
Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy


Abstract
:1. Introduction
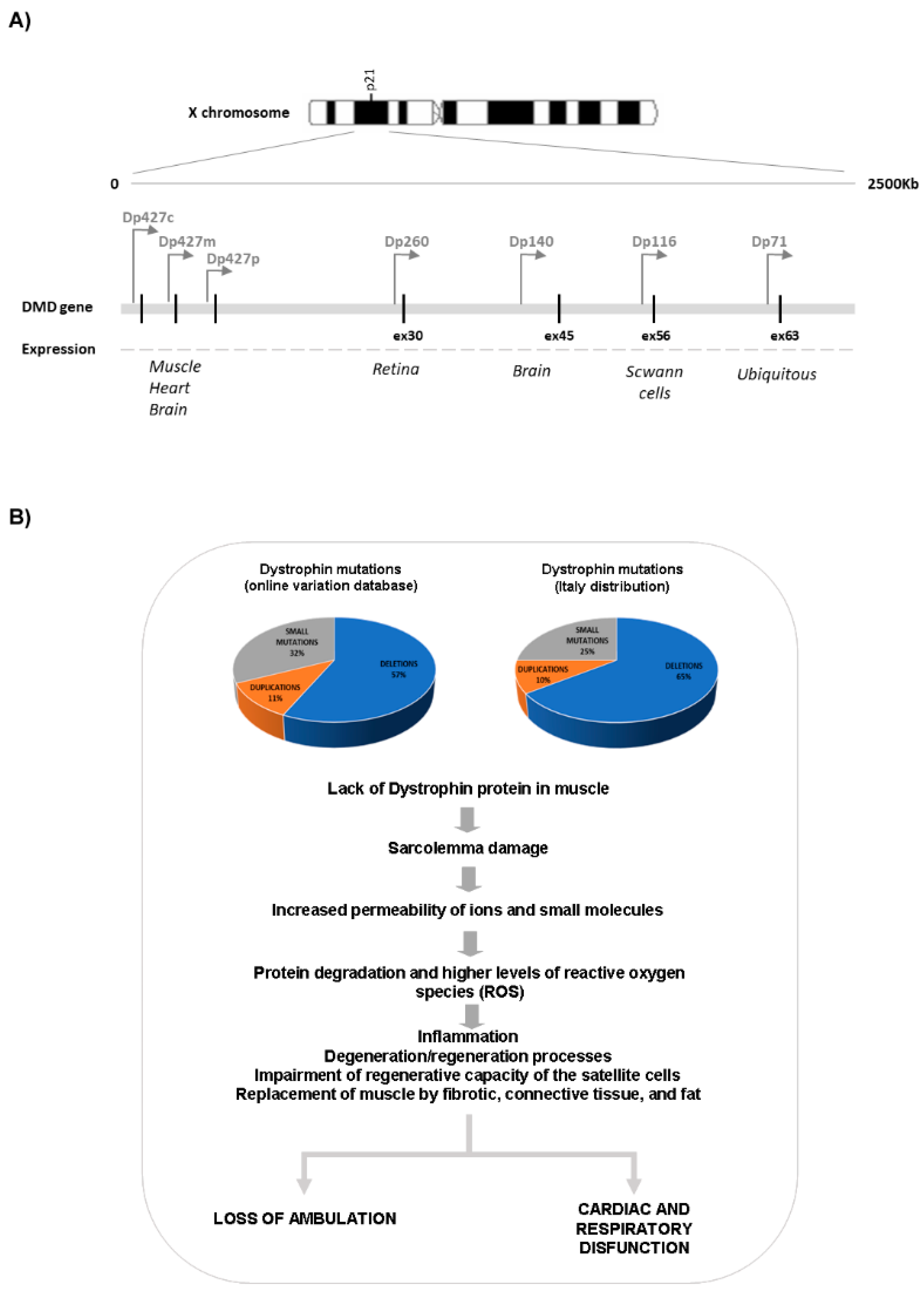
2. Molecular Genetics of DMD
3. Pathophysiology of DMD
3.1. Striated Muscle
3.2. Brain
4. Rationale for Current Therapeutic Interventions in DMD
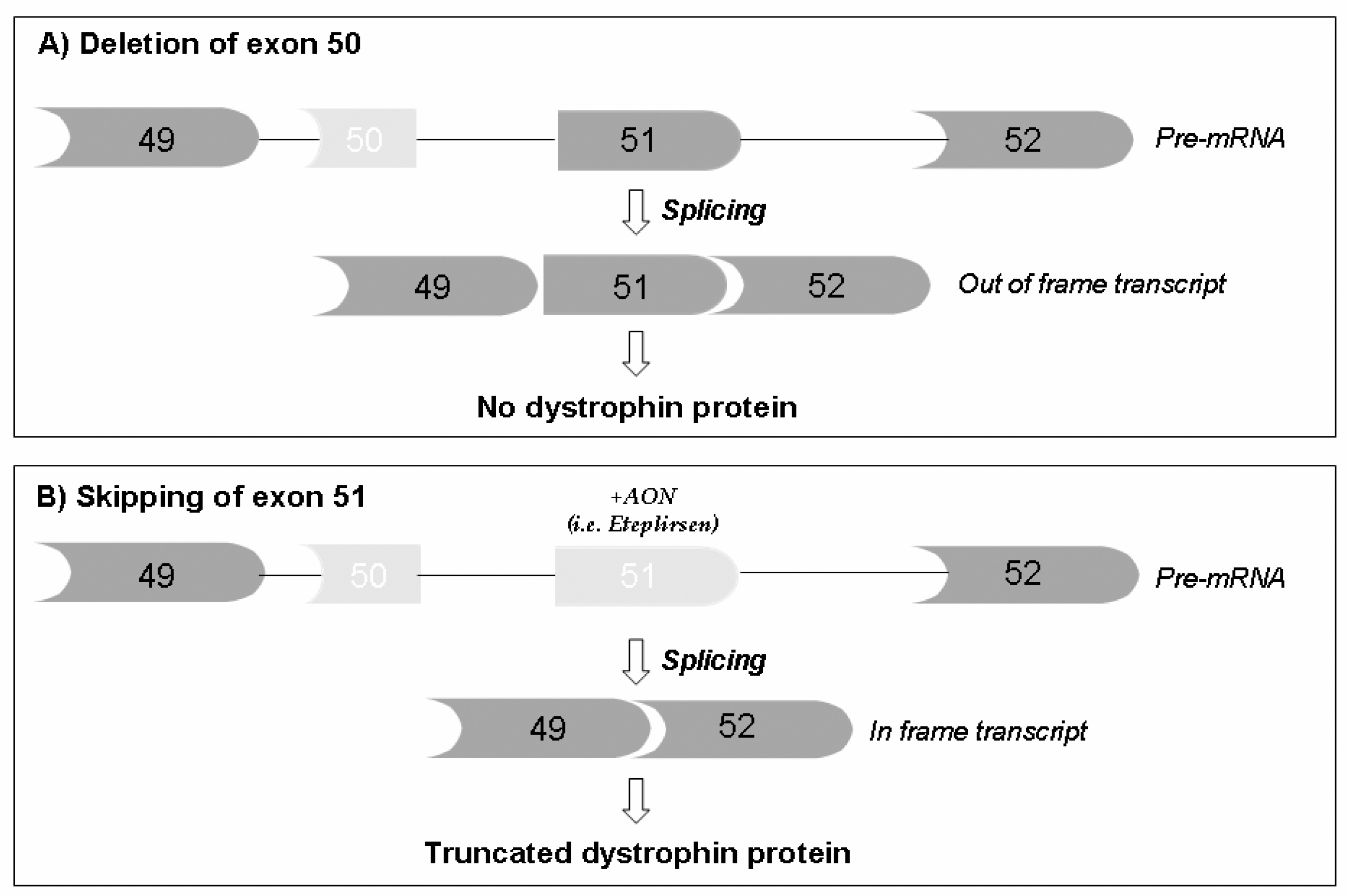
4.1. Restoring Dystrophin Protein Production
4.2. Ataluren as a Read-Through Strategy for Nonsense Mutations
4.3. AAV Gene Therapy
4.4. Future Perspectives
4.4.1. CRISPR/Cas9 Mediated Gene Editing
4.4.2. Stem Cell Therapy
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Neri, M.; Rossi, R.; Trabanelli, C.; Mauro, A.; Selvatici, R.; Falzarano, M.S.; Spedicato, N.; Margutti, A.; Rimessi, P.; Fortunato, F.; et al. The Genetic Landscape of Dystrophin Mutations in Italy: A Nationwide Study. Front. Genet. 2020, 11, 131. [Google Scholar] [CrossRef]
- Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules 2015, 20, 18168–18184. [Google Scholar] [CrossRef] [Green Version]
- Barseghyan, H.; Tang, W.; Wang, R.T.; Almalvez, M.; Segura, E.; Bramble, M.S.; Lipson, A.; Douine, E.D.; Lee, H.; Délot, E.C.; et al. Next-generation mapping: A novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med. 2017, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Parsons, E.P.; Clarke, A.J.; Hood, K.; Lycett, E.; Bradley, D.M. Newborn screening for Duchenne muscular dystrophy: A psychosocial study. Arch. Dis. Child. Fetal Neonatal Ed. 2002, 86, 91F–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, H.G.; Redmond, R.Z.; Scotcher, D.F. Experiences of Women Who Have Had Carrier Testing for Duchenne Muscular Dystrophy and Becker Muscular Dystrophy During Adolescence. J. Genet. Couns. 2018, 27, 1349–1359. [Google Scholar] [CrossRef] [Green Version]
- Breveglieri, G.; D’Aversa, E.; Finotti, A.; Borgatti, M. Non-invasive Prenatal Testing Using Fetal DNA. Mol. Diagn. Ther. 2019, 23, 291–299. [Google Scholar] [CrossRef] [PubMed]
- The DMD Gene Homepage. Available online: https://databases.lovd.nl/shared/genes/DMD (accessed on 18 December 2020).
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- White. S.J.; den Dunnen, J.T. Copy number variation in the genome; the human DMD gene as an example. Cytogenet. Genome Res. 2006, 115, 240–246. [Google Scholar] [CrossRef]
- Batchelor, C.L.; Winder, S.J. Sparks, signals and shock absorbers: How dystrophin loss causes muscular dystrophy. Trends Cell Biol. 2006, 16, 198–205. [Google Scholar] [CrossRef]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol. Neurobiol. 2019, 57, 1748–1767. [Google Scholar] [CrossRef] [Green Version]
- McElhanon, K.E.; Bhattacharya, S. Altered membrane integrity in the progression of muscle diseases. Life Sci. 2018, 192, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Dai, Q.; Huang, H.; Xu, Y.; Zhong, C. An Overview of Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 3–19. [Google Scholar] [CrossRef]
- Boldrin, L.; Zammit, P.S.; Morgan, J.E. Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res. 2015, 14, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.A.; Wang, Y.X.; Von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Kong, J.; Brat, D.J. Cancer Stem Cell Division: When the Rules of Asymmetry Are Broken. Stem Cells Dev. 2015, 24, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Marino-Enriquez, A.; Bennett, R.R.; Zhu, M.; Shen, Y.; Eilers, G.; Lee, J.-C.; Henze, J.; Fletcher, B.S.; Gu, Z.; et al. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat. Genet. 2014, 46, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Ruggieri, S.; De Giorgis, M.; Annese, T.; Tamma, R.; Notarangelo, A.; Marzullo, A.; Senetta, R.; Cassoni, P.; Notarangelo, M.; Ribatti, D.; et al. Dp71 Expression in Human Glioblastoma. Int. J. Mol. Sci. 2019, 20, 5429. [Google Scholar] [CrossRef] [Green Version]
- Gavillet, B.; Rougier, J.-S.; Domenighetti, A.A.; Behar, R.; Boixel, C.; Ruchat, P.; Lehr, H.-A.; Pedrazzini, T.; Abriel, H. Cardiac Sodium Channel Na v 1.5 Is Regulated by a Multiprotein Complex Composed of Syntrophins and Dystrophin. Circ. Res. 2006, 99, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, A.; Doyle, A.D.; Johnson, B.D. Regulation of the cardiac L-type Ca2+ channel by the actin-binding proteins α-actinin and dystrophin. Am. J. Physiol. Cell Physiol. 2002, 282, C1502–C1511. [Google Scholar] [CrossRef] [Green Version]
- Koenig, X.; Dysek, S.; Kimbacher, S.; Mike, A.K.; Cervenka, R.; Lukacs, P.; Nagl, K.; Dang, X.B.; Todt, H.; Bittner, R.E.; et al. Voltage-Gated Ion Channel Dysfunction Precedes Cardiomyopathy Development in the Dystrophic Heart. PLoS ONE 2011, 6, e20300. [Google Scholar] [CrossRef] [Green Version]
- Garbincius, J.F.; Michele, D.E. Dystrophin-glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of AMPK signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 13663–13668. [Google Scholar] [CrossRef] [Green Version]
- Tidball, J.G.; Wehling-Henricks, M. Nitric oxide synthase deficiency and the pathophysiology of muscular dystrophy. J. Physiol. 2014, 592, 4627–4638. [Google Scholar] [CrossRef]
- Lee, A.J.; Buckingham, E.T.; Kauer, A.J.; Mathews, K.D. Descriptive Phenotype of Obsessive-Compulsive Symptoms in Males with Duchenne Muscular Dystrophy. J. Child Neurol. 2018, 33, 572–579. [Google Scholar] [CrossRef]
- Ricotti, V.V.; Mandy, W.P.L.W.; Scoto, M.M.; Pane, M.M.; Deconinck, N.; Messina, S.S.; Mercuri, E.; Skuse, D.D.; Muntoni, F.F. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev. Med. Child Neurol. 2016, 58, 77–84. [Google Scholar] [CrossRef] [PubMed]
- DMD/BMD and the Brain. Available online: https://bindproject.eu/ (accessed on 18 December 2020).
- Doorenweerd, N.; Mahfouz, A.; van Putten, M.; Kaliyaperumal, R.; T’Hoen, P.A.C.; Hendriksen, J.G.M.; Aartsma-Rus, A.M.; Verschuuren, J.J.G.M.; Niks, E.; Reinders, M.J.T.; et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 12575. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.-M.; Panzanelli, P.; Tyagarajan, S.K. Molecular and functional heterogeneity of GABAergic synapses. Cell. Mol. Life Sci. 2012, 69, 2485–2499. [Google Scholar] [CrossRef] [Green Version]
- Daoud, F.; Candelario-Martínez, A.; Billard, J.-M.; Avital, A.; Khelfaoui, M.; Rozenvald, Y.; Guegan, M.; Mornet, D.; Jaillard, D.; Nudel, U.; et al. Role of Mental Retardation-Associated Dystrophin-Gene Product Dp71 in Excitatory Synapse Organization, Synaptic Plasticity and Behavioral Functions. PLoS ONE 2009, 4, e6574. [Google Scholar] [CrossRef]
- Taylor, P.J.; Betts, G.A.; Maroulis, S.; Gilissen, C.; Pedersen, R.L.; Mowat, D.R.; Johnston, H.M.; Buckley, M.F. Dystrophin Gene Mutation Location and the Risk of Cognitive Impairment in Duchenne Muscular Dystrophy. PLoS ONE 2010, 5, e8803. [Google Scholar] [CrossRef]
- D’Angelo, M.G.; Lorusso, M.L.; Civati, F.; Comi, G.P.; Magri, F.; Del Bo, R.; Guglieri, M.; Molteni, M.; Turconi, A.C.; Bresolin, N. Neurocognitive Profiles in Duchenne Muscular Dystrophy and Gene Mutation Site. Pediatr. Neurol. 2011, 45, 292–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, P.B. Emerging Strategies in the Treatment of Duchenne Muscular Dystrophy. Neurotherapeutics 2018, 15, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Takeshima, Y.; Nishio, H.; Sakamoto, H.; Nakamura, H.; Matsuo, M. Modulation of in vitro splicing of the upstream intron by modifying an intra-exon sequence which is deleted from the dystrophin gene in dystrophin Kobe. J. Clin. Investig. 1995, 95, 515–520. [Google Scholar] [CrossRef] [Green Version]
- Pramono, Z.A.; Takeshima, Y.; Alimsardjono, H.; Ishii, A.; Takeda, S.; Matsuo, M. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem. Biophys. Res. Commun. 1996, 226, 445–449. [Google Scholar] [CrossRef]
- Matsuo, M.; Masumura, T.; Nishio, H.; Nakajima, T.; Kitoh, Y.; Takumi, T.; Koga, J.; Nakamura, H. Exon skipping during splicing of dystrophin mRNA precursor due to an intraexon deletion in the dystrophin gene of Duchenne muscular dystrophy kobe. J. Clin. Investig. 1991, 87, 2127–2131. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Masumura, T.; Nakajima, T.; Kitoh, Y.; Takumi, T.; Nishio, H.; Koga, J.; Nakamura, H. A very small frame-shifting deletion within exon 19 of the Duchenne muscular dystrophy gene. Biochem. Biophys. Res. Commun. 1990, 170, 963–967. [Google Scholar] [CrossRef]
- Nicholson, L.V. The “rescue” of dystrophin synthesis in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 1993, 3, 525–531. [Google Scholar] [CrossRef]
- Sherratt, T.G.; Vulliamy, T.; Dubowitz, V.; Sewry, C.A.; Strong, P.N. Exon skipping and translation in patients with frameshift deletions in the dystrophin gene. Am. J. Hum. Genet. 1993, 53, 1007–1015. [Google Scholar]
- Arechavala-Gomeza, V.; Kinali, M.; Feng, L.; Guglieri, M.; Edge, G.; Main, M.; Hunt, D.; Lehovsky, J.; Straub, V.; Bushby, K.; et al. Revertant fibres and dystrophin traces in Duchenne muscular dystrophy: Implication for clinical trials. Neuromuscul. Disord. 2010, 20, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, E.P.; Bronson, A.; Levin, A.A.; Takeda, S.; Yokota, T.; Baudy, A.R.; Connor, E.M. Restoring dystrophin expression in duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. Am. J. Pathol. 2011, 179, 12–22. [Google Scholar] [CrossRef] [Green Version]
- England, S.B.; Nicholson, L.V.B.; Johnson, M.A.; Forrest, S.M.; Love, D.R.; Zubrzycka-Gaarn, E.E.; Bulman, D.E.; Harris, J.B.; Davies, K. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nat. Cell Biol. 1990, 343, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; Van Deutekom, J.; Van Ommen, G.-J.; Den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes 2020, 11, 837. [Google Scholar] [CrossRef]
- Kuntz, N.; Wagner, K.; East, L.; Upadhyay, S.; Han, B.; Koenig, E.; Steiner, D.; Shieh, P. Casimersen treatment in eligible patients with Duchenne muscular dystrophy: Safety, tolerability, and pharmacokinetics over 144 weeks of treatment. Neuromuscul. Disord. 2020, 30, 130–131. [Google Scholar] [CrossRef]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komaki, H.; Takeshima, Y.; Matsumura, T.; Ozasa, S.; Funato, M.; Takeshita, E.; Iwata, Y.; Yajima, H.; Egawa, Y.; Toramoto, T.; et al. Viltolarsen in Japanese Duchenne muscular dystrophy patients: A phase 1/2 study. Ann. Clin. Transl. Neurol. 2020, 7, 2393–2408. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, B.; Shah, S.N.; Lu, P.; Lu, Q. Saponins as Natural Adjuvant for Antisense Morpholino Oligonucleotides Delivery in vitro and in mdx Mice. Mol. Ther. Nucleic Acids 2018, 11, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, J.S.; Hogarth, M.W.; Boehler, J.F.; Nearing, M.; Vila, M.C.; Heredia, R.; Fiorillo, A.A.; Zhang, A.; Hathout, Y.; Hoffman, E.P.; et al. Author Correction: Myoblasts and macrophages are required for therapeutic morpholino antisense oligonucleotide delivery to dystrophic muscle. Nat. Commun. 2018, 9, 1256. [Google Scholar] [CrossRef] [Green Version]
- Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.; Aoki, Y. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine 2019, 45, 630–645. [Google Scholar] [CrossRef] [Green Version]
- Sarepta Therapeutics. Sarepta Therapeutics Announces Positive Clinical Results from MOMENTUM, a Phase 2 Clinical Trial of SRP-5051 in Patients with Duchenne Muscular Dystrophy Amenable to Skipping Exon 51. Available online: https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-announces-positive-clinical-results (accessed on 17 December 2020).
- Sardone, V.; Zhou, H.; Muntoni, F.; Ferlini, A.; Falzarano, M.S. Antisense Oligonucleotide-Based Therapy for Neuromuscular Disease. Molecules 2017, 22, 563. [Google Scholar] [CrossRef] [Green Version]
- Echevarría, L.; Aupy, P.; Relizani, K.; Bestetti, T.; Griffith, G.; Blandel, F.; Komisarski, M.; Haeberli, A.; Svinartchouk, F.; Garcia, L.; et al. Evaluating the Impact of Variable Phosphorothioate Content in Tricyclo-DNA Antisense Oligonucleotides in a Duchenne Muscular Dystrophy Mouse Model. Nucleic Acid Ther. 2019, 29, 148–160. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Wong, B.; Flanigan, K.M.; Wilson, R.; De Kimpe, S.; Lourbakos, A.; Lin, Z.; Campion, G.; DEMAND V Study Group; Iannaccone, S.T.; et al. Placebo-controlled Phase 2 Trial of Drisapersen for Duchenne Muscular Dystrophy. Ann. Clin. Transl. Neurol. 2018, 5, 913–926. [Google Scholar] [CrossRef] [Green Version]
- Goemans, N.; Mercuri, E.; Belousova, E.; Komaki, H.; Dubrovsky, A.; McDonald, C.M.; Kraus, J.E.; Lourbakos, A.; Lin, Z.; Campion, G.; et al. A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.-F.E.; Aartsma-Rus, A. Developments in reading frame restoring therapy approaches for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2020, 19, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Biomarin. FDA Issues Complete Response Letter for KyndrisaTM for Duchenne Muscular Dystrophy Amenable to Exon 51 Skipping. Available online: https://investors.biomarin.com/2016-01-14-FDA-Issues-Complete-Response-Letter-for-KyndrisaTM-for-Duchenne-Muscular-Dystrophy-Amenable-to-Exon-51-Skipping (accessed on 17 December 2020).
- European Medicines Agency. Withdrawal of the Marketing Authorisation Application for Kyndrisa (Drisapersen). Available online: https://www.ema.europa.eu/en/documents/medicine-qa/questions-answers-withdrawal-marketing-authorisation-application-kyndrisa-drisapersen_en.pdf (accessed on 17 December 2020).
- Wan, W.B.; Migawa, M.T.; Vasquez, G.; Murray, H.M.; Nichols, J.G.; Gaus, H.; Berdeja, A.; Lee, S.; Hart, C.E.; Lima, W.F.; et al. Synthesis, biophysical properties and biological activity of second generation antisense oligonucleotides containing chiral phosphorothioate linkages. Nucleic Acids Res. 2014, 42, 13456–13468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, N.; Butler, D.C.D.; Svrzikapa, N.; Mohapatra, S.; Zlatev, I.; Sah, D.W.Y.; Standley, S.M.; Lu, G.; Apponi, L.H.; Frank-Kamenetsky, M.; et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 845–851. [Google Scholar] [CrossRef]
- Wave Life Sciences. Wave Life Sciences Announces Discontinuation of Suvodirsen Development for Duchenne Muscular Dystrophy. Available online: https://ir.wavelifesciences.com/news-releases/news-release-details/wave-life-sciences-announces-discontinuation-suvodirsen (accessed on 17 December 2020).
- Campbell, C.; Barohn, R.J.; Bertini, E.; Chabrol, B.; Comi, G.P.; Darras, B.T.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Iannaccone, S.T.; et al. Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. J. Comp. Eff. Res. 2020, 9, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Li, M.; Berger, S.; Meilak, M.; Rientjes, J.; Currie, P.D. Effect of Ataluren on dystrophin mutations. J. Cell. Mol. Med. 2020, 24, 6680–6689. [Google Scholar] [CrossRef]
- Barthélémy, F.; Wein, N. Personalized gene and cell therapy for Duchenne Muscular Dystrophy. Neuromuscul. Disord. 2018, 28, 803–824. [Google Scholar] [CrossRef]
- Galibert, L.; Merten, O.-W. Latest developments in the large-scale production of adeno-associated virus vectors in insect cells toward the treatment of neuromuscular diseases. J. Invertebr. Pathol. 2011, 107, S80–S93. [Google Scholar] [CrossRef]
- Łoboda, A.; Dulak, J. Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: Past, present, and future. Pharmacol. Rep. 2020, 72, 1227–1263. [Google Scholar] [CrossRef]
- Crudele, J.M.; Chamberlain, J.S. AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet. 2019, 28, R102–R107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, W.; Hsun, Y.-H.; Chang, K.-J.; Yarmishyn, A.A.; Hsiao, Y.-J.; Chien, Y.; Chien, C.S.; Ma, C.; Yang, Y.-P.; Tsai, P.-H.; et al. Current Genetic Survey and Potential Gene-Targeting Therapeutics for Neuromuscular Diseases. Int. J. Mol. Sci. 2020, 21, 9589. [Google Scholar] [CrossRef]
- Amjad, F.; Fatima, T.; Fayyaz, T.; Khan, M.A.; Qadeer, M.I. Novel genetic therapeutic approaches for modulating the severity of β-thalassemia. Biomed. Rep. 2020, 13, 48. [Google Scholar] [CrossRef]
- Ramos, J.N.; Hollinger, K.; Bengtsson, N.E.; Allen, J.M.; Hauschka, S.D.; Chamberlain, J.S. Development of Novel Mi-cro-dystrophins with Enhanced Functionality. Mol. Ther. 2019, 27, 623–635. [Google Scholar] [CrossRef] [Green Version]
- Watchko, J.; O’Day, T.; Wang, B.; Zhou, L.; Tang, Y.; Li, J.; Xiao, X. Adeno-associated virus vector-mediated mini-dystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum. Gene Ther. 2002, 13, 1451–1460. [Google Scholar] [CrossRef]
- Le Guiner, C.; Servais, L.; Montus, M.; Larcher, T.; Fraysse, B.; Moullec, S.; Allais, M.; François, V.; Dutilleul, M.; Malerba, A.; et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 16105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornegay, J.N.; Li, J.; Bogan, J.R.; Bogan, D.J.; Chen, C.; Zheng, H.; Wang, B.; Qiao, C.; Howard, J.F.; Xiao, X. Widespread Muscle Expression of an AAV9 Human Mini-dystrophin Vector After Intravenous Injection in Neonatal Dystrophin-deficient Dogs. Mol. Ther. 2010, 18, 1501–1508. [Google Scholar] [CrossRef]
- Martino, A.T.; Suzuki, M.; Markusic, D.M.; Zolotukhin, I.; Ryals, R.C.; Moghimi, B.; Ertl, H.C.J.; Muruve, D.A.; Lee, B.; Herzog, R.W. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9–dependent innate immune responses in the liver. Blood 2011, 117, 6459–6468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Morales, L.; Malik, A.S.; Mead, A.F.; Greer, C.D.; Mitchell, M.A.; Petrov, M.T.; Su, L.T.; Choi, M.E.; Rosenblum, S.T.; et al. Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat. Med. 2019, 25, 1505–1511. [Google Scholar] [CrossRef]
- Solid Biosciences. Solid Biosciences Announces Clinical Hold On SGT-001 Phase I/II Clinical Trial for Duchenne Muscular Dystrophy. Available online: https://investors.solidbio.com/news-releases/news-release-details/solid-biosciences-announces-clinical-hold-sgt-001-phase-iii (accessed on 19 December 2020).
- Sarepta Therapeutics Reports Sustained Functional Improvement Two Years After Treatment with SRP-9001, its Investigational Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Available online: https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-reports-sustained-functional-improvement (accessed on 17 December 2020).
- Thomas, P.J.; Xu, R.; Martin, P.T. B4GALNT2 (GALGT2) Gene Therapy Reduces Skeletal Muscle Pathology in the FKRP P448L Mouse Model of Limb Girdle Muscular Dystrophy 2I. Am. J. Pathol. 2016, 186, 2429–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.T.; Xu, R.; Rodino-Klapac, L.R.; Oglesbay, E.; Camboni, M.; Montgomery, C.L.; Shontz, K.; Chicoine, L.G.; Clark, K.R.; Sahenk, Z.; et al. Overexpression of Galgt2 in skeletal muscle prevents injury resulting from eccentric contractions in both mdx and wild-type mice. Am. J. Physiol. Physiol. 2009, 296, C476–C488. [Google Scholar] [CrossRef] [Green Version]
- Chicoine, L.G.; Rodino-Klapac, L.R.; Shao, G.; Xu, R.; Bremer, W.G.; Camboni, M.; Golden, B.; Montgomery, C.L.; Shontz, K.; Heller, K.N.; et al. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin α2 surrogates. Mol. Ther. 2014, 22, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Jia, Y.; Zygmunt, D.A.; Martin, P.T. rAAVrh74.MCK.GALGT2 Protects against Loss of Hemodynamic Function in the Aging mdx Mouse Heart. Mol. Ther. 2019, 27, 636–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Epinat, J.C.; Arnould, S.; Chames, P.; Rochaix, P.; Desfontaines, D.; Puzin, C.; Patin, A.; Zanghellini, A.; Pâques, F.; Lacroix, E. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res. 2003, 31, 2952–2962. [Google Scholar] [CrossRef] [Green Version]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless modi-fication of wild-type induced pluripotent stem cells to the natural CCR5Δ32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef] [Green Version]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9, eaan8081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Rivera, R.M.C.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2015, 351, 403–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2015, 351, 407–411. [Google Scholar] [CrossRef] [Green Version]
- El Refaey, M.; Xu, L.; Gao, Y.; Canan, B.D.; Adesanya, T.A.; Warner, S.C.; Akagi, K.; Symer, D.E.; Mohler, P.J.; Ma, J.; et al. In Vivo Genome Editing Restores Dystrophin Expression and Cardiac Function in Dystrophic Mice. Circ. Res. 2017, 121, 923–929. [Google Scholar] [CrossRef]
- Min, Y.-L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019, 5, eaav4324. [Google Scholar] [CrossRef] [Green Version]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.-R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Chemello, F.; Bassel-Duby, R.; Olson, E.N. Correction of muscular dystrophies by CRISPR gene editing. J. Clin. Investig. 2020, 130, 2766–2776. [Google Scholar] [CrossRef]
- Hakim, C.H.; Wasala, N.B.; Nelson, C.E.; Wasala, L.P.; Yue, Y.; Louderman, J.A.; Lessa, T.B.; Dai, A.; Zhang, K.; Jenkins, G.J.; et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.E.; Wu, Y.; Gemberling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Rivera, R.M.C.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.; Mali, P.; Wu, E.Y.; Ng, A.H.; Zhu, K.; Wagers, A.J.; Church, G.M. A multi-functional AAV-CRISPR-Cas9 and its host response. Nat. Methods 2016, 13, 868–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matre, P.R.; Mu, X.; Wu, J.; Danila, D.; Hall, M.A.; Kolonin, M.G.; Darabi, R.; Huard, J. CRISPR/Cas9-Based Dystrophin Restoration Reveals a Novel Role for Dystrophin in Bioenergetics and Stress Resistance of Muscle Progenitors. Stem Cells 2019, 37, 1615–1628. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.C.; Sincennes, M.-C.; Chevalier, F.P.; Brun, C.E.; Lacaria, M.; Segalés, J.; Muñoz-Cánoves, P.; Ming, H.; Rudnicki, M.A. The Dystrophin Glycoprotein Complex Regulates the Epigenetic Activation of Muscle Stem Cell Commitment. Cell Stem Cell 2018, 22, 755–768.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Serra, C.; Lee, G.; Wagner, K.R. Stem cell-based therapies for Duchenne muscular dystrophy. Exp. Neurol. 2020, 323, 113086. [Google Scholar] [CrossRef]
- Fukada, S.I. The roles of muscle stem cells in muscle injury, atrophy and hypertrophy. J. Biochem. 2018, 163, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Huard, J. Stem cells, blood vessels, and angiogenesis as major determinants for musculoskeletal tissue repair. J. Orthop. Res. 2019, 37, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Previtali, S.C.; Napolitano, S.; Cicalese, M.P.; Tedesco, F.S.; Nicastro, F.; Noviello, M.; Roostalu, U.; Sora, M.G.N.; Scarlato, M.; et al. Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol. Med. 2015, 7, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Angelis, M.G.C.-D.; Berghella, L.; Coletta, M.; Lattanzi, L.; Zanchi, M.; Gabriella, M.; Ponzetto, C.; Cossu, G. Skeletal Myogenic Progenitors Originating from Embryonic Dorsal Aorta Coexpress Endothelial and Myogenic Markers and Contribute to Postnatal Muscle Growth and Regeneration. J. Cell Biol. 1999, 147, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Minasi, M.G.; Riminucci, M.; De Angelis, L.; Borello, U.; Berarducci, B.; Innocenzi, A.; Caprioli, A.; Sirabella, D.; Baiocchi, M.; De Maria, R.; et al. The meso-angioblast: A multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 2002, 129, 2773–2783. [Google Scholar]
- Dellavalle, A.; Maroli, G.; Covarello, D.; Azzoni, E.; Innocenzi, A.; Perani, L.; Antonini, S.; Sambasivan, R.; Brunelli, S.; Tajbakhsh, S.; et al. Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat. Commun. 2011, 2, 499. [Google Scholar] [CrossRef] [Green Version]
- Dellavalle, A.; Sampaolesi, M.; Tonlorenzi, R.; Tagliafico, E.; Sacchetti, B.; Perani, L.; Innocenzi, A.; Gálvez, B.G.; Messina, G.; Morosetti, R.; et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 2007, 9, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Sampaolesi, M.; Torrente, Y.; Innocenzi, A.; Tonlorenzi, R.; D’Antona, G.; Pellegrino, M.A.; Barresi, R.; Bresolin, N.; De Angelis, M.G.C.; Campbell, K.P.; et al. Cell Therapy of -Sarcoglycan Null Dystrophic Mice Through Intra-Arterial Delivery of Mesoangioblasts. Science 2003, 301, 487–492. [Google Scholar] [CrossRef] [Green Version]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.-L.; Galvez, B.G.; Barthélémy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nat. Cell Biol. 2006, 444, 574–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.; Jefferies, J.; Byrne, B.; Lima, J.; Ambale-Venkatesh, B.; Ostovaneh, M.R.; Makkar, R.; Goldstein, B.; Smith, R.R.; Fudge, J.; et al. Cardiac and skeletal muscle effects in the randomized HOPE-Duchenne trial. Neurology 2019, 92, e866–e878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagoe-Suzuki, Y.; Takeda, S. Skeletal muscle generated from induced pluripotent stem cells—Induction and application. World J. Stem Cells 2017, 9, 89–97. [Google Scholar] [PubMed]
- Abujarour, R.; Bennett, M.; Valamehr, B.; Lee, T.T.; Robinson, M.; Robbins, D.; Le, T.; Lai, K.; Flynn, P. Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl. Med. 2014, 3, 149–160. [Google Scholar] [CrossRef]
- Shoji, E.; Sakurai, H.; Nishino, T.; Nakahata, T.; Heike, T.; Awaya, T.; Fujii, N.; Manabe, Y.; Matsuo, M.; Sehara-Fujisawa, A. Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep. 2015, 5, 12831. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.Y.; Lim, H.; Estrellas, K.; Mula, J.; Cohen, T.V.; Zhang, Y.; Donnelly, C.J.; Richard, J.P.; Kim, Y.J.; Kim, H.; et al. Concordant but Varied Phenotypes among Duchenne Muscular Dystrophy Patient-Specific Myoblasts Derived using a Human iPSC-Based Model. Cell Rep. 2016, 15, 2301–2312. [Google Scholar] [CrossRef] [Green Version]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Uchimura, T.; Otomo, J.; Sato, M.; Sakurai, H. A human iPS cell myogenic differentiation system permitting high-throughput drug screening. Stem Cell Res. 2017, 25, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Maffioletti, S.M.; Sarcar, S.; Henderson, A.; Mannhardt, I.; Pinton, L.; Moyle, L.A.; Steele-Stallard, H.; Cappellari, O.; Wells, K.E.; Ferrari, G.; et al. Three-Dimensional Human iPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. 2018, 23, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Mondragon-Gonzalez, R.; Perlingeiro, R.C.R. Recapitulating muscle disease phenotypes with myotonic dystrophy 1 induced pluripotent stem cells: A tool for disease modeling and drug discovery. Dis. Model. Mech. 2018, 11, dmm034728. [Google Scholar] [CrossRef] [Green Version]
- Cordova, G.; Negroni, E.; Cabello-Verrugio, C.; Mouly, V.; Trollet, C. Combined Therapies for Duchenne Muscular Dystrophy to Optimize Treatment Efficacy. Front. Genet. 2018, 9, 114. [Google Scholar] [CrossRef] [Green Version]
- Tinsley, J.; Deconinck, N.; Fisher, R.; Kahn, D.; Phelps, S.; Gillis, J.-M.; Davies, K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998, 4, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Squire, S.E.; Edwards, B.; Chen, H.; Burns, D.T.; Shah, N.; Babbs, A.; Davies, S.G.; Wynne, G.M.; Russell, A.J.; et al. Second-generation compound for the modulation of utrophin in the therapy of DMD. Hum. Mol. Genet. 2015, 24, 4212–4224. [Google Scholar] [CrossRef]
- Moorwood, C.; Lozynska, O.; Suri, N.; Napper, A.D.; Diamond, S.L.; Khurana, T.S. Drug Discovery for Duchenne Muscular Dystrophy via Utrophin Promoter Activation Screening. PLoS ONE 2011, 6, e26169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinazzola, J.M.; Kunkel, L.M. Pharmacological therapeutics targeting the secondary defects and downstream pathology of Duchenne muscular dystrophy. Expert Opin. Orphan Drugs 2016, 4, 1179–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiraud, S.; Davies, K.E. Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Chemical Modification | Therapeutic Molecule | Company | Skipped Exon | Clinical Trial | Phase | Duration |
|---|---|---|---|---|---|---|
| Phosphorodi-amidate morpholino oligomers (PMO) | Eteplirsen | Sarepta Ther. | Exon 51 | NCT03218995 NCT04179409 NCT03992430 NCT03985878 | Phase 2 Phase 2 Phase 3 Phase 2 | 2017–2021 2020–2022 2020–2026 2019–2027 |
| Golodirsen | Sarepta Ther. | Exon 53 | NCT04179409 NCT02500381 NCT03532542 | Phase 2 Phase 3 Phase 3 | 2020–2022 2016–2023 2018–2026 | |
| Casimersen | Sarepta Ther. | Exon 45 | NCT04179409 NCT03532542 | Phase 2 Phase 3 | 2020–2022 2018–2026 | |
| Viltolarsen | NS Pharma, Inc. | Exon 53 | NCT03167255 NCT04060199 | Phase 2 Phase 3 | 2017–2021 2020–2024 | |
| 2′-O-Methyl-phosphorothioates (2’OMePS) | Drisapersen | BioMarin Pharmaceutical | Exon 51 | NCT02636686 | Phase 3 | 2015–2018 |
| DS-5141b | Daiichi Sankyo Co., Ltd. | Exon 45 | NCT04433234 | Phase 2 | 2020–2022 | |
| Peptide phosphorodiamidate morpholino oligomer (PPMO) | SRP-5051 | Sarepta Ther. | Exon 51 | NCT03675126 NCT04004065 | Phase 1/2 Phase 2 | 2018–2024 2019–2022 |
| Stereopure | Suvodirsen | Wave Life Sciences Ltd. | Exon 51 | NCT03907072 | Phase 2/3 | 2019–2020 |
| Name | Chemical modification | Properties |
|---|---|---|
| Phosphorodiamidate morpholino oligomers (PMO) |
| Advantages
|
| Peptide phosphorodiamidate morpholino oligomer (PPMO) | Conjugation with the cell-penetrating peptide (CPP) | Advantages
Nephrotoxicity correlated with arginine content of the CPP |
| 2’-O-methyl-phosphorothioates (2’OMePS) |
| Advantages
Toxicity and adverse effects due to retention in the kidneys and liver |
| Stereopure 2’OMePS | Stereochemical and chemical purity (defined stereochemistry at each PS linkage) | Advantages
No induction of dystrophin expression in vivo |
| Sponsor | Clinical Trials. Gov Identifier | Trial Name | Study Phase | Drug Name | AAV-Serotype | Primary Outcome | Secondary Outcome | Side Effects |
|---|---|---|---|---|---|---|---|---|
| Solid Biosciences, LLC | NCT03368742 | Micro-dystrophin Gene Transfer Study in Adolescents and Children With DMD (IGNITE DMD) | Phase I and II, open-label, randomized, controlled | SGT-001 | AAV-9 Muscle (skeletal and cardiac) tissue tropism | Safety and microdystrophin expression in biopsy | / | A serious adverse event (SAE) characterized by complement activation, thrombocytopenia, a decrease in red blood cell count, acute kidney injury, and cardio-pulmonary insufficiency |
| Sarepta Therapeutics, Inc. | NCT03375164 | Systemic Gene Delivery Clinical Trial for Duchenne Muscular Dystrophy (DMD) | Phase I and II, open-label, non-randomized | rAAVrh74.MHCK7. Micro-dystrophin | AAV-rh74 Muscle (skeletal and cardiac) tissue tropism | Safety | Microdystrophin expression in biopsy and motor performances | No SAEs; Adverse events reported: elevated γ-glutamil transpeptidase; transient nausea |
| Pfizer | NCT03362502 | A Study to Evaluate the Safety and Tolerability of PF-06939926 Gene Therapy in Duchenne Muscular Dystrophy | Phase Ib, open-label, non-randomized | PF-06939926 | AAV-9 Muscle (skeletal and cardiac) tissue tropism | Safety and tolerability | Micro-dystrophin expression in biopsy | Three SAEs fully recovered: persistent vomiting; acute kidney injury with atypical hemolytic uremic syndrome (aHUS)-like complement activation; thrombocytopenia with aHUS-like complement activation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fortunato, F.; Rossi, R.; Falzarano, M.S.; Ferlini, A. Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy. J. Clin. Med. 2021, 10, 820. https://doi.org/10.3390/jcm10040820
Fortunato F, Rossi R, Falzarano MS, Ferlini A. Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy. Journal of Clinical Medicine. 2021; 10(4):820. https://doi.org/10.3390/jcm10040820
Chicago/Turabian StyleFortunato, Fernanda, Rachele Rossi, Maria Sofia Falzarano, and Alessandra Ferlini. 2021. "Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy" Journal of Clinical Medicine 10, no. 4: 820. https://doi.org/10.3390/jcm10040820
APA StyleFortunato, F., Rossi, R., Falzarano, M. S., & Ferlini, A. (2021). Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy. Journal of Clinical Medicine, 10(4), 820. https://doi.org/10.3390/jcm10040820