis: A single-center analysis. Clin. Transplant. 2013, 27 (Suppl. S25), 40–48. [Google Scholar] [CrossRef]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizk, M.; Tuzmen, S. Update on the clinical utility of an RNA interference-based treatment: Focus on Patisiran. Pharmgenom. Pers. Med. 2017, 10, 267–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonezawa, S.; Koide, H.; Asai, T. Recent advances in siRNA delivery mediated by lipid-based nanoparticles. Adv. Drug Deliv. Rev. 2020, 154–155, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Adams, D.; Kristen, A.; Grogan, M.; González-Duarte, A.; Maurer, M.S.; Merlini, G.; Damy, T.; Slama, M.S.; Brannagan, I.I.I.; et al. Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients With Hereditary Transthyretin-Mediated Amyloidosis. Circulation 2019, 139, 431–443. [Google Scholar] [CrossRef]
- Fontana, M.; Martinez-Naharro, A.; Chacko, L.; Rowczenio, D.; Gilbertson, J.A.; Whelan, C.J.; Strehina, S.; Lane, T.; Moon, J.; Hutt, D.F.; et al. Reduction in CMR Derived Extracellular Volume With Patisiran Indicates Cardiac Amyloid Regression. JACC Cardiovasc. Imaging 2021, 14, 189–199. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Dasgupta, N.R.; Rissing, S.M.; Smith, J.; Jung, J.; Benson, M.D. Inotersen therapy of transthyretin amyloid cardiomyopathy. Amyloid 2020, 27, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, T.S.; Karsten, V.; Chan, A.; Chiesa, J.; Boyce, M.; Bettencourt, B.R.; Hutabarat, R.; Nochur, S.; Vaishnaw, A.; Gollob, J. Clinical Proof of Concept for a Novel Hepatocyte-Targeting GalNAc-siRNA Conjugate. Mol. Ther. 2017, 25, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Judge, D.P.; Kristen, A.V.; Grogan, M.; Maurer, M.S.; Falk, R.H.; Hanna, M.; Gillmore, J.; Garg, P.; Vaishnaw, A.K.; Harrop, J.; et al. Phase 3 Multicenter Study of Revusiran in Patients with Hereditary Transthyretin-Mediated (hATTR) Amyloidosis with Cardiomyopathy (ENDEAVOUR). Cardiovasc. Drugs Ther. 2020, 34, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.-J. Therapeutic siRNA: State of the art. Signal. Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, B.A.; Karsten, V.; Attarwala, H.; Goel, V.; Melch, M.; Clausen, V.A.; Garg, P.; Vaishnaw, A.K.; Sweetser, M.T.; Robbie, G.J.; et al. Single-Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting N-acetylgalactosamine-Small Interfering Ribonucleic Acid Conjugate, Vutrisiran, in Healthy Subjects. Clin. Pharmacol. Ther. 2021, 109, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Alnylam Pharmaceuticals. Alnylam Presents Positive 18-Month Results from HELIOS-A Phase 3 Study of Investigational Vutrisiran in Patients with hATTR Amyloidosis with Polyneuropathy. Available online: https://investors.alnylam.com/press-release?id=26396 (accessed on 21 January 2022).
- Coelho, T.; Ando, Y.; Benson, M.D.; Berk, J.L.; Waddington-Cruz, M.; Dyck, P.J.; Gillmore, J.D.; Khella, S.L.; Litchy, W.J.; Obici, L.; et al. Design and Rationale of the Global Phase 3 NEURO-TTRansform Study of Antisense Oligonucleotide AKCEA-TTR-LRx (ION-682884-CS3) in Hereditary Transthyretin-Mediated Amyloid Polyneuropathy. Neurol. Ther. 2021, 10, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Landmark CRISPR trial shows promise against deadly disease. Nature 2021. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Chukwudi, C.U. rRNA Binding Sites and the Molecular Mechanism of Action of the Tetracyclines. Antimicrob. Agents Chemother 2016, 60, 4433–4441. [Google Scholar] [CrossRef] [Green Version]
- Medina, L.; González-Lizárraga, F.; Dominguez-Meijide, A.; Ploper, D.; Parrales, V.; Sequeira, S.; Cima-Omori, M.-S.; Zweckstetter, M.; Del Bel, E.; Michel, P.P.; et al. Doxycycline Interferes With Tau Aggregation and Reduces Its Neuronal Toxicity. Front. Aging Neurosci. 2021, 13, 635760. [Google Scholar] [CrossRef]
- Cardoso, I.; Martins, D.; Ribeiro, T.; Merlini, G.; Saraiva, M.J. Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: Studies in FAP mouse models. J. Transl. Med. 2010, 8, 74. [Google Scholar] [CrossRef] [Green Version]
- Obici, L.; Cortese, A.; Lozza, A.; Lucchetti, J.; Gobbi, M.; Palladini, G.; Perlini, S.; Saraiva, M.J.; Merlini, G. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 2012, 19 (Suppl. S1), 34–36. [Google Scholar] [CrossRef]
- Wixner, J.; Pilebro, B.; Lundgren, H.-E.; Olsson, M.; Anan, I. Effect of doxycycline and ursodeoxycholic acid on transthyretin amyloidosis. Amyloid 2017, 24 (Suppl. S1), 78–79. [Google Scholar] [CrossRef]
- Higaki, J.N.; Chakrabartty, A.; Galant, N.J.; Hadley, K.C.; Hammerson, B.; Nijjar, T.; Torres, R.; Tapia, J.R.; Salmans, J.; Barbour, R.; et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of transthyretin. Amyloid 2016, 23, 86–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalon, A.; Hagenbuch, A.; Huy, C.; Varela, E.; Combaluzier, B.; Damy, T.; Suhr, O.B.; Saraiva, M.J.; Hock, C.; Nitsch, R.M.; et al. A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells. Nat. Commun. 2021, 12, 3142. [Google Scholar] [CrossRef] [PubMed]
- Yadav, J.D.; Othee, H.; Chan, K.A.; Man, D.C.; Belliveau, P.P.; Towle, J. Transthyretin Amyloid Cardiomyopathy-Current and Future Therapies. Ann. Pharmacother. 2021, 55, 1502–1514. [Google Scholar] [CrossRef] [PubMed]
- Abou-El-Enein, M.; Elsanhoury, A.; Reinke, P. Overcoming Challenges Facing Advanced Therapies in the EU Market. Cell Stem Cell 2016, 19, 293–297. [Google Scholar] [CrossRef] [Green Version]
- Kazi, D.S.; Bellows, B.K.; Baron, S.J.; Shen, C.; Cohen, D.J.; Spertus, J.A.; Yeh, R.W.; Arnold, S.V.; Sperry, B.W.; Maurer, M.S.; et al. Cost-Effectiveness of Tafamidis Therapy for Transthyretin Amyloid Cardiomyopathy. Circulation 2020, 141, 1214–1224. [Google Scholar] [CrossRef]
- Rapezzi, C.; Elliott, P.; Damy, T.; Nativi-Nicolau, J.; Berk, J.L.; Velazquez, E.J.; Boman, K.; Gundapaneni, B.; Patterson, T.A.; Schwartz, J.H.; et al. Efficacy of Tafamidis in Patients With Hereditary and Wild-Type Transthyretin Amyloid Cardiomyopathy: Further Analyses From ATTR-ACT. JACC Heart Fail. 2021, 9, 115–123. [Google Scholar] [CrossRef]
- Monteiro, C.; Martins da Silva, A.; Ferreira, N.; Mesgarzadeh, J.; Novais, M.; Coelho, T.; Kelly, J.W. Cerebrospinal fluid and vitreous body exposure to orally administered tafamidis in hereditary ATTRV30M (p.TTRV50M) amyloidosis patients. Amyloid 2018, 25, 120–128. [Google Scholar] [CrossRef]
- Dohrn, M.F.; Medina, J.; Dague, K.R.O.; Hund, E. Are we creating a new phenotype? Physiological barriers and ethical considerations in the treatment of hereditary transthyretin-amyloidosis. Neurol. Res. Pract. 2021, 3, 57. [Google Scholar] [CrossRef]
- Magrinelli, F.; Fabrizi, G.M.; Santoro, L.; Manganelli, F.; Zanette, G.; Cavallaro, T.; Tamburin, S. Pharmacological treatment for familial amyloid polyneuropathy. Cochrane Database Syst. Rev. 2020, 4, CD012395. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Yes | Sometimes | No | |
|---|---|---|---|
| Diuretics ± aldosterone antagonists |  | ||
| Renin-angiotensin system inhibitors |  | ||
| Beta-adrenoreceptor blockers | | ||
| Alpha-1-adrenoreceptor agonists | | ||
| Calcium channel blockers |  | ||
| Digoxin * | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tschöpe, C.; Elsanhoury, A. Treatment of Transthyretin Amyloid Cardiomyopathy: The Current Options, the Future, and the Challenges. J. Clin. Med. 2022, 11, 2148. https://doi.org/10.3390/jcm11082148
Tschöpe C, Elsanhoury A. Treatment of Transthyretin Amyloid Cardiomyopathy: The Current Options, the Future, and the Challenges. Journal of Clinical Medicine. 2022; 11(8):2148. https://doi.org/10.3390/jcm11082148
Chicago/Turabian StyleTschöpe, Carsten, and Ahmed Elsanhoury. 2022. "Treatment of Transthyretin Amyloid Cardiomyopathy: The Current Options, the Future, and the Challenges" Journal of Clinical Medicine 11, no. 8: 2148. https://doi.org/10.3390/jcm11082148
APA StyleTschöpe, C., & Elsanhoury, A. (2022). Treatment of Transthyretin Amyloid Cardiomyopathy: The Current Options, the Future, and the Challenges. Journal of Clinical Medicine, 11(8), 2148. https://doi.org/10.3390/jcm11082148