
The Clinical and Neuropathological Features of Sporadic (Late-Onset) and Genetic Forms of Alzheimer’s Disease

 , , ,
, , ,
Abstract
:1. Introduction and Background
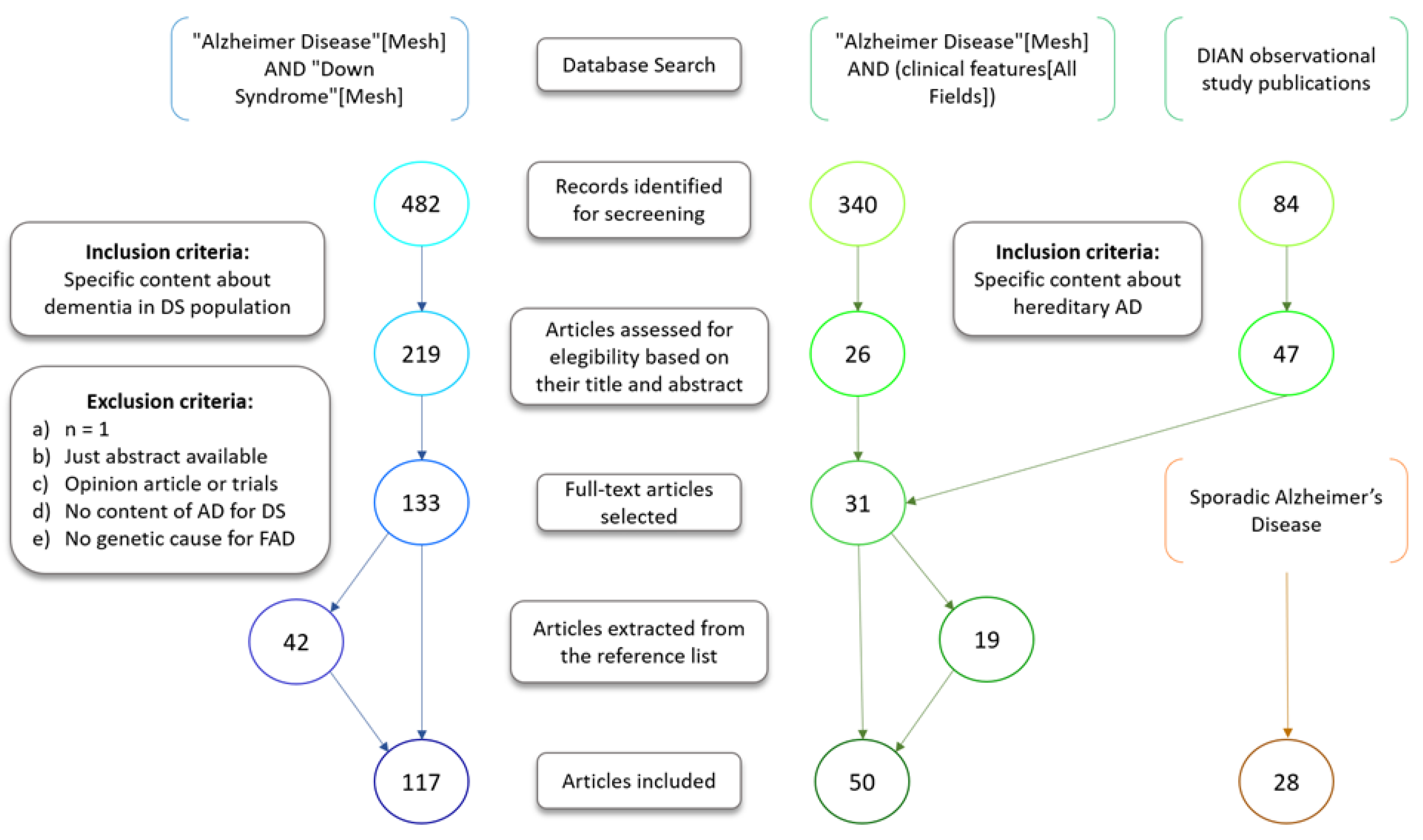
2. Methods
3. Clinical Features
3.1. Diagnosis
3.2. Age of Onset
3.3. Vasculature
3.4. Amnestic Phenotypes
3.5. Non-Amnestic Phenotypes
3.6. Neurological Symptoms
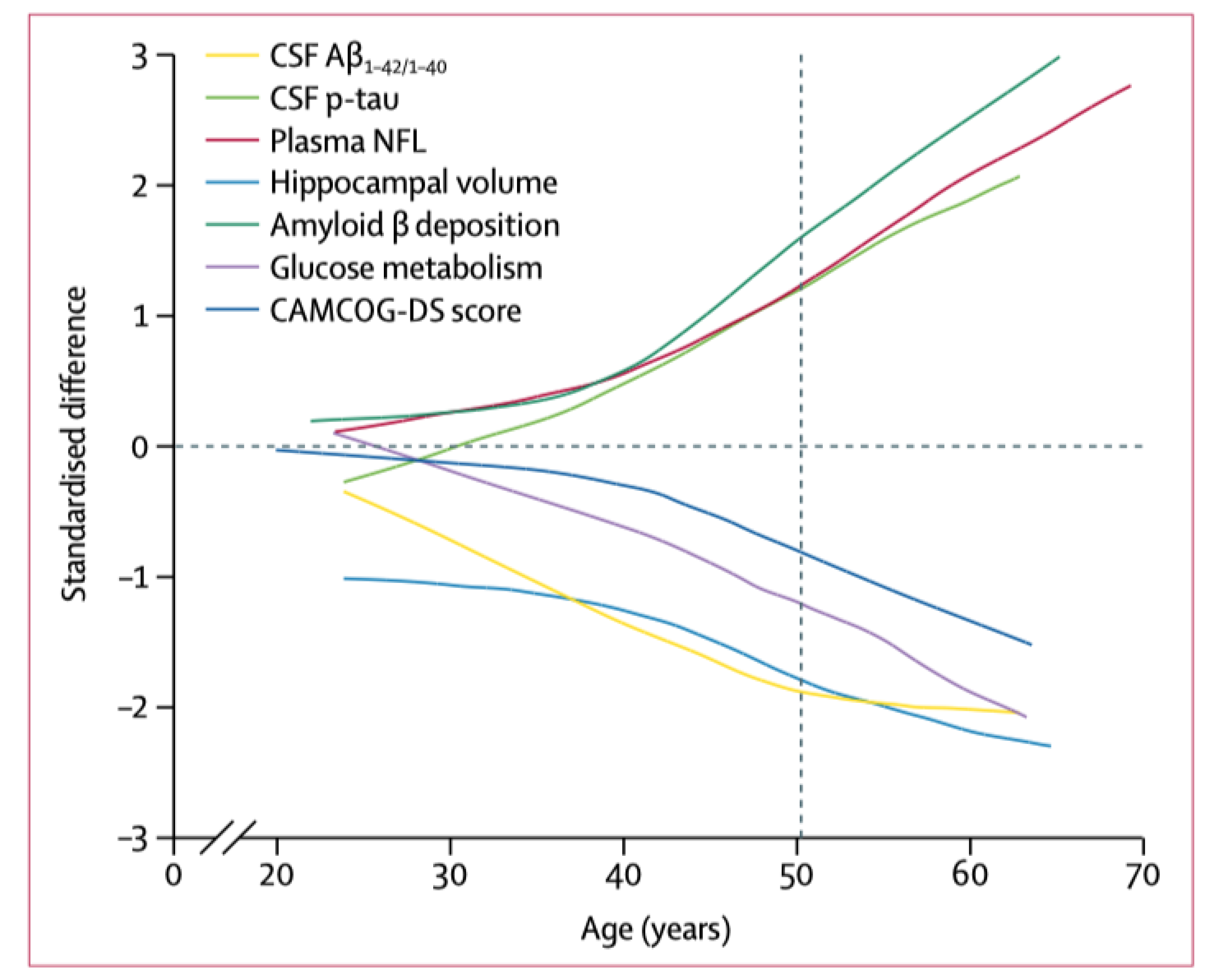
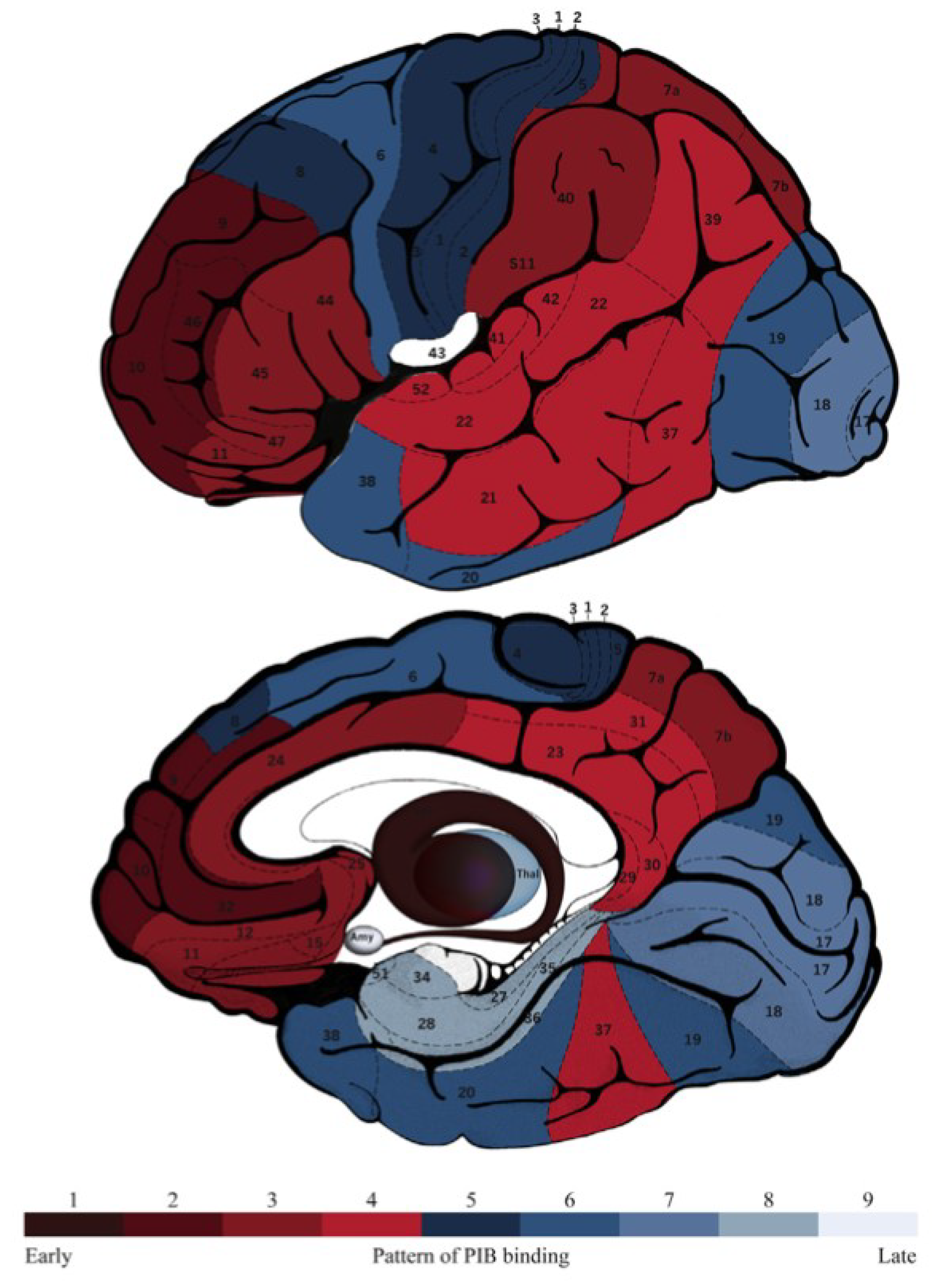
4. Neuroimaging and Neuropathology
5. Co-Pathologies
5.1. Lewy Body Pathology
5.2. TDP-43 Pathology
6. Other Similarities and Differences
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte biomarkers in Alzheimer’s disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [Green Version]
- WHO. World Health Organization. Available online: https://www.alzint.org/resource/numbers-of-people-with-dementia-worldwide/ (accessed on 30 September 2021).
- Zhu, X.-C.; Tan, L.; Wang, H.-F.; Jiang, T.; Cao, L.; Wang, C.; Wang, J.; Tan, C.-C.; Meng, X.-F.; Yu, J.-T. Rate of early onset Alzheimer’s disease: A systematic review and meta-analysis. Ann. Transl. Med. 2015, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Malt, E.A.; Dahl, R.C.; Haugsand, T.M.; Ulvestad, I.H.; Emilsen, N.M.; Hansen, B.; Cardenas, Y.E.G.; Skøld, R.O.; Thorsen, A.T.B.; Davidsen, E.M.M. Health and disease in adults with Down syndrome. Tidsskr Den Nor Legeforening 2013, 133, 290–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Rasmussen, S.; Friedman, J. Mortality associated with Down′s syndrome in the USA from 1983 to 1997: A population-based study. Lancet 2002, 359, 1019–1025. [Google Scholar] [CrossRef]
- Thase, M.E. Longevity and mortality in Down′s syndrome. J. Intellect. Disabil. Res. 2008, 26, 177–192. [Google Scholar] [CrossRef]
- Englund, A.; Jonsson, B.; Zander, C.S.; Gustafsson, J.; Annerén, G. Changes in mortality and causes of death in the Swedish Down syndrome population. Am. J. Med. Genet. Part A 2013, 161, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Oliver, C.; Holland, A.J. Down′s Syndrome and Alzheimer’s disease: A review. Psychol. Med. 1986, 16, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Handen, B.L.; Lott, I.T.; Christian, B.T.; Schupf, N.; Obryant, S.; Mapstone, M.; Fagan, A.M.; Lee, J.H.; Tudorascu, D.; Wang, M.; et al. The Alzheimer’s Biomarker Consortium-Down Syndrome: Rationale and methodology. Alzheimer Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12065. [Google Scholar] [CrossRef]
- Head, E.; Lott, I.T.; Wilcock, D.M.; Lemere, C.A. Aging in Down syndrome and the development of Alzheimer’s disease neuropathology. Curr. Alzheimer Res. 2015, 13, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Evans, E. Dementia in People with Intellectual Disability: Guidelines for Australian GPs. Ph.D. Thesis, University of New South Wales, Sydney, Australia, 2018. [Google Scholar]
- Scheepers, M. Faculty for People with Intellectual Disabilities Dementia and People with Intellectual Disabilities Guidance on the Assessment, Diagnosis, Interventions and Support of People with Intellectual Disabilities Who Develop Dementia; Jessica Kingsley Publishers: London, UK, 2014. [Google Scholar]
- McCarron, M.; McCallion, P.; Reilly, E.; Mulryan, N. A prospective 14-year longitudinal follow-up of dementia in persons with Down syndrome. J. Intellect. Disabil. Res. 2013, 58, 61–70. [Google Scholar] [CrossRef]
- McCarron, M.; McCallion, P.; Reilly, E.; Dunne, P.; Carroll, R.; Mulryan, N. A prospective 20-year longitudinal follow-up of dementia in persons with Down syndrome. J. Intellect. Disabil. Res. 2017, 61, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Orphanet. Available online: https://www.orpha.net/consor/cgi-bin/index.php (accessed on 1 July 2021).
- Buss, L.; Fisher, E.; Hardy, J.; Nizetic, D.; Groet, J.; Pulford, L.; Strydom, A. Intracerebral haemorrhage in Down syndrome: Protected or predisposed? F1000Research 2016, 5, 876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimer Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, N.S.; Rossor, M.N. Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark. Med. 2010, 4, 99–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, R.J.; Plascencia-Villa, G.; Perry, G. The amyloid cascade and Alzheimer’s disease therapeutics: Theory versus observation. Lab. Investig. 2019, 99, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.S.; Nicholas, J.M.; Weston, P.; Liang, Y.; Lashley, T.; Guerreiro, R.; Adamson, G.; Kenny, J.; Beck, J.; Chavez-Gutierrez, L.; et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: A case series. Lancet Neurol. 2016, 15, 1326–1335. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Liu, C.; Che, C.; Huang, H. Clinical genetics of Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 291862. [Google Scholar] [CrossRef]
- Storandt, M.; Balota, D.A.; Aschenbrenner, A.J.; Morris, J.C. Clinical and psychological characteristics of the initial cohort of the Dominantly Inherited Alzheimer Network (DIAN). Neuropsychology 2014, 28, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Mann, D.M.; Pickering-Brown, S.M.; Takeuchi, A.; Iwatsubo, T. Members of the familial Alzheimer’s disease pathology study group. Amyloid angiopathy and variability in amyloid beta deposition is determined by mutation position in presenilin-1-linked Alzheimer’s disease. Am. J. Pathol. 2001, 158, 2165–2175. [Google Scholar] [CrossRef]
- Alberici, A.; Benussi, A.; Premi, E.; Borroni, B.; Padovani, A. Clinical, genetic, and neuroimaging features of early onset Alzheimer disease: The challenges of diagnosis and treatment. Curr. Alzheimer Res. 2014, 11, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Dams-O′Connor, K.; Guetta, G.; Hahn-Ketter, A.E.; Fedor, A. Traumatic brain injury as a risk factor for Alzheimer’s disease: Current knowledge and future directions. Neurodegener. Dis. Manag. 2016, 6, 417–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzmeier, N.; Düzel, E.; Jessen, F.; Buerger, K.; Levin, J.; Duering, M.; Dichgans, M.; Haass, C.; Suárez-Calvet, M.; Fagan, A.M.; et al. Left frontal hub connectivity delays cognitive impairment in autosomal-dominant and sporadic Alzheimer’s disease. Brain 2018, 141, 1186–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; Van Der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B.; et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef]
- Lai, F.; Kammann, E.; Rebeck, G.W.; Anderson, A.; Chen, Y.; Nixon, R.A. APOE genotype and gender effects on Alzheimer disease in 100 adults with Down syndrome. Neurology 1999, 53, 331. [Google Scholar] [CrossRef]
- Prasher, V.P.; Sajith, S.G.; Rees, S.D.; Patel, A.; Tewari, S.; Schupf, N.; Zigman, W.B. Significant effect of APOE epsilon 4 genotype on the risk of dementia in Alzheimer’s disease and mortality in persons with Down syndrome. Int. J. Geriatr. Psychiatry 2008, 23, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Coppus, A.; Evenhuis, H.; Verberne, G.-J.; Visser, F.; Arias-Vasquez, A.; Sayed-Tabatabaei, F.; Vergeer-Drop, J.; Eikelenboom, P.; van Gool, W.; van Duijn, C. The impact of apolipoprotein E on dementia in persons with Down′s syndrome. Neurobiol. Aging 2008, 29, 828–835. [Google Scholar] [CrossRef]
- Benejam, B.; Videla, L.; Barroeta, I.; Fernandez, S. Association of apolipoprotein E ε 4 allele with clinical and multimodal biomarker changes of Alzheimer disease in adults with down syndrome. JAMA Neurol. 2021, 78, 937–947. [Google Scholar]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef]
- McNaughton, D.; Knight, W.; Guerreiro, R.; Ryan, N.; Lowe, J.; Poulter, M.; Nicholl, D.J.; Hardy, J.; Revesz, T.; Lowe, J.; et al. Duplication of Amyloid Precursor Protein (APP), but not Prion Protein (PRNP) gene is a significant cause of early onset dementia in a large UK series. Neurobiol. Aging 2012, 33, 426.e13–426.e21. [Google Scholar] [CrossRef] [Green Version]
- Mok, K.Y.; Jones, E.L.; Hanney, M.; Harold, D.; Sims, R.; Williams, J.; Ballard, C.; Hardy, J. Polymorphisms in BACE2 may affect the age of onset Alzheimer’s dementia in Down syndrome. Neurobiol. Aging 2013, 35, 1513.e1–1513.e5. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, F.K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V.L.J.; Fisher, E.M.C.; Strydom, A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16, 564–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryoo, S.R.; Cho, H.J.; Lee, H.W.; Jeong, H.K.; Radnaabazar, C.; Kim, Y.S.; Kim, M.J.; Son, M.Y.; Seo, H.; Chung, S.H.; et al. Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phos-phorylation of amyloid precursor protein: Evidence for a functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 2008, 104, 1333–1344. [Google Scholar] [CrossRef]
- Wegiel, J.; Dowjat, K.; Kaczmarski, W.; Kuchna, I.; Nowicki, K.; Frackowiak, J.; Kolecka, B.M.; Wegiel, J.; Silverman, W.P.; Reisberg, B.; et al. The role of overexpressed DYRK1A protein in the early onset of neurofibrillary degeneration in Down syndrome. Acta Neuropathol. 2008, 116, 391–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, M.; Kinjo, A.; Kimura, S.; Mori, R.; Kawakubo, T.; Shirotani, K.; Yagishita, S.; Maruyama, K.; Iwata, N. Perturbed cal-cineurin-NFAT signaling is associated with the development of Alzheimer’s disease. Biol. Pharm. Bull. 2016, 39, 1646–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; He, G.; Song, W. BACE2, as a novel APP θ-secretase, is not responsible for the pathogenesis of Alzheimer’s disease in Down syndrome. FASEB J. 2006, 20, 1369–1376. [Google Scholar] [CrossRef]
- Sun, X.; Tong, Y.; Qing, H.; Chen, C.-H.; Song, W. Increased BACE1 maturation contributes to the pathogenesis of Alzheimer’s disease in Down syndrome. FASEB J. 2006, 20, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Kerkel, K.; Schupf, N.; Hatta, K.; Pang, D.; Salas, M.; Kratz, A.; Minden, M.; Murty, V.; Zigman, W.; Mayeux, R.P.; et al. Altered DNA methylation in leukocytes with trisomy 21. PLoS Genet. 2010, 6, e1001212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Salvioli, S.; Gentilini, D.; Di Blasio, A.M.; Giulina, C.; Tung, S.; Vinters, H.V.; et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 2015, 14, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Panegyres, P.K.; Chen, H.-Y. Differences between early and late onset Alzheimer’s disease. Am. J. Neurodegener. Dis. 2013, 2, 300–306. [Google Scholar] [PubMed]
- Deb, S.; Hare, M.; Prior, L. Symptoms of dementia among adults with Down′s syndrome: A qualitative study. J. Intellect. Disabil. Res. 2007, 51, 726–739. [Google Scholar] [CrossRef]
- Zis, P.; Strydom, A. Clinical aspects and biomarkers of Alzheimer’s disease in Down syndrome. Free Radic. Biol. Med. 2018, 114, 3–9. [Google Scholar] [CrossRef]
- Shamseer, L.; Moher, D.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; The PRISMA-P Group. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: Elaboration and explanation. BMJ 2015, 349, g7647. [Google Scholar] [CrossRef] [Green Version]
- DIAN Observational Study. The Dominantly Inherited Alzheimer Network. Available online: https://dian.wustl.edu/ (accessed on 1 July 2021).
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging—Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Schupf, N. Genetic and host factors for dementia in Down′s syndrome. Br. J. Psychiatry 2002, 180, 405–410. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Lee, A.J.; Dang, L.-H.; Pang, D.; Kisselev, S.; Krinsky-McHale, S.J.; Zigman, W.B.; Luchsinger, J.; Silverman, W.; Tycko, B.; et al. Candidate gene analysis for Alzheimer’s disease in adults with Down syndrome. Neurobiol. Aging 2017, 56, 150–158. [Google Scholar] [CrossRef]
- Patel, A.; Rees, S.D.; Kelly, M.A.; Bain, S.C.; Barnett, A.H.; Thalitaya, D.; Prasher, V.P. Association of variants within APOE, SORL1, RUNX1, BACE1 and ALDH18A1 with dementia in Alzheimer’s disease in subjects with Down syndrome. Neurosci. Lett. 2011, 487, 144–148. [Google Scholar] [CrossRef]
- Jones, E.L.; Mok, K.; Hanney, M.; Harold, D.; Sims, R.; Williams, J.; Ballard, C. Evidence that PICALM affects age at onset of Alzheimer’s dementia in Down syndrome. Neurobiol. Aging 2013, 34, 2441.e1–2441.e5. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.L.; Margallo-Lana, M.; Prasher, V.P.; Ballard, C.G. The extended tau haplotype and the age of onset of dementia in Down syndrome. Dement. Geriatr. Cogn. Disord. 2008, 26, 199–202. [Google Scholar] [CrossRef]
- Raha-Chowdhury, R.; Henderson, J.W.; Raha, A.A.; Vuono, R.; Bickerton, A.; Jones, E.; Fincham, R.; Allinson, K.; Holland, A.; Zaman, S.H. Choroid plexus acts as gatekeeper for TREM2, abnormal accumulation of ApoE, and fibrillary tau in Alzheimer’s disease and in Down syndrome dementia. J. Alzheimer Dis. 2019, 69, 91–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinai, A.; Mokrysz, C.; Bernal, J.; Bohnen, I.; Bonell, S.; Courtenay, K.; Dodd, K.; Gazizova, D.; Hassiotis, A.; Hillier, R.; et al. Predictors of age of diagnosis and survival of Alzheimer’s disease in Down syndrome. J. Alzheimer Dis. 2017, 61, 717–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lao, P.J.; Gutierrez, J.; Keator, D.; Rizvi, B.; Banerjee, A.; Igwe, K.C.; Laing, K.K.; Sathishkumar, M.; Moni, F.; Andrews, H.; et al. Alzheimer-related cerebrovascular disease in Down syndrome. Ann. Neurol. 2020, 88, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lee, J.H.; Pang, D.; Temkin, A.; Park, N.; Janicki, S.C.; Zigman, W.B.; Silverman, W.; Tycko, B.; Schupf, N. Estrogen receptor-beta variants are associated with increased risk of Alzheimer’s disease in women with down syndrome. Dement. Geriatr. Cogn. Disord. 2011, 32, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Schupf, N.; Lee, J.H.; Wei, M.; Pang, D.; Chace, C.; Cheng, R.; Zigman, W.B.; Tycko, B.; Silverman, W. Estrogen receptor-α variants increase risk of Alzheimer’s disease in women with Down syndrome. Dement. Geriatr. Cogn. Disord. 2008, 25, 476–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattila, K.M.; Axelman, K.; Rinne, J.O.; Blomberg, M.; Lehtimäki, T.; Laippala, P.; Röyttä, M.; Viitanen, M.; Wahlund, L.-O.; Winblad, B.; et al. Interaction between estrogen receptor 1 and the ε4 allele of apolipoprotein E increases the risk of familial Alzheimer’s disease in women. Neurosci. Lett. 2000, 282, 45–48. [Google Scholar] [CrossRef]
- Jayadev, S.; Leverenz, J.; Steinbart, E.; Stahl, J.; Klunk, W.; Yu, C.-E.; Bird, T.D. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 2010, 133, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- Ganguli, M.; Dodge, H.H.; Shen, C.; Pandav, R.S.; DeKosky, S. Alzheimer disease and mortality. Arch. Neurol. 2005, 62, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Shea, Y.-F.; Chu, L.-W.; Chan, A.O.-K.; Ha, J.; Li, Y.; Song, Y.-Q. A systematic review of familial Alzheimer’s disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J. Formos. Med. Assoc. 2016, 115, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanleenuwat, P.; Iwanowski, P.; Kozubski, W. Alzheimer’s dementia: Pathogenesis and impact of cardiovascular risk factors on cognitive decline. Postgrad. Med. 2019, 131, 415–422. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Schmitt, F.A.; Head, E. Cerebrovascular contributions to aging and Alzheimer’s disease in Down syndrome. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Prasher, V.P.; Airuehia, E.; Patel, A.; Haque, M.S. Total serum cholesterol levels and Alzheimer’s dementia in patients with down syndrome. Int. J. Geriatr. Psychiatry 2008, 23, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Zigman, W.B.; Schupf, N.; Jenkins, E.C.; Urv, T.K.; Tycko, B.; Silverman, W. Cholesterol level, statin use and Alzheimer’s disease in adults with Down syndrome. Neurosci. Lett. 2007, 416, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drachman, D.A.; Smith, T.W.; Alkamachi, B.; Kane, K. Microvascular changes in Down syndrome with Alzheimer’s-type pathology: Insights into a potential vascular mechanism for Down syndrome and Alzheimer’s disease. Alzheimer Dement. 2017, 13, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Viqar, F.; Zimmerman, M.E.; Narkhede, A.; Tosto, G.; Benzinger, T.L.; Marcus, D.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; et al. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann. Neurol. 2016, 79, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Head, E.; Phelan, M.J.; Doran, E.; Kim, R.C.; Poon, W.; Schmitt, F.A.; Lott, I.T. Cerebrovascular pathology in Down syndrome and Alzheimer disease. Acta Neuropathol. Commun. 2017, 5, 93. [Google Scholar] [CrossRef]
- Carmona-Iragui, M.; Balasa, M.; Benejam, B.; Alcolea, D.; Fernández, S.; Videla, L.; Sala, I.; Sánchez-Saudinós, M.B.; Morenas-Rodriguez, E.; Ribosa-Nogué, R.; et al. Cerebral amyloid angiopathy in Down syndrome and sporadic and autosomal-dominant Alzheimer’s disease. Alzheimer Dement. 2017, 13, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringman, J.M.; Monsell, S.; Ng, D.W.; Zhou, Y.; Nguyen, A.; Coppola, G.; Van Berlo, V.; Mendez, M.F.; Tung, S.; Weintraub, S.; et al. Neuropathology of autosomal dominant Alzheimer disease in the National Alzheimer Coordinating Center Database. J. Neuropathol. Exp. Neurol. 2016, 75, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Basun, H.; Bogdanovic, N.; Ingelsson, M.; Almkvist, O.; Näslund, J.; Axelman, K.; Bird, T.D.; Nochlin, D.; Schellenberg, G.D.; Wahlund, L.-O.; et al. Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch. Neurol. 2008, 65, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Singh, S.; Cras, P.; Wang, R.; Kros, J.M.; van Swieten, J.; Lübke, U.; Ceuterick, C.; Serneels, S.; Vennekens, K.L.; Timmermans, J.P.; et al. Dense-core senile plaques in the Flemish variant of Alzheimer’s disease are vas-ocentric. Am. J. Pathol. 2002, 161, 507–520. [Google Scholar] [CrossRef]
- Yanai, K.; Ishida, Y.; Nishido, H.; Miyamoto, S.; Yamazaki, K.; Hoya, K. Multiple cerebral hemorrhagic lesions depicted by susceptibility-weighted imaging in a patient with Down syndrome: Case report. J. Stroke Cerebrovasc. Dis. 2019, 28, e37–e38. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Iragui, M.; Videla, L.; Lleó, A.; Fortea, J. Down syndrome, Alzheimer disease, and cerebral amyloid angiopathy: The complex triangle of brain amyloidosis. Dev. Neurobiol. 2019, 79, 716–737. [Google Scholar] [CrossRef] [PubMed]
- Helman, A.M.; Siever, M.; McCarty, K.L.; Lott, I.T.; Doran, E.; Abner, E.L.; Schmitt, F.A.; Head, E. Microbleeds and cerebral amyloid angiopathy in the brains of people with Down syndrome with Alzheimer’s disease. J. Alzheimer Dis. 2019, 67, 103–112. [Google Scholar] [CrossRef]
- Rovelet-Lecrux, A.; Hannequin, D.; Raux, G.; Le Meur, N.; Laquerrière, A.; Vital, A.; Dumanchin, C.; Feuillette, S.; Brice, A.; Vercelletto, M.; et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006, 38, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.; Davidson, Y.S.; Robinson, A.C.; Allen, N.; Hashimoto, T.; Richardson, A.; Jones, M.; Snowden, J.S.; Pendleton, N.; Potier, M.C.; et al. Patterns and severity of vascular amyloid in Alzheimer’s disease associated with duplications and missense mutations in APP gene, Down syndrome and sporadic Alzheimer’s disease. Acta Neuropathol 2018, 136, 569–587. [Google Scholar] [CrossRef]
- Barnes, J.; Dickerson, B.C.; Frost, C.; Jiskoot, L.C.; Wolk, D.; van der Flier, W.M. Alzheimer’s disease first symptoms are age dependent: Evidence from the NACC dataset. Alzheimer Dement. 2015, 11, 1349–1357. [Google Scholar] [CrossRef] [Green Version]
- Lautarescu, B.A.; Holland, A.J.; Zaman, S.H. The early presentation of dementia in people with Down syndrome: A systematic review of longitudinal studies. Neuropsychol. Rev. 2017, 27, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Benejam, B.; Videla, L.; Vilaplana, E.; Barroeta, I.; Carmona-Iragui, M.; Altuna-Azkargorta, M.; Valldeneu, S.; Fernandez, S.; Giménez, S.; Iulita, F.; et al. Diagnosis of prodromal and Alzheimer’s disease dementia in adults with Down syndrome using neuropsychological tests. Alzheimer Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12047. [Google Scholar] [CrossRef]
- Dekker, A.D.; Sacco, S.; Carfi, A.; Benejam, B.; Vermeiren, Y.; Beugelsdijk, G.; Schippers, M.; Hassefras, L.; Eleveld, J.; Grefelman, S.; et al. The behavioral and psychological symptoms of dementia in Down syndrome (BPSD-DS) scale: Comprehensive assessment of psychopathology in Down syndrome. J. Alzheimer Dis. 2018, 63, 797–819. [Google Scholar] [CrossRef] [Green Version]
- Giménez, S.; Videla, L.; Romero, S.; Benejam, B.; Clos, S.; Fernández, S.; Martínez, M.; Carmona-Iragui, M.; Antonijoan, R.M.; Mayos, M.; et al. Prevalence of sleep disorders in adults with Down syndrome: A comparative study of self-reported, actigraphic, and polysomnographic findings. J. Clin. Sleep Med. 2018, 14, 1725–1733. [Google Scholar] [CrossRef]
- Cody, K.A.; Piro-Gambetti, B.; Zammit, M.D.; Christian, B.T.; Handen, B.L.; Klunk, W.E.; Zaman, S.; Johnson, S.C.; Plante, D.T.; Hartley, S.L. Association of sleep with cognition and beta amyloid accumulation in adults with Down syndrome. Neurobiol. Aging 2020, 93, 44–51. [Google Scholar] [CrossRef]
- Temple, V.; Jozsvai, E.; Konstantareas, M.M.; Hewitt, T.-A. Alzheimer dementia in Down′s syndrome: The relevance of cognitive ability. J. Intellect. Disabil. Res. 2001, 45, 47–55. [Google Scholar] [CrossRef]
- Blok, J.B.; Scheirs, J.G.M.; Thijm, N.S. Personality and behavioural changes do not precede memory problems as possible signs of dementia in ageing people with Down syndrome. Int. J. Geriatr. Psychiatry 2017, 32, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Hartley, S.L.; Handen, B.L.; Devenny, D.; Tudorascu, D.; Piro-Gambetti, B.; Zammit, M.D.; Laymon, C.M.; Klunk, W.E.; Zaman, S.; Cohen, A.; et al. Cognitive indicators of transition to preclinical and prodromal stages of Alzheimer’s disease in Down syndrome. Alzheimer Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12096. [Google Scholar] [CrossRef]
- Hithersay, R.; Baksh, R.A.; Startin, C.M.; Wijeratne, P.; Hamburg, S.; Carter, B.; Strydom, A.; The LonDownS Consortium. Optimal age and outcome measures for Alzheimer’s disease prevention trials in people with Down syndrome. Alzheimer Dement. 2021, 17, 595–604. [Google Scholar] [CrossRef]
- Firth, N.C.; Startin, C.M.; Hithersay, R.; Hamburg, S.; Wijeratne, P.A.; Mok, K.Y.; Hardy, J.; Alexander, D.C.; Strydom, A.; The LonDownS Consortium; et al. Aging related cognitive changes associated with Alzheimer’s disease in Down syndrome. Ann. Clin. Transl. Neurol. 2018, 5, 741–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, S.L.; Handen, B.L.; Devenny, D.; Mihaila, I.; Hardison, R.; Lao, P.J.; Klunk, W.E.; Bulova, P.; Johnson, S.C.; Christian, B.T. Cognitive decline and brain amyloid-β accumulation across 3 years in adults with Down syndrome. Neurobiol. Aging 2017, 58, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Startin, C.M.; Hamburg, S.; Hithersay, R.; Al-Janabi, T.; Mok, K.Y.; Hardy, J.; Strydom, A.; LonDownS Consortium. Cognitive markers of preclinical and prodromal Alzheimer’s disease in Down syndrome. Alzheimer Dement. 2019, 15, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Reisberg, B.; Zaudig, M.; Petersen, R.C.; Ritchie, K.; Broich, K.; Belleville, S.; Brodaty, H.; Bennett, D.; Chertkow, H.; et al. Mild cognitive impairment. Lancet 2006, 367, 1262–1270. [Google Scholar] [CrossRef]
- Papp, K.V.; Amariglio, R.E.; Mormino, E.C.; Hedden, T.; Dekhytar, M.; Johnson, K.A.; Sperling, R.A.; Rentz, D.M. Free and cued memory in relation to biomarker-defined abnormalities in clinically normal older adults and those at risk for Alzheimer’s dis-ease. Neuropsychologia 2015, 73, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almkvist, O.; Rodriguez-Vieitez, E.; Thordardottir, S.; Amberla, K.; Axelman, K.; Basun, H.; Kinhult-Ståhlbom, A.; Lilius, L.; Remes, A.; Wahlund, L.-O.; et al. Predicting cognitive decline across four decades in mutation carriers and non-carriers in autosomal-dominant Alzheimer’s disease. J. Int. Neuropsychol. Soc. 2017, 23, 195–203. [Google Scholar] [CrossRef]
- Acosta-Baena, N.; Sepulveda-Falla, D.; Lopera-Gómez, C.M.; Elorza, M.C.J.; Moreno, S.; Aguirre-Acevedo, D.C.; Saldarriaga, A.; Lopera, F. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: A retrospective cohort study. Lancet Neurol. 2011, 10, 213–220. [Google Scholar] [CrossRef]
- Kinnunen, K.M.; Cash, D.M.; Poole, T.; Frost, C.; Benzinger, T.L.; Ahsan, R.L.; Leung, K.K.; Cardoso, M.J.; Modat, M.; Malone, I.B.; et al. Presymptomatic atrophy in autosomal dominant Alzheimer’s disease: A serial magnetic resonance imaging study. Alzheimer Dement. 2018, 14, 43–53. [Google Scholar] [CrossRef]
- Parizkova, M.; Lerch, O.; Moffat, S.D.; Andel, R.; Mazancova, A.F.; Nedelska, Z.; Vyhnalek, M.; Hort, J.; Laczó, J. The effect of Alzheimer’s disease on spatial navigation strategies. Neurobiol. Aging 2018, 64, 107–115. [Google Scholar] [CrossRef]
- Godbolt, A.K.; Cipolotti, L.; Watt, H.; Fox, N.C.; Janssen, J.C.; Rossor, M.N. The natural history of Alzheimer disease. Arch. Neurol. 2004, 61, 1743–1748. [Google Scholar] [CrossRef] [Green Version]
- Lenoir, H.; Siéroff, É. Visual perceptual disorders in Alzheimer’s disease. Geriatr. Psychol. Neuropsychiatry Vieil. 2019, 17, 307–316. [Google Scholar]
- Krinsky-McHale, S.J.; Silverman, W.; Gordon, J.; Devenny, D.A.; Oley, N.; Abramov, I. Vision deficits in adults with Down syndrome. J. Appl. Res. Intellect. Disabil. 2013, 27, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Del Viva, M.M.; Tozzi, A.; Bargagna, S.; Cioni, G. Motion perception deficit in Down Syndrome. Neuropsychologia 2015, 75, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Krinsky-McHale, S.J.; Devenny, D.A.; Kittler, P.; Silverman, W. Selective attention deficits associated with mild cognitive impairment and early stage Alzheimer’s disease in adults with Down syndrome. Am. J. Ment. Retard. 2008, 113, 369–386. [Google Scholar] [CrossRef]
- Medina, L.D.; Woo, E.; Rodriguez-Agudelo, Y.; Maldonado, H.C.; Yi, D.; Coppola, G.; Zhou, Y.; Chui, H.C.; Ringman, J.M. Reaction time and response inhibition in autosomal dominant Alzheimer’s disease. Brain Cogn. 2021, 147, 105656. [Google Scholar] [CrossRef]
- Carvalho, C.L.; Belan, A.F.R.; De Castro, L.R.; Radanovic, M. Analysis of the linguistic profile in down syndrome using the arizona battery for communication disorders of dementia—A pilot study. CoDAS 2018, 30. [Google Scholar] [CrossRef]
- Pulsifer, M.B.; Evans, C.L.; Hom, C.; Krinsky-McHale, S.J.; Silverman, W.; Lai, F.; Lott, I.; Schupf, N.; Wen, J.; Rosas, H.D. Language skills as a predictor of cognitive decline in adults with Down syndrome. Alzheimer Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12080. [Google Scholar] [CrossRef]
- Lindquist, S.G.; Hasholt, L.; Bahl, J.M.C.; Heegaard, N.H.H.; Andersen, B.B.; Nørremølle, A.; Stokholm, J.; Schwartz, M.; Batbayli, M.; Laursen, H.; et al. A novel presenilin 2 mutation (V393M) in early-onset dementia with profound language impairment. Eur. J. Neurol. 2008, 15, 1135–1139. [Google Scholar] [CrossRef]
- Godbolt, A.K.; Beck, J.A.; Collinge, J.; Garrard, P.; Warren, J.D.; Fox, N.C.; Rossor, M.N. A presenilin 1 R278I mutation presenting with language impairment. Neurology 2004, 63, 1702–1704. [Google Scholar] [CrossRef]
- Fonseca, L.M.; Padilla, C.; Jones, E.; Neale, N.; Haddad, G.G.; Mattar, G.P.; Barros, E.; Clare, I.C.H.; Busatto, G.F.; Bottino, C.M.D.C.; et al. Amnestic and non-amnestic symptoms of dementia: An international study of Alzheimer’s disease in people with Down′s syndrome. Int. J. Geriatr. Psychiatry 2020, 35, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Startin, C.M.; Lowe, B.; Hamburg, S.; Hithersay, R.; Strydom, A.; Fisher, E.; Nizetic, D.; Hardy, J.; Tybulewicz, V.; Karmiloff-Smith, A. Validating the cognitive scale for down syndrome (CS-DS) to detect longitudinal cognitive decline in adults with down syndrome. Front. Psychiatry 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krinsky-McHale, S.J.; Zigman, W.B.; Lee, J.H.; Schupf, N.; Pang, D.; Listwan, T.; Kovacs, C.; Silverman, W. Promising outcome measures of early Alzheimer’s dementia in adults with Down syndrome. Alzheimer Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12044. [Google Scholar] [CrossRef]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol. 2011, 10, 785–796. [Google Scholar] [CrossRef] [Green Version]
- Graff-Radford, J.; Yong, K.X.X.; Apostolova, L.G.; Bouwman, F.H.; Carrillo, M.; Dickerson, B.C.; Rabinovici, G.D.; Schott, J.M.; Jones, D.T.; Murray, M.E. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021, 20, 222–234. [Google Scholar] [CrossRef]
- Swanberg, M.M.; Tractenberg, R.E.; Mohs, R.; Thal, L.J.; Cummings, J.L. Executive dysfunction in Alzheimer disease. Arch. Neurol. 2004, 61, 556–560. [Google Scholar] [CrossRef] [Green Version]
- Rowe, J.; Lavender, A.; Turk, V. Cognitive executive function in Down′s syndrome. Br. J. Clin. Psychol. 2006, 45, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.L.; Holland, A.J.; Treppner, P.; Watson, P.; Huppert, F. Executive dysfunction and its association with personality and behaviour changes in the development of Alzheimer’s disease in adults with Down syndrome and mild to moderate learning disabilities. Br. J. Clin. Psychol. 2008, 47, 1–29. [Google Scholar] [CrossRef]
- Finkel, I.S. Behavioral and psychological symptoms of dementia: A current focus for clinicians, researchers, and caregivers. J. Clin. Psychiatry 2001, 62, 3–6. [Google Scholar]
- Dekker, A.D.; Strydom, A.; Coppus, A.M.; Nizetic, D.; Vermeiren, Y.; Naudé, P.J.; Van Dam, D.; Potier, M.-C.; Fortea, J.; De Deyn, P.P. Behavioural and psychological symptoms of dementia in Down syndrome: Early indicators of clinical Alzheimer’s disease? Cortex 2015, 73, 36–61. [Google Scholar] [CrossRef]
- Ball, S.L.; Holland, A.J.; Hon, J.; Huppert, F.A.; Treppner, P.; Watson, P.C. Personality and behaviour changes mark the early stages of Alzheimer’s disease in adults with Down′s syndrome: Findings from a prospective population-based study. Int. J. Geriatr. Psychiatry 2006, 21, 661–673. [Google Scholar] [CrossRef]
- Balsis, S.; Carpenter, B.D.; Storandt, M. Personality change precedes clinical diagnosis of dementia of the Alzheimer type. J. Gerontol. Ser. B 2005, 60, P98–P101. [Google Scholar] [CrossRef] [Green Version]
- Ringman, J.M.; Liang, L.-J.; Zhou, Y.; Vangala, S.; Teng, E.; Kremen, S.; Wharton, D.; Goate, A.; Marcus, D.S.; Farlow, M.; et al. Early behavioural changes in familial Alzheimer’s disease in the Dominantly Inherited Alzheimer Network. Brain 2015, 138, 1036–1045. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.; Oliver, C. The relationship between acquired impairments of executive function and behaviour change in adults with Down syndrome. J. Intellect. Disabil. Res. 2010, 54, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Dykens, E.M. Psychiatric and behavioral disorders in persons with Down syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2007, 13, 272–278. [Google Scholar] [CrossRef]
- Määttä, T.; Tervo-Määttä, T.; Taanila, A.; Kaski, M.; Iivanainen, M. Adaptive behaviour change and health in adults with Down syndrome: A prospective clinical follow-up study. In Pharmacology and Nutritional Intervention in the Treatment of Disease; InTech Open: Rijeka, Croatia, 2014. [Google Scholar]
- Nelson, L.D.; Orme, D.; Osann, K.; Lott, I.T. Neurological changes and emotional functioning in adults with Down syndrome. J. Intellect. Disabil. Res. 2001, 45, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, L.M.; Yokomizo, J.E.; Bottino, C.M.; Fuentes, D. Frontal lobe degeneration in adults with Down syndrome and Alzheimer’s disease: A review. Dement. Geriatr. Cogn. Disord. 2016, 41, 123–136. [Google Scholar] [CrossRef]
- Beacher, F.; Daly, E.; Simmons, A.; Prasher, V.; Morris, R.; Robinson, C.; Lovestone, S.; Murphy, K.; Murphy, D. Brain anatomy and ageing in non-demented adults with Down′s syndrome: An in vivo MRI study. Psychol. Med. 2009, 40, 611–619. [Google Scholar] [CrossRef]
- Powell, D.; Caban-Holt, A.; Jicha, G.; Robertson, W.; Davis, R.; Gold, B.T.; Schmitt, F.A.; Head, E. Frontal white matter integrity in adults with Down syndrome with and without dementia. Neurobiol. Aging 2014, 35, 1562–1569. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, L.M.; de Oliveira, M.C.; de Figueiredo Ferreira Guilhoto, L.M.; Cavalheiro, E.A.; Bottino, C.M. Bereavement and be-havioral changes as risk factors for cognitive decline in adults with Down syndrome. Neuropsychiatr. Dis. Treat. 2014, 10, 2209–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, L.M.; Mattar, G.P.; Haddad, G.G.; Gonçalves, A.S.; Miguel, A.D.Q.C.; Guilhoto, L.M.; Zaman, S.; Holland, A.J.; Bottino, C.M.D.C.; Hoexter, M.Q. Frontal-subcortical behaviors during Alzheimer’s disease in individuals with Down syndrome. Neurobiol. Aging 2019, 78, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Stout, J.C.; Wyman, M.F.; Johnson, S.A.; Peavy, G.M.; Salmon, D.P. Frontal behavioral syndromes and functional status in probable Alzheimer disease. Am. J. Geriatr. Psychiatry 2003, 11, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.S.; Kumar, S.; Demichele-Sweet, M.A.A.; Sweet, R.A. Psychosis in Alzheimer’s disease. Biol. Psychiatry 2014, 75, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Vöglein, J.; Paumier, K.; Jucker, M.; Preische, O.; McDade, E.; Hassenstab, J.; Benzinger, T.L.; Noble, J.; Berman, S.B.; Graff-Radford, N.R.; et al. Clinical, pathophysiological and genetic features of motor symptoms in autosomal dominant Alzheimer’s disease. Brain 2019, 142, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Karlstrom, H.; Brooks, W.S.; Kwok, J.B.J.; Broe, G.A.; Kril, J.J.; McCann, H.; Halliday, G.M.; Schofield, P.R. Variable phenotype of Alzheimer’s disease with spastic paraparesis. J. Neurochem. 2007, 104, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; Baker, M.; McGowan, E.; Lewis, P.; Hutton, M.; Crook, R.; Wood, N.W.; Kumar-Singh, S.; Geddes, J.; Swash, M.; et al. Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann. Neurol. 2000, 48, 806–808. [Google Scholar] [CrossRef]
- Soosman, S.K.; Joseph-Mathurin, N.; Braskie, M.N.; Bordelon, Y.M.; Wharton, D.; Casado, M.; Coppola, G.; McCallum, H.; Nuwer, M.; Coutin-Churchman, P.; et al. Widespread white matter and conduction defects in PSEN1-related spastic paraparesis. Neurobiol. Aging 2016, 47, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, M.; Ryman, D.C.; McDade, E.; Jasielec, M.S.; Buckles, V.D.; Cairns, N.J.; Fagan, A.M.; Goate, A.; Marcus, D.S.; Xiong, C.; et al. Neurological manifestations of autosomal dominant familial Alzheimer’s disease: A comparison of the published literature with the Dominantly Inherited Alzheimer Network observational study (DIAN-OBS). Lancet Neurol. 2016, 15, 1317–1325. [Google Scholar] [CrossRef] [Green Version]
- Cabrejo, L.; Guyant-Maréchal, L.; Laquerrière, A.; Vercelletto, M.; De La Fournière, F.; Thomas-Antérion, C.; Verny, C.; Letournel, F.; Pasquier, F.; Vital, A.; et al. Phenotype associated with APP duplication in five families. Brain 2006, 129, 2966–2976. [Google Scholar] [CrossRef] [Green Version]
- Meeus, M.; Kenis, S.; Wojciechowski, M.; Ceulemans, B. Epilepsy in children with Down syndrome: Not so benign as generally accepted. Acta Neurol. Belg. 2015, 115, 569–573. [Google Scholar] [CrossRef]
- Gholipour, T.; Mitchell, S.; Sarkis, R.A.; Chemali, Z. The clinical and neurobehavioral course of Down syndrome and dementia with or without new-onset epilepsy. Epilepsy Behav. 2017, 68, 11–16. [Google Scholar] [CrossRef]
- Altuna, M.; Giménez, S.; Fortea, J. Epilepsy in Down syndrome: A highly prevalent comorbidity. J. Clin. Med. 2021, 10, 2776. [Google Scholar] [CrossRef]
- Xu, Y.; Lavrencic, L.; Radford, K.; Booth, A.; Yoshimura, S.; Anstey, K.J.; Anderson, C.S.; Peters, R. Systematic review of coexistent epileptic seizures and Alzheimer’s disease: Incidence and prevalence. J. Am. Geriatr. Soc. 2021. [Google Scholar] [CrossRef]
- Alexander, M.; Petri, H.; Ding, Y.; Wandel, C.; Khwaja, O.; Foskett, N. Morbidity and medication in a large population of individuals with Down syndrome compared to the general population. Dev. Med. Child Neurol. 2015, 58, 246–254. [Google Scholar] [CrossRef]
- Menéndez, M. Down syndrome, Alzheimer’s disease and seizures. Brain Dev. 2005, 27, 246–252. [Google Scholar] [CrossRef]
- Aller-Alvarez, J.S.; Menéndez-González, M.; Ribacoba-Montero, R.; Salvado, M.; Vega, V.; Suárez-Moro, R.; Sueiras, M.; Toledo, M.; Salas-Puig, J.; Álvarez-Sabin, J. Epilepsia mioclónica en el síndrome de Down y en la enfermedad de Alzheimer. Neurologia 2017, 32, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palop, J.J.; Mucke, L. Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [Green Version]
- De Simone, R.; Puig, X.S.; Gélisse, P.; Crespel, A.; Genton, P. Senile myoclonic epilepsy: Delineation of a common condition associated with Alzheimer’s disease in Down syndrome. Seizure 2010, 19, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Rissman, R.A.; De Blas, A.L.; Armstrong, D.M. GABAA receptors in aging and Alzheimer’s disease. J. Neurochem. 2007, 103, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A.; Magara, S.; Cancedda, L. The GABAergic hypothesis for cognitive disabilities in Down syndrome. Front. Cell. Neurosci. 2017, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- Anderson-Mooney, A.J.; Schmitt, F.A.; Head, E.; Lott, I.T.; Heilman, K.M. Gait dyspraxia as a clinical marker of cognitive decline in Down syndrome: A review of theory and proposed mechanisms. Brain Cogn. 2016, 104, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Chhatwal, J.P.; Schultz, A.P.; Johnson, K.; Benzinger, T.L.; Jack, C.; Ances, B.M.; Thompson, P.; Saykin, A.J.; Correia, S.; Marcus, D.S.; et al. Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology 2013, 81, 736–744. [Google Scholar] [CrossRef]
- Thomas, J.B.; Brier, M.R.; Bateman, R.J.; Snyder, A.Z.; Benzinger, T.L.; Xiong, C.; Raichle, M.; Holtzman, D.M.; Sperling, R.A.; Mayeux, R.; et al. Functional connectivity in autosomal dominant and late-onset Alzheimer disease. JAMA Neurol. 2014, 71, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, L.R.; Vatansever, D.; Annus, T.; Williams, G.B.; Hong, Y.T.; Fryer, T.D.; Nestor, P.J.; Holland, A.J.; Zaman, S.H. Differential effects of Down′s syndrome and Alzheimer’s neuropathology on default mode connectivity. Hum. Brain. Mapp. 2019, 40, 4551–4563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araque Caballero, M.Á.; Suárez-Calvet, M.; Duering, M.; Franzmeier, N.; Benzinger, T.; Fagan, A.M.; Bateman, R.J.; Jack, C.R.; Levin, J.; Dichgans, M.; et al. White matter diffusion alterations precede symptom onset in autosomal dominant Alzheimer’s disease. Brain 2018, 141, 3065–3080. [Google Scholar] [CrossRef] [PubMed]
- Haier, R.; Head, K.; Head, E.; Lott, I. Neuroimaging of individuals with Down′s syndrome at-risk for dementia: Evidence for possible compensatory events. NeuroImage 2008, 39, 1324–1332. [Google Scholar] [CrossRef] [Green Version]
- Haier, R.J.; Alkire, M.T.; White, N.S.; Uncapher, M.R.; Head, E.; Lott, I.T.; Cotman, C.W. Temporal cortex hypermetabolism in Down syndrome prior to the onset of dementia. Neurology 2003, 61, 1673–1679. [Google Scholar] [CrossRef]
- Clifford, R.J., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Alcolea, D.; Delaby, C.; Muñoz, L.; Torres, S.; Estellés, T.; Zhu, N.; Barroeta, I.; Carmona-Iragui, M.; Illán-Gala, I.; Santos-Santos, M.Á.; et al. Use of plasma biomarkers for AT(N) classification of neurodegenerative dementias. J. Neurol. Neurosurg. Psychiatry 2021. [Google Scholar] [CrossRef]
- Fortea, J.; Vilaplana, E.; Carmona-Iragui, M.; Benejam, B.; Videla, L.; Barroeta, I.; Fernández, S.; Altuna-Azkargorta, M.; Pegueroles, J.; Montal, V.; et al. Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet 2020, 395, 1988–1997. [Google Scholar] [CrossRef]
- Benzinger, T.L.S.; Blazey, T.; Jack, C.R.; Koeppe, R.A.; Su, Y.; Xiong, C.; Raichle, M.E.; Snyder, A.Z.; Ances, B.; Bateman, R.; et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, E4502–E4509. [Google Scholar] [CrossRef] [Green Version]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Villemagne, V.L.; Ataka, S.; Mizuno, T.; Brooks, W.S.; Wada, Y.; Kondo, M.; Jones, G.; Watanabe, Y.; Mulligan, R.; Nakagawa, M.; et al. High striatal amyloid β-peptide deposition across different autosomal Alzheimer disease mutation types. Arch. Neurol. 2009, 66, 1537–1544. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Chen, K.; Quiroz, Y.T.; Jakimovich, L.J.; Gomez, M.G.; Langois, C.M.; Langbaum, J.B.; Ayutyanont, N.; Roontiva, A.; Thiyyagura, P.; et al. Florbetapir PET analysis of amyloid-β deposition in the presenilin 1 E280A au-tosomal dominant Alzheimer’s disease kindred: A cross-sectional study. Lancet Neurol. 2012, 11, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.D.; McDade, E.; Christian, B.; Price, J.; Mathis, C.; Klunk, W.; Handen, B.L. Early striatal amyloid deposition distin-guishes Down syndrome and autosomal dominant Alzheimer’s disease from late-onset amyloid deposition. Alzheimer Dement. 2018, 14, 743–750. [Google Scholar] [CrossRef]
- Nelson, L.D.; Siddarth, P.; Kepe, V.; Scheibel, K.E.; Huang, S.C.; Barrio, J.R.; Small, G.W. Positron emission tomography of brain β-amyloid and tau levels in adults with Down syndrome. Arch. Neurol. 2011, 68, 768–774. [Google Scholar] [CrossRef]
- Annus, T.; Wilson, L.R.; Hong, Y.T.; Acosta–Cabronero, J.; Fryer, T.D.; Cardenas–Blanco, A.; Smith, R.; Boros, I.; Coles, J.P.; Aigbirhio, F.I.; et al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimer Dement. 2016, 12, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grothe, M.J.; Barthel, H.; Sepulcre, J.; Dyrba, M.; Sabri, O.; Teipel, S.J.; Alzheimer’s Disease Neuroimaging Initiative. In Vivo staging of regional amyloid deposition. Neurology 2017, 89, 2031–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aß-deposition in the human brain and its relevance for the development of AD. Neurology 2020, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Head, E.; Doran, E.; Nistor, M.; Hill, M.; Schmitt, F.A.; Haier, R.J.; Lott, I.T. Plasma amyloid-β as a function of age, level of intellectual disability, and presence of dementia in Down syndrome. J. Alzheimer Dis. 2011, 23, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Schupf, N.; Patel, B.; Pang, D.; Zigman, W.; Silverman, W.; Mehta, P.D.; Mayeux, R. Elevated plasma β-amyloid peptide Aβ42 levels, incident dementia, and mortality in Down syndrome. Arch. Neurol. 2007, 64, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.L.; Hanney, M.; Francis, P.T.; Ballard, C.G. Amyloid β concentrations in older people with Down syndrome and dementia. Neurosci. Lett. 2009, 451, 162–164. [Google Scholar] [CrossRef]
- Quiroz, Y.T.; Schultz, A.P.; Chen, K.; Protas, H.D.; Brickhouse, M.; Fleisher, A.S.; Langbaum, J.B.; Thiyyagura, P.; Fagan, A.M.; Shah, A.R.; et al. Brain imaging and blood biomarker abnormalities in children with autosomal dominant Alzheimer disease a cross-sectional study. JAMA Neurol. 2015, 72, 912–919. [Google Scholar] [CrossRef]
- Startin, C.M.; LonDownS Consortium; Ashton, N.J.; Hamburg, S.; Hithersay, R.; Wiseman, F.K.; Mok, K.Y.; Hardy, J.; Lleó, A.; Lovestone, S.; et al. Plasma biomarkers for amyloid, tau, and cytokines in Down syndrome and sporadic Alzheimer’s disease. Alzheimer Res. Ther. 2019, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Mayeux, R.; Honig, L.S.; Tang, M.X.; Manly, J.; Stern, Y.; Schupf, N.; Mehta, P.D. Plasma Aβ40 and Aβ42 and Alzheimer’s disease: Relation to age, mortality, and risk. Neurology 2003, 61, 1185–1190. [Google Scholar] [CrossRef]
- Van Oijen, M.; Hofman, A.; Soares, H.D.; Koudstaal, P.J.; Breteler, M.M. Plasma Aβ1-40 and Aβ1-42 and the risk of dementia: A prospective case-cohort study. Lancet Neurol 2006, 5, 655–660. [Google Scholar] [CrossRef]
- Fagan, A.M.; Xiong, C.; Jasielec, M.S.; Bateman, R.; Goate, A.; Benzinger, T.; Ghetti, B.; Martins, R.; Masters, C.L.; Mayeux, R.; et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci. Transl. Med. 2014, 6, 226ra30. [Google Scholar] [CrossRef] [Green Version]
- Schupf, N.; Zigman, W.B.; Tang, M.X.; Pang, D.; Mayeux, R.; Mehta, P.; Silverman, W. Change in plasma Aβ peptides and onset of dementia in adults with Down syndrome. Neurology 2010, 75, 1639–1644. [Google Scholar] [CrossRef] [Green Version]
- Prasher, V.P.; Sajith, S.G.; Mehta, P.; Zigman, W.B.; Schupf, N. Plasma β-amyloid and duration of Alzheimer’s disease in adults with Down syndrome. Int. J. Geriatr. Psychiatry 2010, 25, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Coppus, A.M.; Schuur, M.; Vergeer, J.; Janssens, A.C.J.; Oostra, B.A.; Verbeek, M.M.; van Duijn, C.M. Plasma β amyloid and the risk of Alzheimer’s disease in Down syndrome. Neurobiol. Aging 2012, 33, 1988–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhajraf, F.; Ness, D.; Hye, A.; Strydom, A. Plasma amyloid and tau as dementia biomarkers in Down syndrome: Systematic review and meta-analyses. Dev. Neurobiol. 2019, 79, 684–698. [Google Scholar] [CrossRef]
- Englund, H.; Annerén, G.; Gustafsson, J.; Wester, U.; Wiltfang, J.; Lannfelt, L.; Blennow, K.; Höglund, K. Increase in β-amyloid levels in cerebrospinal fluid of children with Down syndrome. Dement. Geriatr. Cogn. Disord. 2007, 24, 369–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, A.; Fortea, J.; Blesa, R.; De Deyn, P.P. Cerebrospinal fluid biomarkers for Alzheimer’s disease in Down syndrome. Alzheimer Dementia Diagn. Assess. Dis. Monit. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Hyman, B.T.; West, H.L.; Rebeck, G.W.; Buldyrev, S.V.; Mantegna, R.N.; Ukleja, M.; Havlin, S.; Stanley, H.E. Quantitative analysis of senile plaques in Alzheimer disease: Observation of log-normal size distribution and molecular epidemiology of differences associated with apolipoprotein E genotype and trisomy 21 (Down syndrome). Proc. Natl. Acad. Sci. USA 1995, 92, 3586–3590. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, C.; McCann, H.; Halliday, G.M. Variations in the neuropathology of familial Alzheimer’s disease. Acta Neuropathol. 2009, 118, 37–52. [Google Scholar] [CrossRef]
- Armstrong, A.R. Size frequency distributions of β-amyloid (Aβ) deposits: A comparative study of four neurodegenerative disorders. Folia Neuropathol. 2012, 3, 240–249. [Google Scholar] [CrossRef]
- Ohara, P.T. Electron microscopical study of the brain in Down′s syndrome. Brain 1972, 95, 681–684. [Google Scholar] [CrossRef]
- Gross, T.J.; Doran, E.; Cheema, A.K.; Head, E.; Lott, I.T.; Mapstone, M. Plasma metabolites related to cellular energy metabolism are altered in adults with Down syndrome and Alzheimer’s disease. Dev. Neurobiol. 2019, 79, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Ishii, K.; Sasaki, M.; Hosaka, K.; Mori, T.; Matsui, M.; Hirono, N.; Mori, E. Differences in cerebral metabolic im-pairment between early and late onset types of Alzheimer’s disease. J. Neurol. Sci. 2002, 200, 27–32. [Google Scholar] [CrossRef]
- Lao, P.J.; Brickman, A.M. Multimodal neuroimaging study of cerebrovascular disease, amyloid deposition, and neuro-degeneration in Alzheimer’s disease progression. Alzheimer Dement. Diagn. Assess. Dis. Monit. 2018, 10, 638–646. [Google Scholar]
- Zammit, M.D.; Laymon, C.M.; Betthauser, T.J.; Cody, K.A.; Tudorascu, D.L.; Minhas, D.S.; Sabbagh, M.N.; Johnson, S.C.; Zaman, S.H.; Mathis, C.A.; et al. Amyloid accumulation in Down syndrome measured with amyloid load. Alzheimer Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12020. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.C.; Lukic, A.S.; Andrews, R.D.; Marendic, B.; Brewer, J.; Rissman, R.A.; Mosconi, L.; Strother, S.C.; Wernick, M.N.; Mobley, W.C.; et al. Dissociation of Down syndrome and Alzheimer’s disease effects with imaging. Alzheimer Dement. Transl. Res. Clin. Interv. 2016, 2, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengyel, Z.; Balogh, E.; Emri, M.; Szikszai, E.; Kollár, J.; Sikula, J.; Ésik, O.; Trón, L.; Oláh, É. Pattern of increased cerebral FDG uptake in Down syndrome patients. Pediatr. Neurol. 2006, 34, 270–275. [Google Scholar] [CrossRef]
- Chan, D.; Janssen, J.C.; Whitwell, J.L.; Watt, H.C.; Jenkins, R.; Frost, C.; Rossor, M.N.; Fox, N.C. Change in rates of cerebral atrophy over time in early-onset Alzheimer’s disease: Longitudinal MRI study. Lancet 2003, 362, 1121–1122. [Google Scholar] [CrossRef]
- Rafii, M.S.; Wishnek, H.; Brewer, J.B.; Donohue, M.C.; Ness, S.; Mobley, W.C.; Aisen, P.S.; Rissman, R.A. The Down Syndrome Biomarker Initiative (DSBI) pilot: Proof of concept for deep phenotyping of Alzheimer’s disease biomarkers in Down syndrome. Front. Behav. Neurosci. 2015, 9, 239. [Google Scholar] [CrossRef] [Green Version]
- Neale, N.; Padilla, C.; Fonseca, L.; Holland, T.; Zaman, S. Neuroimaging and other modalities to assess Alzheimer’s disease in Down syndrome. NeuroImage Clin. 2018, 17, 263–271. [Google Scholar] [CrossRef]
- Annus, T.; Wilson, L.R.; Acosta-Cabronero, J.; Cardenas-Blanco, A.; Hong, Y.T.; Fryer, T.D.; Coles, J.P.; Menon, D.K.; Zaman, S.H.; Holland, A.J.; et al. The Down syndrome brain in the presence and absence of fibrillar β-amyloidosis. Neurobiol. Aging 2017, 53, 11–19. [Google Scholar] [CrossRef]
- Weis, S.; Weber, G.; Neuhold, A.; Rett, A. Down syndrome: MR quantification of brain structures and comparison with normal control subjects. AJNR Am. J. Neuroradiol. 1991, 12, 1207–1211. [Google Scholar] [PubMed]
- Abrahamson, E.E.; Head, E.; Lott, I.T.; Handen, B.L.; Mufson, E.J.; Christian, B.T.; Klunk, W.E.; Ikonomovic, M.D. Neuropathological correlates of amyloid PET imaging in Down syndrome. Dev. Neurobiol. 2019, 79, 750–766. [Google Scholar] [CrossRef] [PubMed]
- Tudorascu, D.L.; Laymon, C.M.; Zammit, M.; Minhas, D.S.; Anderson, S.J.; Ellison, P.A.; Zaman, S.; Ances, B.M.; Sabbagh, M.; Johnson, S.C.; et al. Relationship of amyloid beta and neurofibrillary tau deposition in Neu-rodegeneration in Aging Down Syndrome (NiAD) study at baseline. Alzheimer Dement. Transl. Res. Clin. Interv. 2020, 6, 1–8. [Google Scholar]
- Rafii, M.S.; Lukic, A.S.; Andrews, R.D.; Brewer, J.; Rissman, R.A.; Strother, S.C.; Wernick, M.N.; Pennington, C.; Mobley, W.C.; Ness, S.; et al. PET imaging of tau pathology and relationship to amyloid, longitudinal MRI, and cognitive change in Down syndrome: Results from the Down Syndrome Biomarker Initiative (DSBI). J. Alzheimer Dis. 2017, 60, 439–450. [Google Scholar] [CrossRef]
- Gordon, B.; Blazey, T.M.; Christensen, J.; Dincer, A.; Flores, S.; Keefe, S.; Chen, C.; Su, Y.; McDade, E.M.; Wang, G.; et al. Tau PET in autosomal dominant Alzheimer’s disease: Relationship with cognition, dementia and other biomarkers. Brain 2019, 142, 1063–1076. [Google Scholar] [CrossRef]
- Molina, L.; Touchon, J.; Herpé, M.; Lefranc, D.; Duplan, L.; Cristol, J.P.; Sabatier, R.; Vermersch, P.; Pau, B.; Mourton-Gilles, C. Tau and apo E in CSF: Potential aid for discriminating Alzheimer’s disease from other dementias. Neuroreport 1999, 10, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- McDade, E.; Wang, G.; Gordon, B.A.; Hassenstab, J.; Benzinger, T.L.; Buckles, V.; Fagan, A.M.; Holtzman, D.M.; Cairns, N.J.; Goate, A.M.; et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology 2018, 91, e1295–e1306. [Google Scholar] [CrossRef]
- Fortea, J.; Carmona-Iragui, M.; Benejam, B.; Fernández, S.; Videla, L.; Barroeta, I.; Alcolea, D.; Pegueroles, J.; Muñoz, L.; Belbin, O.; et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet Neurol. 2018, 17, 860–869. [Google Scholar] [CrossRef]
- Lleó, A.; Zetterberg, H.; Pegueroles, J.; Karikari, T.K.; Carmona-Iragui, M.; Ashton, N.J.; Montal, V.; Barroeta, I.; Lantero-Rodríguez, J.; Videla, L.; et al. Phosphorylated tau181 in plasma as a potential biomarker for Alzheimer’s disease in adults with Down syndrome. Nat. Commun. 2021, 12, 1–8. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- McDade, E.; Bateman, R.J. Tau positron emission tomography in autosomal dominant Alzheimer disease small windows, big picture. JAMA Neurol. 2018, 75, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Margallo-Lana, M.L.; Moore, P.B.; Kay, D.W.K.; Perry, R.H.; Reid, B.E.; Berney, T.P.; Tyrer, S.P. Fifteen-year follow-up of 92 hospi-talized adults with Down′s syndrome: Incidence of cognitive decline, its relationship to age and neuropathology. J. Intellect. Disabil. Res. 2007, 51, 463–477. [Google Scholar] [CrossRef]
- Jellinger, K.A. Pathobiological subtypes of Alzheimer disease. Dement. Geriatr. Cogn. Disord. 2020, 49, 321–333. [Google Scholar] [CrossRef]
- Vogel, J.W.; Initiative, T.A.D.N.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 27, 871–881. [Google Scholar] [CrossRef]
- Young, A.L.; Marinescu, R.V.; Oxtoby, N.P.; Bocchetta, M.; Yong, K.; Firth, N.C.; Cash, D.M.; Thomas, D.L.; Dick, K.M.; Cardoso, J.; et al. Uncovering the heterogeneity and temporal complexity of neurodegen-erative diseases with subtype and stage inference. Nat. Commun. 2018, 9, 4273. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A.; Attems, J. Challenges of multimorbidity of the aging brain: A critical update. J. Neural Transm. 2015, 122, 505–521. [Google Scholar] [CrossRef]
- Davidson, Y.S.; Robinson, A.; Prasher, V.P.; Mann, D.M.A. The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer’s disease in individuals with Down syndrome. Acta Neuropathol. Commun. 2018, 6, 56. [Google Scholar] [CrossRef]
- Prasher, V.P.; Airuehia, E.; Carey, M. The first confirmed case of Down syndrome with dementia with lewy bodies. J. Appl. Res. Intellect. Disabil. 2010, 23, 296–300. [Google Scholar] [CrossRef]
- Cairns, N.J.; Perrin, R.J.; Franklin, E.E.; Carter, D.; Vincent, B.; Xie, M.; Bateman, R.; Benzinger, T.; Friedrichsen, K.; Brooks, W.S.; et al. Neuropathologic assessment of participants in two multi-center longitudinal observational studies: The Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology 2015, 35, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahimi, J.; Kovacs, G.G. Prevalence of mixed pathologies in the aging brain. Alzheimer Res. Ther. 2014, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Davidson, Y.S.; Raby, S.; Foulds, P.G.; Robinson, A.; Thompson, J.C.; Sikkink, S.; Yusuf, I.; Amin, H.; DuPlessis, D.; Troakes, C.; et al. TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down′s syndrome: Association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol. 2011, 122, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Lippa, C.F.; Rosso, A.; Stutzbach, L.D.; Neumann, M.; Lee, V.M.-Y.; Trojanowski, J.Q. Transactive response DNA-binding protein 43 burden in familial Alzheimer disease and Down syndrome. Arch. Neurol. 2009, 66, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant Age-related TDP-43 Encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, C.; Fahimi, A.; Das, D.; SMojabi, F.; Ponnusamy, R.; Salehi, A. Noradrenergic system in Down syndrome and Alz-heimer′s disease a target for therapy. Curr. Alzheimer Res. 2015, 13, 68–83. [Google Scholar] [CrossRef]
- Hartley, D.; Blumenthal, T.; Carrillo, M.; DiPaolo, G.; Esralew, L.; Gardiner, K.; Granholm, A.-C.; Iqbal, K.; Krams, M.; Lemere, C.; et al. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimer Dement. 2015, 11, 700–709. [Google Scholar] [CrossRef] [Green Version]
- Iulita, M.F.; Cuello, A.C. Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol. Sci. 2014, 35, 338–348. [Google Scholar] [CrossRef]
- Florencia Iulita, M.; Claudio Cuello, A. The NGF metabolic pathway in the CNS and its dysregulation in Down syndrome and Alzheimer’s disease. Curr. Alzheimer Res. 2015, 13, 53–67. [Google Scholar] [CrossRef]
- Barone, E.; Head, E.; Butterfield, D.A.; Perluigi, M. HNE-modified proteins in Down syndrome: Involvement in development of Alzheimer disease neuropathology. Free Radic. Biol. Med. 2017, 111, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Zana, M.; Janka, Z.; Kálmán, J. Oxidative stress: A bridge between Down′s syndrome and Alzheimer’s disease. Neurobiol. Aging 2007, 28, 648–676. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Griffin, W.S.T. Down′s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflamm. 2013, 10, 864. [Google Scholar] [CrossRef] [Green Version]
- Griffin, W.S.T. Inflammation and neurodegenerative diseases. Am. J. Clin. Nutr. 2006, 83, 470S–474S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, A.; Wolf, T.; Sitzmann, A.; Willette, A.A. Neuroinflammation in Alzheimer’s disease: Pleiotropic roles for cytokines and neuronal pentraxins. Behav. Brain Res. 2018, 347, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.G.; Serra, P.; Ghiani, A.; Manca, E.; Hardoy, M.C.; Del Giacco, G.S.; Diaz, G.; Carpiniello, B.; Manconi, P.E. Chemokines and pro-inflammatory cytokines in Down′s syndrome: An early marker for Alzheimer-type dementia? Psychother. Psychosom. 2002, 71, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Head, E.; Azizeh, B.Y.; Lott, I.T.; Tenner, A.J.; Cotman, C.W.; Cribbs, D.H. Complement association with neurons and β-amyloid deposition in the brains of aged individuals with Down syndrome. Neurobiol. Dis. 2001, 8, 252–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portelius, E.; Soininen, H.; Andreasson, U.; Zetterberg, H.; Persson, R.; Karlsson, G.; Blennow, K.; Herukka, S.-K.; Mattsson, N. Exploring Alzheimer molecular pathology in Down′s syndrome cerebrospinal fluid. Neurodegener. Dis. 2014, 14, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Hurban, J.; Helman, A.M.; Sudduth, T.L.; McCarty, K.L.; Beckett, T.L.; Ferrell, J.C.; Murphy, M.P.; Abner, E.L.; Schmitt, F.A.; et al. Down syndrome individuals with Alzheimer’s disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2468–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppus, A.M.; Fekkes, D.; Verhoeven, W.M.; Evenhuis, H.M.; Van Duijn, C.M. Neopterin and the risk of dementia in persons with Down syndrome. Neurosci. Lett. 2009, 458, 60–64. [Google Scholar] [CrossRef]
- Zis, P.; Strydom, A.; Buckley, D.; Adekitan, D.; McHugh, P. Cognitive ability in Down syndrome and its relationship to urinary neopterin, a marker of activated cellular immunity. Neurosci. Lett. 2016, 636, 254–257. [Google Scholar] [CrossRef]
- Verstegen, R.H.J.; Chang, K.J.J.; Kusters, M.A.A. Clinical implications of immune-mediated diseases in children with Down syndrome. Pediatr. Allergy Immunol. 2019, 31, 117–123. [Google Scholar] [CrossRef]
- Barnhart, R.C.; Connolly, B. Aging and Down syndrome: Implications for physical therapy. Phys. Ther. 2007, 87, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Chaushu, S.; Zigmond, M.; Yefenof, E.; Stabholz, A.; Shapira, J.; Merrick, J.; Bachrach, G. Age-dependent deficiency in saliva and salivary antibodies secretion in Down′s syndrome. Arch. Oral Biol. 2007, 52, 1088–1096. [Google Scholar] [CrossRef]
- Murakami, J.; Kato, T.; Kawai, S.; Akiyama, S.; Amano, A.; Morisaki, I. Cellular motility of Down syndrome gingival fibro-blasts is susceptible to impairment by Porphyromonas gingivalis invasion. J. Periodontol. 2008, 79, 721–727. [Google Scholar] [CrossRef]
- Carrada, C.F.; Scalioni, F.A.R.; Cesar, D.E.; Devito, K.; Ribeiro, L.C.; Ribeiro, R.A. Salivary Periodontopathic bacteria in children and adolescents with Down syndrome. PLoS ONE 2016, 11, e0162988. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [Green Version]
- Kamer, A.R.; Fortea, J.O.; Videla, S.; Mayoral, A.; Janal, M.; Iragui, M.C.; Benejam, B.; Craig, R.G.; Saxena, D.; Corby, P.; et al. Periodontal disease′s contribution to Alzheimer’s disease progression in Down syndrome. Alzheimer Dement. Diagn. Assess. Dis. Monit. 2016, 2, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, F.; Pupo, G.; Tramutola, A.; Giorgi, A.; Schininà, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; Perluigi, M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: Clues for understanding the development of Alz-heimer disease. Free Radic. Biol. Med. 2014, 71, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zis, P.; Dickinson, M.; Shende, S.; Walker, Z.; Strydom, A. Oxidative stress and memory decline in adults with Down syndrome: Longitudinal study. J. Alzheimer Dis. 2012, 31, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Zis, P.; McHugh, P.; McQuillin, A.; Praticò, D.; Dickinson, M.; Shende, S.; Walker, Z.; Strydom, A. Memory decline in Down syndrome and its relationship to iPF2alpha, a urinary marker of oxidative stress. PLoS ONE 2014, 9, e97709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, E.; Lott, I.T. Down syndrome and beta-amyloid deposition. Curr. Opin. Neurol. 2004, 17, 95–100. [Google Scholar] [CrossRef]
- Di Domenico, F.; Coccia, R.; Cocciolo, A.; Murphy, M.P.; Cenini, G.; Head, E.; Butterfield, D.A.; Giorgi, A.; Schinina, M.E.; Mancuso, C.; et al. Impairment of proteostasis network in Down syndrome prior to the devel-opment of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Coskun, P.E.; Wyrembak, J.; Derbereva, O.; Melkonian, G.; Doran, E.; Lott, I.T.; Head, E.; Cotman, C.W.; Wallace, D.C. Systemic mitochondrial dysfunction and the etiology of Alzheimer’s disease and Down syndrome dementia. J. Alzheimer Dis. 2010, 20, S293–S310. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LOAD | ADAD | DSAD | ||||
|---|---|---|---|---|---|---|
| Clinical Features | ||||||
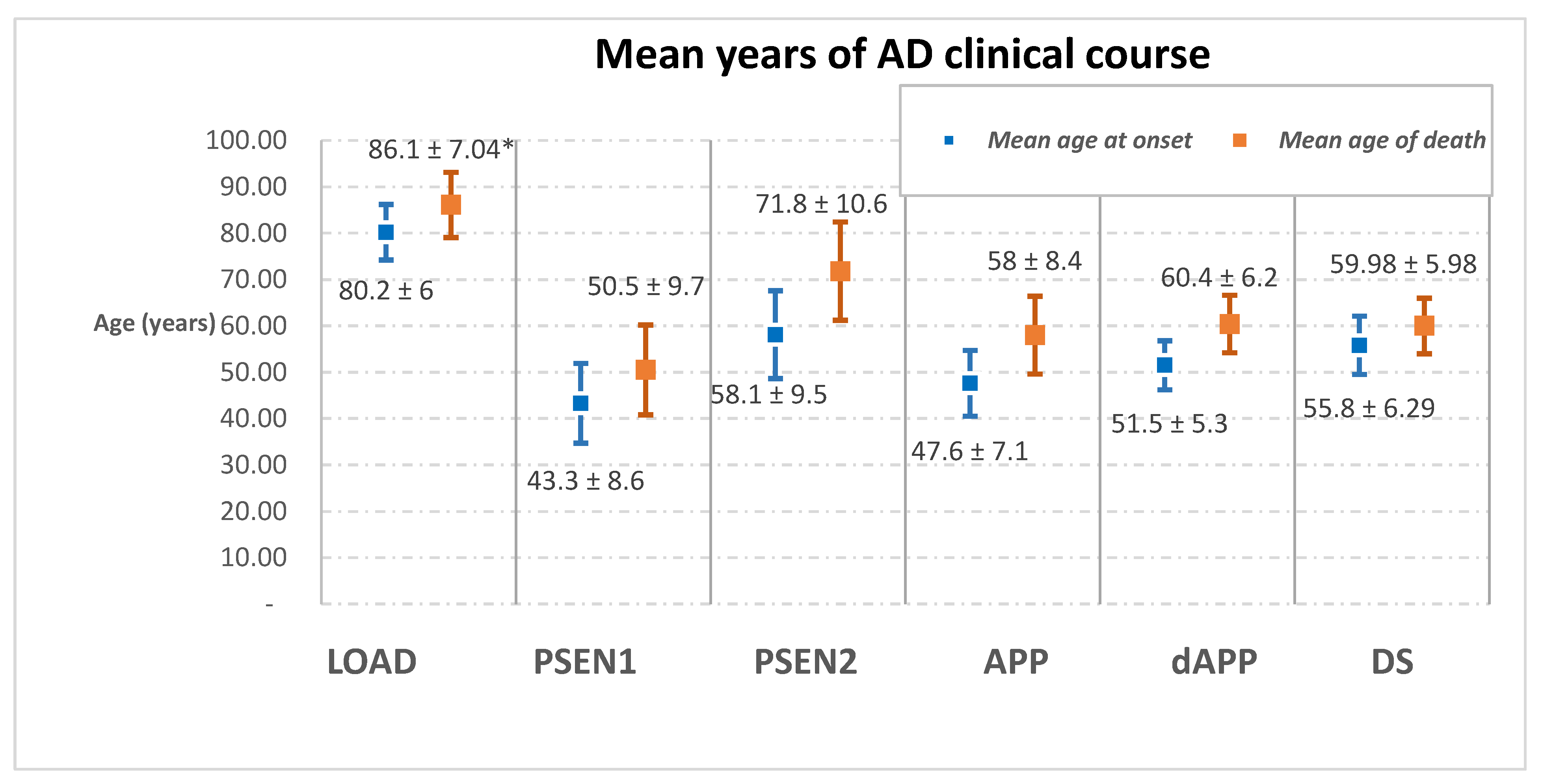
| Mean age of onset | 80.2 ± 6 | PSEN1 | PSEN2 | APP | dAPP | 55.8 ± 6.29 |
| 43.3 ± 8.6 | 58.1 ± 9.5 | 47.6 ± 7.1 | 51.5 ± 5.3 | |||
| Mean age of death | 86.1 ± 7.04 | 50.5 ± 9.7 | 71.8 ± 10.6 | 58 ± 8.4 | 60.4 ± 6.2 | 59.98 ± 5.98 |
| Cerebral Amyloid Angiopathy | Low | High (with higher prevalence in dAPP) | High | |||
| Amnestic phenotypes | Early episodic memory loss Early loss of visuoperceptual skills Attention deficits Language deterioration | Early episodic memory loss Late loss of visuoperceptual skills Attention deficits Language deterioration (depending on the particular mutation) | Early episodic memory loss Early loss of visuoperceptual skills Attention deficits Language deterioration Decline in functional skills | |||
| Non amnestic phenotypes (e.g: behavioural changes and executive dysfunction) | Less Common | More Common | More Common | |||
| Neurological symptoms (e.g: epilepsy, myoclonus, spastic paraparesis, cerebellar signs) | Less Common | More Common | More Common | |||
| Neuroimaging and Neuropathology | ||||||
| Altered default mode connectivity | Present | Present | Present | |||
| Biochemical changes | Similar in magnitude and direction | Similar in magnitude and direction | Similar in magnitude and direction | |||
| Aβ deposition map | Similar | Similar | Similar | |||
| Initial Striatal Aβ | Absent | Present | Present | |||
| Hypometabolism map | Similar | Similar | Similar | |||
| Atrophy map | Similar | Similar (but accelerated) | Similar | |||
| Distribution of tau | Similar | Similar | Similar | |||
| CO-PATHOLOGIES | ||||||
| Lewy body pathology | Common | Most common | Rare | |||
| TDP-43 Pathology | More common Similar distribution | Less Common Similar distribution | Less Common Similar distribution | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rujeedawa, T.; Carrillo Félez, E.; Clare, I.C.H.; Fortea, J.; Strydom, A.; Rebillat, A.-S.; Coppus, A.; Levin, J.; Zaman, S.H. The Clinical and Neuropathological Features of Sporadic (Late-Onset) and Genetic Forms of Alzheimer’s Disease. J. Clin. Med. 2021, 10, 4582. https://doi.org/10.3390/jcm10194582
Rujeedawa T, Carrillo Félez E, Clare ICH, Fortea J, Strydom A, Rebillat A-S, Coppus A, Levin J, Zaman SH. The Clinical and Neuropathological Features of Sporadic (Late-Onset) and Genetic Forms of Alzheimer’s Disease. Journal of Clinical Medicine. 2021; 10(19):4582. https://doi.org/10.3390/jcm10194582
Chicago/Turabian StyleRujeedawa, Tanzil, Eva Carrillo Félez, Isabel C. H. Clare, Juan Fortea, Andre Strydom, Anne-Sophie Rebillat, Antonia Coppus, Johannes Levin, and Shahid H. Zaman. 2021. "The Clinical and Neuropathological Features of Sporadic (Late-Onset) and Genetic Forms of Alzheimer’s Disease" Journal of Clinical Medicine 10, no. 19: 4582. https://doi.org/10.3390/jcm10194582
APA StyleRujeedawa, T., Carrillo Félez, E., Clare, I. C. H., Fortea, J., Strydom, A., Rebillat, A. -S., Coppus, A., Levin, J., & Zaman, S. H. (2021). The Clinical and Neuropathological Features of Sporadic (Late-Onset) and Genetic Forms of Alzheimer’s Disease. Journal of Clinical Medicine, 10(19), 4582. https://doi.org/10.3390/jcm10194582