
A Systematic Review and Meta-Analysis of Enzyme Replacement Therapy in Late-Onset Pompe Disease

 , ,
, ,
Abstract
:1. Introduction
2. Methodology
2.1. Information Sources and Search Strategy
2.2. Eligibility Criteria and Study Selection
2.3. Data Collection
2.4. Statistical Analysis
2.5. Evaluation of the Quality of Included Studies
3. Results
3.1. Characteristics of Included Studies
3.2. Assessment of Functional Capacity
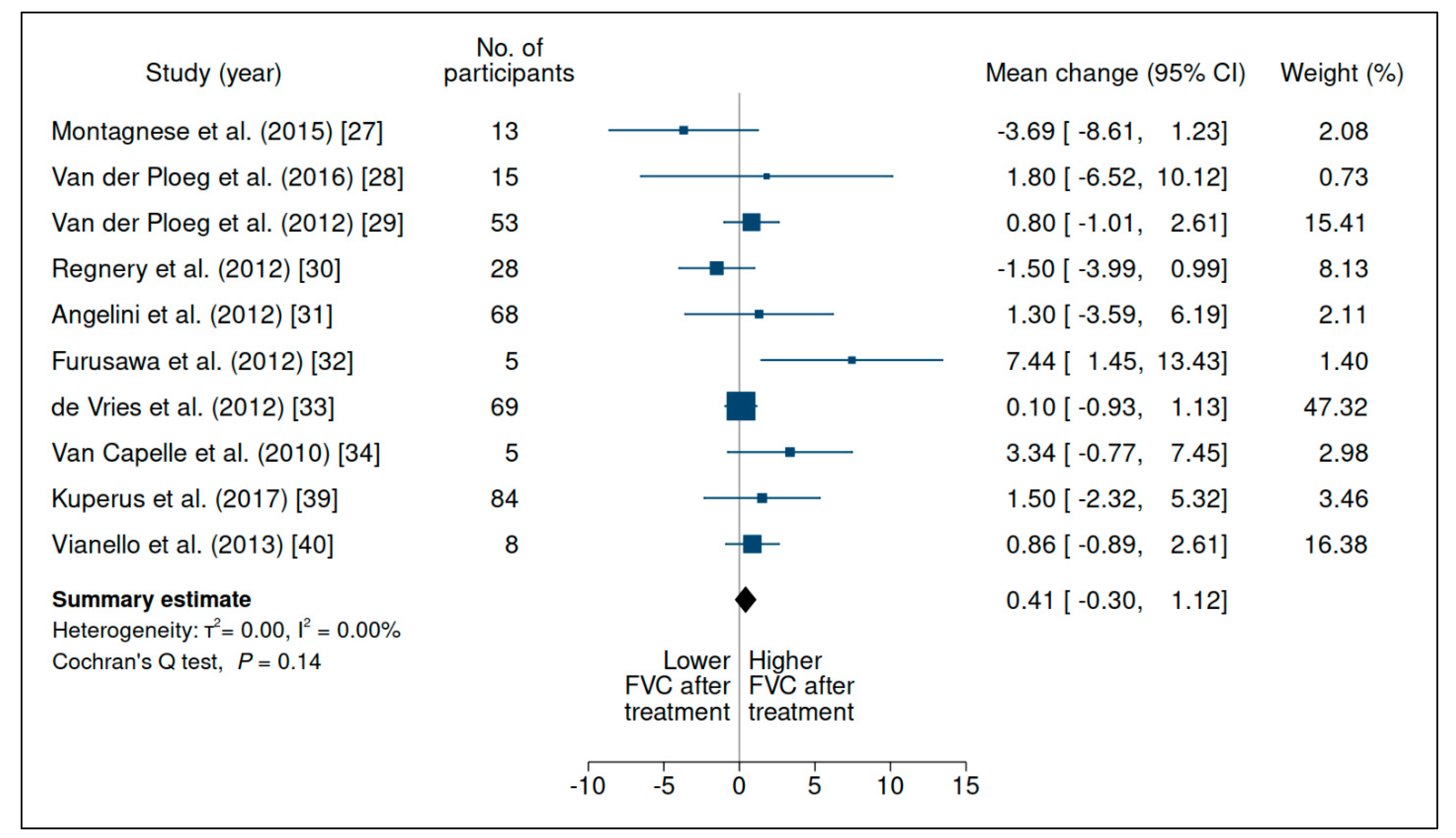
3.2.1. Forced Vital Capacity
3.2.2. Six-Minute Walking Test
3.2.3. Walton and Gardner-Medwin Scale (WGMS)
3.3. Upper-Limb Strength
3.4. Quality of Life
3.5. Time on Ventilation
3.6. Safety
3.6.1. Adverse Events
3.6.2. Mortality
3.6.3. Anti-Alglucosidase Alfa Antibodies
3.7. Risk of Bias and Quality of Included Studies
3.8. Certainty of Evidence by Outcomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 6MWT | 6 min walking test |
| ABC | approximate Bayesian computation model |
| AE | adverse event |
| CI | confidence interval |
| OPD | infantile-onset Pompe disease |
| ERT | enzyme replacement therapy |
| FVC | forced vital capacity |
| GAA | acid alpha-glucosidase |
| GLMM | generalized linear mixed model |
| IAR | infusion-associated reaction |
| IgG | immunoglobulin G |
| IR | incidence rate |
| LOPD | late-onset Pompe disease |
| LOTS | late-onset treatment study |
| ML | maximum likelihood |
| MRC | Medical Research Council |
| NB | newborn |
| NRSI | non-randomized studies of interventions |
| PD | Pompe disease |
| PedsQL | Pediatric Quality of Life Inventory™ |
| PICO | patients, intervention, control, outcome |
| QOL | quality of life |
| RCT | randomized clinical trial |
| REML | restricted maximum-likelihood estimator |
| RoB | Risk of Bias tool |
| ROBINS-I | Risk Of Bias In Non-randomized Studies of Interventions tool |
| SM | Supplementary Materials |
| TOV | time on ventilatory support |
| WGMS | Walton and Gardner-Medwin Scale |
References
- Llerena, J.C.; Horovitz, D.M.; Marie, S.K.; Porta, G.; Giugliani, R.; Rojas, M.V.; Martins, A.M.; Brazilian Network for Studies in Pompe Disease (ReBrPOM). The Brazilian consensus on the management of Pompe disease. J. Pediatr. 2009, 155, S47–S56. [Google Scholar] [CrossRef]
- Levine, J.C.; Kishnani, P.S.; Chen, Y.T.; Herlong, J.R.; Li, J.S. Cardiac remodeling after enzyme replacement therapy with acid alpha-glucosidase for infants with Pompe disease. Pediatr. Cardiol. 2008, 29, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Slonim, A.E.; Bulone, L.; Goldberg, T.; Minikes, J.; Slonim, E.; Galanko, J.; Martiniuk, F. Modification of the natural history of adult-onset acid maltase deficiency by nutrition and exercise therapy. Muscle Nerve 2007, 35, 70–77. [Google Scholar] [CrossRef]
- Geel, T.M.; McLaughlin, P.M.; de Leij, L.F.; Ruiters, M.H.; Niezen-Koning, K.E. Pompe disease: Current state of treatment modalities and animal models. Mol. Genet. Metab. 2007, 92, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Winchester, B.; Bali, D.; Bodamer, O.A.; Caillaud, C.; Christensen, E.; Cooper, A.; Cupler, E.; Deschauer, M.; Fumić, K.; Jackson, M.; et al. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: Report from an international consensus meeting. Mol. Genet. Metab. 2008, 93, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Bembi, B.; Cerini, E.; Danesino, C.; Donati, M.A.; Gasperini, S.; Morandi, L.; Musumeci, O.; Parenti, G.; Ravaglia, S.; Seidita, F.; et al. Diagnosis of glycogenosis type II. Neurology 2008, 71, S4–S11. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, L.; Quan, S. Enzyme replacement therapy for infantile-onset Pompe disease. Cochrane Database Syst. Rev. 2017, 11, CD011539. [Google Scholar] [CrossRef]
- Llerena Junior, J.C.; Nascimento, O.J.; Oliveira, A.S.; Dourado Junior, M.E.; Marrone, C.D.; Siqueira, H.H.; Sobreira, C.F.; Dias-Tosta, E.; Werneck, L.C. Guidelines for the diagnosis, treatment and clinical monitoring of patients with juvenile and adult Pompe disease. Arq Neuropsiquiatr. 2016, 74, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Fleischer, J.; Grange, D.K.; Braddock, S.R.; Hitchins, L.; Hickey, R.; Christensen, K.M.; Groepper, D.; et al. Newborn Screening for Pompe Disease in Illinois: Experience with 684,290 Infants. Int. J. Neonatal Screen. 2020, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Ficicioglu, C.; Ahrens-Nicklas, R.C.; Barch, J.; Cuddapah, S.R.; DiBoscio, B.S.; DiPerna, J.C.; Gordon, P.L.; Henderson, N.; Menello, C.; Luongo, N.; et al. Newborn Screening for Pompe Disease: Pennsylvania Experience. Int. J. Neonatal Screen. 2020, 6, 89. [Google Scholar] [CrossRef]
- Klug, T.L.; Swartz, L.B.; Washburn, J.; Brannen, C.; Kiesling, J.L. Lessons Learned from Pompe Disease Newborn Screening and Follow-up. Int. J. Neonatal Screen. 2020, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Momosaki, K.; Kido, J.; Yoshida, S.; Sugawara, K.; Miyamoto, T.; Inoue, T.; Okumiya, T.; Matsumoto, S.; Endo, F.; Hirose, S.; et al. Newborn screening for Pompe disease in Japan: Report and literature review of mutations in the GAA gene in Japanese and Asian patients. J. Hum. Genet. 2019, 64, 741–755. [Google Scholar] [CrossRef]
- Strothotte, S.; Strigl-Pill, N.; Grunert, B.; Kornblum, C.; Eger, K.; Wessig, C.; Deschauer, M.; Breunig, F.; Glocker, F.X.; Vielhaber, S.; et al. Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J. Neurol. 2010, 257, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Toscano, A.; Schoser, B. Enzyme replacement therapy in late-onset Pompe disease: A systematic literature review. J. Neurol. 2013, 260, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Int. J. Surg. 2021, 88, 105906. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.R.; Davis, S.D.; Robertson, K.A.; Akshintala, S.; Allen, J.; Fisher, M.J.; Blakeley, J.O.; Widemann, B.C.; Ferner, R.E.; Marcus, C.L.; et al. Sleep and pulmonary outcomes for clinical trials of airway plexiform neurofibromas in NF1. Neurology 2016, 87, S13–S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozov, T.; Silva, F.A.; Santana, M.A.; Adde, F.V.; Mendes, R.H.; Group, B.C.F.M.S. A first-year dornase alfa treatment impact on clinical parameters of patients with cystic fibrosis: The Brazilian cystic fibrosis multicenter study. Rev. Paul Pediatr. 2013, 31, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Schrover, R.; Evans, K.; Giugliani, R.; Noble, I.; Bhattacharya, K. Minimal clinically important difference for the 6-min walk test: Literature review and application to Morquio A syndrome. Orphanet J. Rare Dis. 2017, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, G.; Chemaitelly, H.; Abu-Raddad, L.J.; Rücker, G. Seriously misleading results using inverse of Freeman-Tukey double arcsine transformation in meta-analysis of single proportions. Res. Synth. Methods 2019, 10, 476–483. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.; Reis, I.M. Simulation-based estimation of mean and standard deviation for meta-analysis via Approximate Bayesian Computation (ABC). BMC Med. Res. Methodol. 2015, 15, 61. [Google Scholar] [CrossRef] [Green Version]
- Pereira, T.V.; Patsopoulos, N.A.; Salanti, G.; Ioannidis, J.P. Critical interpretation of Cochran’s Q test depends on power and prior assumptions about heterogeneity. Res. Synth. Methods 2010, 1, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Sterne, J.A.C.; Savović, J.; Page, M.J.; Elbers, R.G.; Blencowe, N.S.; Boutron, I.; Cates, C.J.; Cheng, H.Y.; Corbett, M.S.; Eldridge, S.M.; et al. RoB 2: A revised tool for assessing risk of bias in randomised trials. BMJ 2019, 366, l4898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterne, J.A.; Hernán, M.A.; Reeves, B.C.; Savović, J.; Berkman, N.D.; Viswanathan, M.; Henry, D.; Altman, D.G.; Ansari, M.T.; Boutron, I.; et al. ROBINS-I: A tool for assessing risk of bias in non-randomised studies of interventions. BMJ 2016, 355, i4919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balshem, H.; Helfand, M.; Schünemann, H.J.; Oxman, A.D.; Kunz, R.; Brozek, J.; Vist, G.E.; Falck-Ytter, Y.; Meerpohl, J.; Norris, S.; et al. GRADE guidelines: 3. Rating the quality of evidence. J. Clin. Epidemiol. 2011, 64, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Guyatt, G.H.; Oxman, A.D.; Vist, G.E.; Kunz, R.; Falck-Ytter, Y.; Alonso-Coello, P.; Schünemann, H.J.; Group, G.W. GRADE: An emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008, 336, 924–926. [Google Scholar] [CrossRef] [Green Version]
- Guyatt, G.H.; Oxman, A.D.; Schünemann, H.J.; Tugwell, P.; Knottnerus, A. GRADE guidelines: A new series of articles in the Journal of Clinical Epidemiology. J. Clin. Epidemiol. 2011, 64, 380–382. [Google Scholar] [CrossRef]
- Montagnese, F.; Barca, E.; Musumeci, O.; Mondello, S.; Migliorato, A.; Ciranni, A.; Rodolico, C.; De Filippi, P.; Danesino, C.; Toscano, A. Clinical and molecular aspects of 30 patients with late-onset Pompe disease (LOPD): Unusual features and response to treatment. J. Neurol. 2015, 262, 968–978. [Google Scholar] [CrossRef]
- van der Ploeg, A.; Carlier, P.G.; Carlier, R.Y.; Kissel, J.T.; Schoser, B.; Wenninger, S.; Pestronk, A.; Barohn, R.J.; Dimachkie, M.M.; Goker-Alpan, O.; et al. Prospective exploratory muscle biopsy, imaging, and functional assessment in patients with late-onset Pompe disease treated with alglucosidase alfa: The EMBASSY Study. Mol. Genet. Metab. 2016, 119, 115–123. [Google Scholar] [CrossRef]
- van der Ploeg, A.T.; Barohn, R.; Carlson, L.; Charrow, J.; Clemens, P.R.; Hopkin, R.J.; Kishnani, P.S.; Laforêt, P.; Morgan, C.; Nations, S.; et al. Open-label extension study following the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol. Genet. Metab. 2012, 107, 456–461. [Google Scholar] [CrossRef]
- Regnery, C.; Kornblum, C.; Hanisch, F.; Vielhaber, S.; Strigl-Pill, N.; Grunert, B.; Müller-Felber, W.; Glocker, F.X.; Spranger, M.; Deschauer, M.; et al. 36 months observational clinical study of 38 adult Pompe disease patients under alglucosidase alfa enzyme replacement therapy. J. Inherit. Metab. Dis. 2012, 35, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Semplicini, C.; Ravaglia, S.; Bembi, B.; Servidei, S.; Pegoraro, E.; Moggio, M.; Filosto, M.; Sette, E.; Crescimanno, G.; et al. Observational clinical study in juvenile-adult glycogenosis type 2 patients undergoing enzyme replacement therapy for up to 4 years. J. Neurol. 2012, 259, 952–958. [Google Scholar] [CrossRef]
- Furusawa, Y.; Mori-Yoshimura, M.; Yamamoto, T.; Sakamoto, C.; Wakita, M.; Kobayashi, Y.; Fukumoto, Y.; Oya, Y.; Fukuda, T.; Sugie, H.; et al. Effects of enzyme replacement therapy on five patients with advanced late-onset glycogen storage disease type II: A 2-year follow-up study. J. Inherit. Metab. Dis. 2012, 35, 301–310. [Google Scholar] [CrossRef]
- de Vries, J.M.; van der Beek, N.A.; Hop, W.C.; Karstens, F.P.; Wokke, J.H.; de Visser, M.; van Engelen, B.G.; Kuks, J.B.; van der Kooi, A.J.; Notermans, N.C.; et al. Effect of enzyme therapy and prognostic factors in 69 adults with Pompe disease: An open-label single-center study. Orphanet J. Rare Dis. 2012, 7, 73. [Google Scholar] [CrossRef] [Green Version]
- van Capelle, C.I.; van der Beek, N.A.; Hagemans, M.L.; Arts, W.F.; Hop, W.C.; Lee, P.; Jaeken, J.; Frohn-Mulder, I.M.; Merkus, P.J.; Corzo, D.; et al. Effect of enzyme therapy in juvenile patients with Pompe disease: A three-year open-label study. Neuromuscul. Disord. 2010, 20, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Ravaglia, S.; Pichiecchio, A.; Ponzio, M.; Danesino, C.; Saeidi Garaghani, K.; Poloni, G.U.; Toscano, A.; Moglia, A.; Carlucci, A.; Bini, P.; et al. Changes in skeletal muscle qualities during enzyme replacement therapy in late-onset type II glycogenosis: Temporal and spatial pattern of mass vs. strength response. J. Inherit. Metab. Dis. 2010, 33, 737–745. [Google Scholar] [CrossRef] [PubMed]
- van der Ploeg, A.T.; Clemens, P.R.; Corzo, D.; Escolar, D.M.; Florence, J.; Groeneveld, G.J.; Herson, S.; Kishnani, P.S.; Laforet, P.; Lake, S.L.; et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N. Engl. J. Med. 2010, 362, 1396–1406. [Google Scholar] [CrossRef] [Green Version]
- Angelini, C.; Semplicini, C.; Tonin, P.; Filosto, M.; Pegoraro, E.; Sorarù, G.; Fanin, M. Progress in Enzyme Replacement Therapy in Glycogen Storage Disease Type II. Ther. Adv. Neurol. Disord. 2009, 2, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkowski, G.; Konopko, M.; Rola, R.; Ługowska, A.; Ryglewicz, D.; Sienkiewicz-Jarosz, H. Enzymatic replacement therapy in patients with late-onset Pompe disease—6-Year follow up. Neurol. Neurochir. Pol. 2018, 52, 465–469. [Google Scholar] [CrossRef]
- Kuperus, E.; Kruijshaar, M.E.; Wens, S.C.A.; de Vries, J.M.; Favejee, M.M.; van der Meijden, J.C.; Rizopoulos, D.; Brusse, E.; van Doorn, P.A.; van der Ploeg, A.T.; et al. Long-term benefit of enzyme replacement therapy in Pompe disease: A 5-year prospective study. Neurology 2017, 89, 2365–2373. [Google Scholar] [CrossRef]
- Vianello, A.; Semplicini, C.; Paladini, L.; Concas, A.; Ravaglia, S.; Servidei, S.; Toscano, A.; Mongini, T.; Angelini, C.; Pegoraro, E. Enzyme replacement therapy improves respiratory outcomes in patients with late-onset type II glycogenosis and high ventilator dependency. Lung 2013, 191, 537–544. [Google Scholar] [CrossRef]
- Ravaglia, S.; De Filippi, P.; Pichiecchio, A.; Ponzio, M.; Saeidi Garaghani, K.; Poloni, G.U.; Bini, P.; Danesino, C. Can genes influencing muscle function affect the therapeutic response to enzyme replacement therapy (ERT) in late-onset type II glycogenosis? Mol. Genet. Metab. 2012, 107, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Bembi, B.; Pisa, F.E.; Confalonieri, M.; Ciana, G.; Fiumara, A.; Parini, R.; Rigoldi, M.; Moglia, A.; Costa, A.; Carlucci, A.; et al. Long-term observational, non-randomized study of enzyme replacement therapy in late-onset glycogenosis type II. J. Inherit. Metab. Dis. 2010, 33, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Orlikowski, D.; Pellegrini, N.; Prigent, H.; Laforêt, P.; Carlier, R.; Carlier, P.; Eymard, B.; Lofaso, F.; Annane, D. Recombinant human acid alpha-glucosidase (rhGAA) in adult patients with severe respiratory failure due to Pompe disease. Neuromuscul. Disord. 2011, 21, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Forsha, D.; Li, J.S.; Smith, P.B.; van der Ploeg, A.T.; Kishnani, P.; Pasquali, S.K.; Investigators, L.-O.T.S. Cardiovascular abnormalities in late-onset Pompe disease and response to enzyme replacement therapy. Genet. Med. 2011, 13, 625–631. [Google Scholar] [CrossRef] [Green Version]
- de Vries, J.M.; Kuperus, E.; Hoogeveen-Westerveld, M.; Kroos, M.A.; Wens, S.C.; Stok, M.; van der Beek, N.A.; Kruijshaar, M.E.; Rizopoulos, D.; van Doorn, P.A.; et al. Pompe disease in adulthood: Effects of antibody formation on enzyme replacement therapy. Genet. Med. 2017, 19, 90–97. [Google Scholar] [CrossRef]
- Papadimas, G.K.; Spengos, K.; Konstantinopoulou, A.; Vassilopoulou, S.; Vontzalidis, A.; Papadopoulos, C.; Michelakakis, H.; Manta, P. Adult Pompe disease: Clinical manifestations and outcome of the first Greek patients receiving enzyme replacement therapy. Clin. Neurol. Neurosurg. 2011, 113, 303–307. [Google Scholar] [CrossRef]
- Güngör, D.; Kruijshaar, M.E.; Plug, I.; Rizopoulos, D.; Kanters, T.A.; Wens, S.C.; Reuser, A.J.; van Doorn, P.A.; van der Ploeg, A.T. Quality of life and participation in daily life of adults with Pompe disease receiving enzyme replacement therapy: 10 years of international follow-up. J. Inherit. Metab. Dis. 2016, 39, 253–260. [Google Scholar] [CrossRef] [Green Version]
- van Gelder, C.M.; Hoogeveen-Westerveld, M.; Kroos, M.A.; Plug, I.; van der Ploeg, A.T.; Reuser, A.J. Enzyme therapy and immune response in relation to CRIM status: The Dutch experience in classic infantile Pompe disease. J. Inherit. Metab. Dis. 2015, 38, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Kishnani, P.S.; Nicolino, M.; Voit, T.; Rogers, R.C.; Tsai, A.C.; Waterson, J.; Herman, G.E.; Amalfitano, A.; Thurberg, B.L.; Richards, S.; et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J. Pediatr. 2006, 149, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Schoser, B.; Stewart, A.; Kanters, S.; Hamed, A.; Jansen, J.; Chan, K.; Karamouzian, M.; Toscano, A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: A systematic review and meta-analysis. J. Neurol. 2017, 264, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Güngör, D.; Kruijshaar, M.E.; Plug, I.; D’Agostino, R.B.; Hagemans, M.L.; van Doorn, P.A.; Reuser, A.J.; van der Ploeg, A.T. Impact of enzyme replacement therapy on survival in adults with Pompe disease: Results from a prospective international observational study. Orphanet J. Rare Dis. 2013, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- Anderson, L.J.; Henley, W.; Wyatt, K.M.; Nikolaou, V.; Waldek, S.; Hughes, D.A.; Lachmann, R.H.; Logan, S. Effectiveness of enzyme replacement therapy in adults with late-onset Pompe disease: Results from the NCS-LSD cohort study. J. Inherit. Metab. Dis. 2014, 37, 945–952. [Google Scholar] [CrossRef]
- Herbert, M.; Kazi, Z.B.; Richards, S.; Rosenberg, A.S.; Kishnani, P.S. Response to de Vries et al. Genet. Med. 2017, 19, 1281–1282. [Google Scholar] [CrossRef]
- Harlaar, L.; Hogrel, J.Y.; Perniconi, B.; Kruijshaar, M.E.; Rizopoulos, D.; Taouagh, N.; Canal, A.; Brusse, E.; van Doorn, P.A.; van der Ploeg, A.T.; et al. Large variation in effects during 10 years of enzyme therapy in adults with Pompe disease. Neurology 2019, 93, e1756–e1767. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Search Query |
|---|---|
| MEDLINE (via PubMed) | “Glycogen Storage Disease Type II” [Mesh] AND “alpha-Glucosidases” [Mesh] AND “humans” [MeSH] |
| Embase | “glycogen storage disease type 2”/exp AND “recombinant glucan 1, 4 alpha glucosidase”/exp OR “recombinant glucan 1,4 alpha glucosidase” |
| Outcome | Number of Articles | References |
|---|---|---|
| Assessment of functional capacity: | ||
| -FVC | 15 | [13,27,28,29,30,31,32,33,34,35,36,37,38,39,40] |
| -6MWT | 14 | [13,27,28,29,30,31,34,35,36,37,38,39,41,42] |
| -WGMS | 6 | [13,27,30,31,40,43] |
| Safety | 14 | [13,28,29,30,31,33,34,36,39,40,42,43,44,45] |
| Upper-limb strength | 9 | [13,28,29,30,33,34,36,39,46] |
| Quality of life | 6 | [13,28,30,36,43,47] |
| Time on ventilation | 6 | [30,31,33,40,42,43] |
| Survival | 0 | - |
| Sleep quality | 0 | - |
| Swallowing disorder | 0 | - |
| Author | Patients (n/Male) | Design | Age at Onset of ERT—yo–μ (sd) (Range) | Follow-Up Duration | Control | Patients on Ventilation (n) |
|---|---|---|---|---|---|---|
| Angelini et al. (2009) [37] | 11/3 | Cohort | 31.1 (8) | * N/A | - | 1/11 |
| Angelini et al. (2012) [31] | 68/33 | Cohort | 43 (15.4) (7 to 72) | 36 months | - | 27/68 |
| Bembi et al. (2010) [42] | 24/14 | NRSI | Young: 12 (3.3) Adults: 47.6 (10.7) | 36 months | - | 9/24 |
| de Vries et al. (2012) [33] | 49/21 | Cohort | 52.1 (median) (26.2 to 76.3) | 23 months | - | 13/49 |
| de Vries et al. (2017) [45] | 73/37 | NRSI | 52 (26 to 74) | 36 months | - | 22/73 |
| Forsha et al. (2011) [44] | 87/44 | Post-hoc analysis of RCT | 44 (39 to 52) | 19.5 months | Placebo | N/A |
| Furusawa et al. (2011) [32] | 5/2 | Case series | 47 (13.6) (32 to 66) | 24 months | - | 5/5 |
| Gungor et al. (2016) [47] | 174/81 | Cohort | 50 (median) (24 to 76) | *120 months | - | 84/174 |
| Kuperus et al. (2017) [39] | 88/45 | Cohort | 52 (median) (24 to 76) | 73.2 months (median) | - | 21/88 |
| Montagnese et al. (2015) [27] | 14/N/A | Cohort | 53.2 (11.1) (36 to 72) | 31 months (mean) | - | N/A |
| Orlikowski et al. (2011) [43] | 5/2 | NRSI | 47.8 (14.4) (28 to 62) | 12 months | - | 5/5 |
| Papadimas et al. (2011) [46] | 5/1 | Cohort | 46.8 (14.4) (40 to 73) | 12 months | - | N/A |
| Ravaglia et al. (2010) [35] | 11/6 | NRSI | 54.2 (11.2) | at least 24 months | - | N/A |
| Ravaglia et al. (2012) [41] | 16/7 | NRSI | 54.5 (15.1) | at least 24 months | - | N/A |
| Regnery et al. (2012) [30] | 38/18 | NRSI | 53.1 (27 to 73) | 36 months | - | 13/38 |
| Strothotte et al. (2010) [13] | 44/24 | NRSI | 48.9 (12.9) (21 to 69) | 12 months | - | 16/44 |
| van Capelle et al. (2010) [34] | 5/3 | Phase II open study, followed by an extension period | 11.1 (3.7) (5.9 to 15.2) | 36 months | - | 1/5 |
| van der Ploeg et al. (2010) [36] | 90/45 | RCT (LOTS) | 45.3 (12.4) (15.9 to 70) | 19.5 months | Placebo | ERT = 20/60 Placebo = 11/30 |
| van der Ploeg et al. (2012) [29] | 60/34 | Open study (LOTS extension) | 45.3 (12.4) (15.9 to 79) | 26 months | - | 20/60 |
| van der Ploeg et al. (2016) [28] | 16/7 | NRSI | 51.6 (13.7) (24.5 to 70.7) | 6 months | - | 0/16 |
| Vianello et al. (2013) [40] | Group A: 8/5 Group B: 6/1 | Cohort with historical control | Group A: 51.5 (12.2) (29 to 65) Group B: 43.8 (15.8) (18 to 59) | Group A = 35.8 months (mean) | Group B (Historical control without ERT) = 52.6 months (mean) | Group A=8/8 Group B=6/6 |
| Witkowski et al. (2018) [38] | 5/2 | Case series | 35.8 (26 to 41) | 72 months | - | N/A |
| TOTAL | 896/388 | - | 42.8 (7 to 72.3) | 32.5 months | - | 265 |
| Summary IR (95% CI) | ||||||
|---|---|---|---|---|---|---|
| Outcome | Studies | Participants | PQ | I2 | Random-Effects Model | Fixed-Effects Model |
| Mortality | 9 | 675 | 0.66 | 38.9 | 0.44 (0.15 to 1.28) | 0.56 (0.31 to 1.01) |
| AB+ | 7 | 323 | <0.001 | 94.7 | 42.63 (24.07 to 75.49) | 35.28 (31.41 to 39.62) |
| AE | 3 | 139 | <0.001 | 97.4 | 30.93 (2.96 to 323.51) | 26.59 (21.0 to 33.67) |
| SAE | 5 | 367 | <0.001 | 89.7 | 4.19 (0.63 to 27.69) | 2.32 (1.52 to 3.57) |
| IAR | 4 | 43 | <0.001 | 97.2 | 3.03 (0.03 to 305.58) | 23.71 (16.26 to 34.58) |
| Patients with IAR | 7 | 274 | <0.001 | 94.1 | 6.58 (1.67 to 25.93) | 6.69 (5.20 to 8.60) |
| Study | N of Patients | N Ab Titer ≥ 1:250 | Method of Measuring Ab | Reduced Response to Treatment | AEs Attributable to Ab Presence |
|---|---|---|---|---|---|
| Angelini et al. (2012) [31] | 15 | 11 | N/A | N/E | Yes (n = 1) |
| de Vries et al. (2017) [45] | 73 | 46 | Van Gelder et al. (2014) [48] | Yes (n = 1) | Yes |
| Kuperus et al. (2017) [39] | 73 | 44 | Van Gelder et al. (2014) [48] | Yes (n = 1) | N/E |
| Orlikowski et al. (2011) [43] | 5 | 5 | N/A | No | N/E |
| Regnery et al. (2012) [30] | 38 | 38 | N/A | Yes (n = 1) | N/E |
| van Capelle et al. (2010) [34] | 5 | 5 | N/A | No | N/E |
| Van der Ploeg et al. (2010) [36] | 59 | 59 | Kishnani et al. (2006) [49] | N/E | No |
| Van der Ploeg et al. (2012) [29] | 59 | 59 | Kishnani et al. (2006) [49] | Yes (n = 2) | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dornelles, A.D.; Junges, A.P.P.; Pereira, T.V.; Krug, B.C.; Gonçalves, C.B.T.; Llerena, J.C., Jr.; Kishnani, P.S.; de Oliveira, H.A., Jr.; Schwartz, I.V.D. A Systematic Review and Meta-Analysis of Enzyme Replacement Therapy in Late-Onset Pompe Disease. J. Clin. Med. 2021, 10, 4828. https://doi.org/10.3390/jcm10214828
Dornelles AD, Junges APP, Pereira TV, Krug BC, Gonçalves CBT, Llerena JC Jr., Kishnani PS, de Oliveira HA Jr., Schwartz IVD. A Systematic Review and Meta-Analysis of Enzyme Replacement Therapy in Late-Onset Pompe Disease. Journal of Clinical Medicine. 2021; 10(21):4828. https://doi.org/10.3390/jcm10214828
Chicago/Turabian StyleDornelles, Alícia Dorneles, Ana Paula Pedroso Junges, Tiago Veiga Pereira, Bárbara Corrêa Krug, Candice Beatriz Treter Gonçalves, Juan Clinton Llerena, Jr., Priya Sunil Kishnani, Haliton Alves de Oliveira, Jr., and Ida Vanessa Doederlein Schwartz. 2021. "A Systematic Review and Meta-Analysis of Enzyme Replacement Therapy in Late-Onset Pompe Disease" Journal of Clinical Medicine 10, no. 21: 4828. https://doi.org/10.3390/jcm10214828
APA StyleDornelles, A. D., Junges, A. P. P., Pereira, T. V., Krug, B. C., Gonçalves, C. B. T., Llerena, J. C., Jr., Kishnani, P. S., de Oliveira, H. A., Jr., & Schwartz, I. V. D. (2021). A Systematic Review and Meta-Analysis of Enzyme Replacement Therapy in Late-Onset Pompe Disease. Journal of Clinical Medicine, 10(21), 4828. https://doi.org/10.3390/jcm10214828