
Skeletal Muscle Subpopulation Rearrangements upon Rhabdomyosarcoma Development through Single-Cell Mass Cytometry

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Strains and Animal Procedures
2.2. In Vivo Fluorescence Molecular Tomography
2.3. Histological Analysis
2.4. Immunofluorescence
2.5. Muscle Mononuclear and eRMS Cell Purification
2.6. Flow Cytometry
2.7. Cell Culture
2.8. In Vitro Infection of Sorted Cells
2.9. RNA Extraction, Retro-Transcription and Real-Time PCR
2.10. Sphere Formation Assay
2.11. Tumor-Induction Assay
2.12. Single-Cell Mass Cytometry
2.13. CyTOF Data Processing
2.14. Statistical Analysis
3. Results
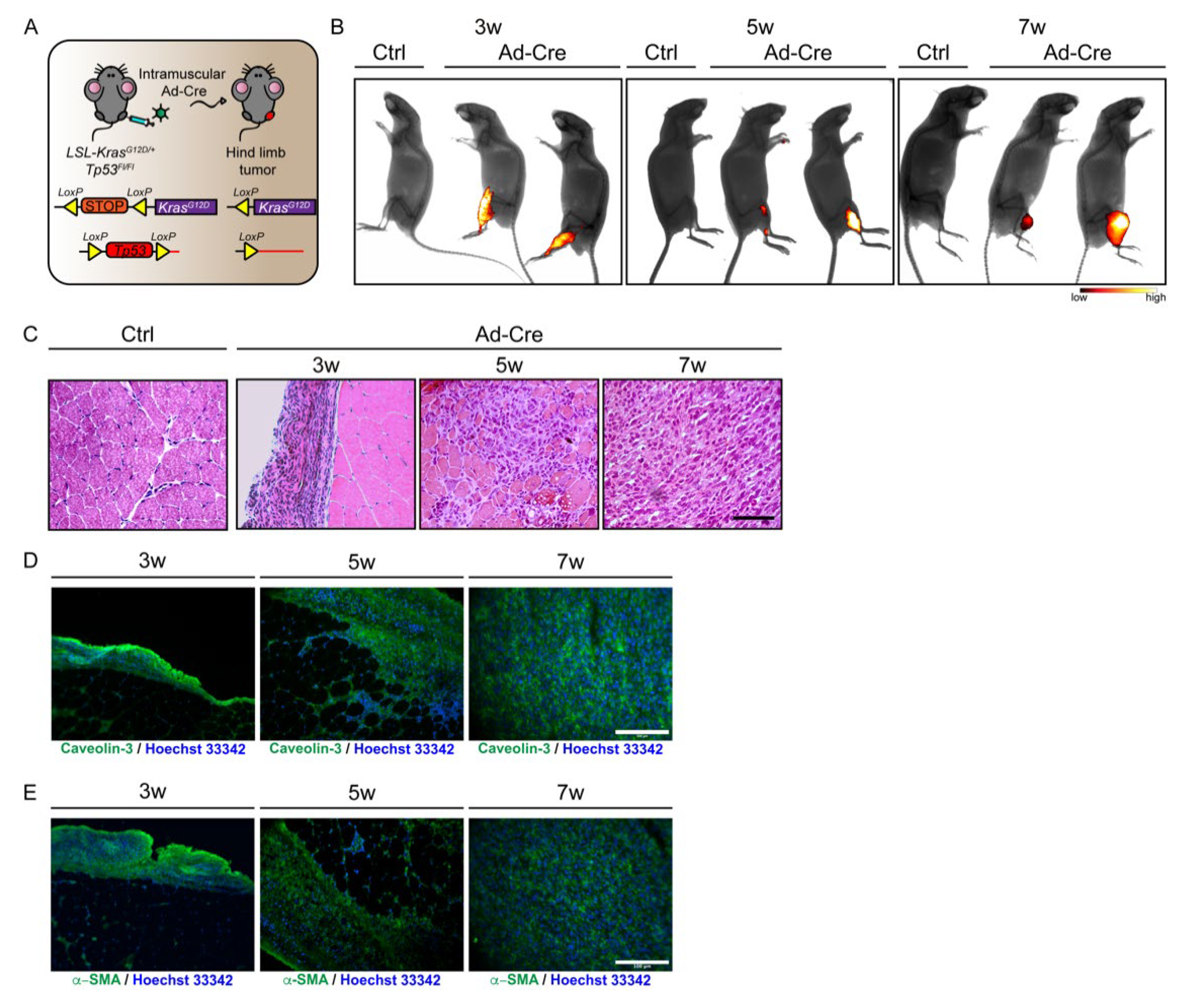
3.1. Generation of eRMS in LSL-KrasG12D/+;Tp53Fl/Fl Mice
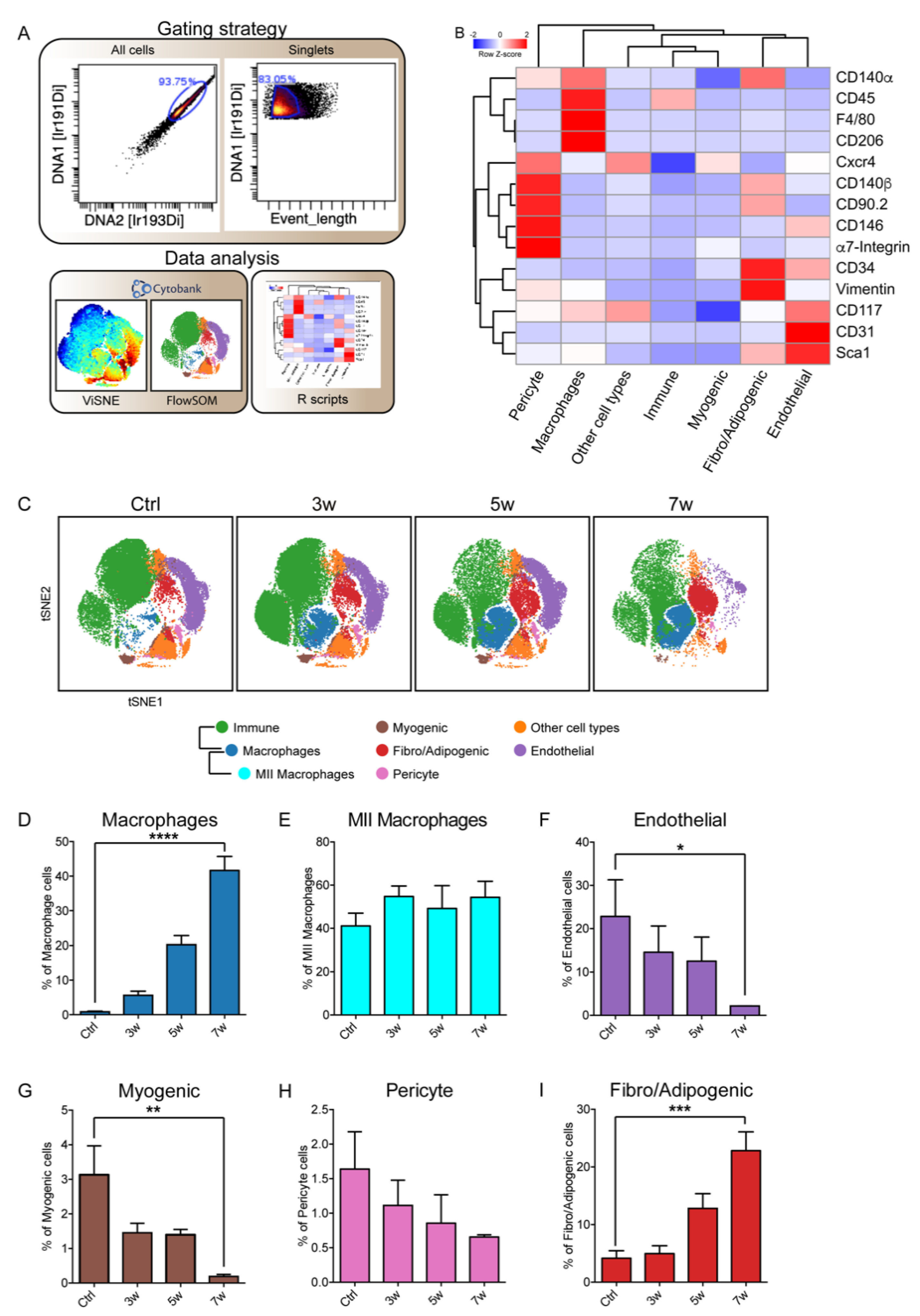
3.2. Immunophenotyping of Skeletal-Muscle Mononuclear Cell Populations upon eRMS Induction
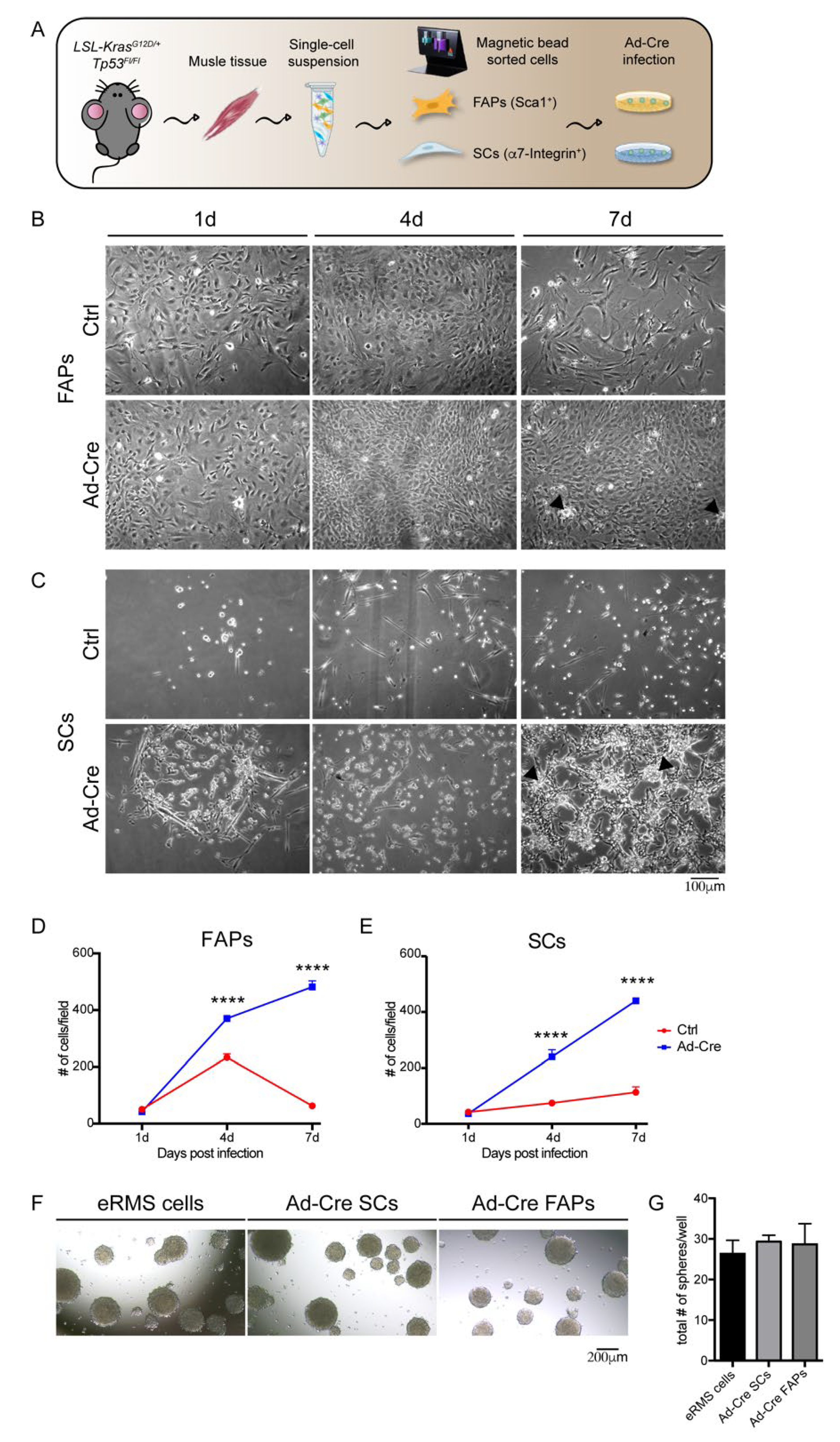
3.3. Skeletal-Muscle Progenitor Populations (FAPs and SCs) Isolated from LSL-KrasG12D/+;Tp53Fl/Fl Mice Promote Tumor Transformation
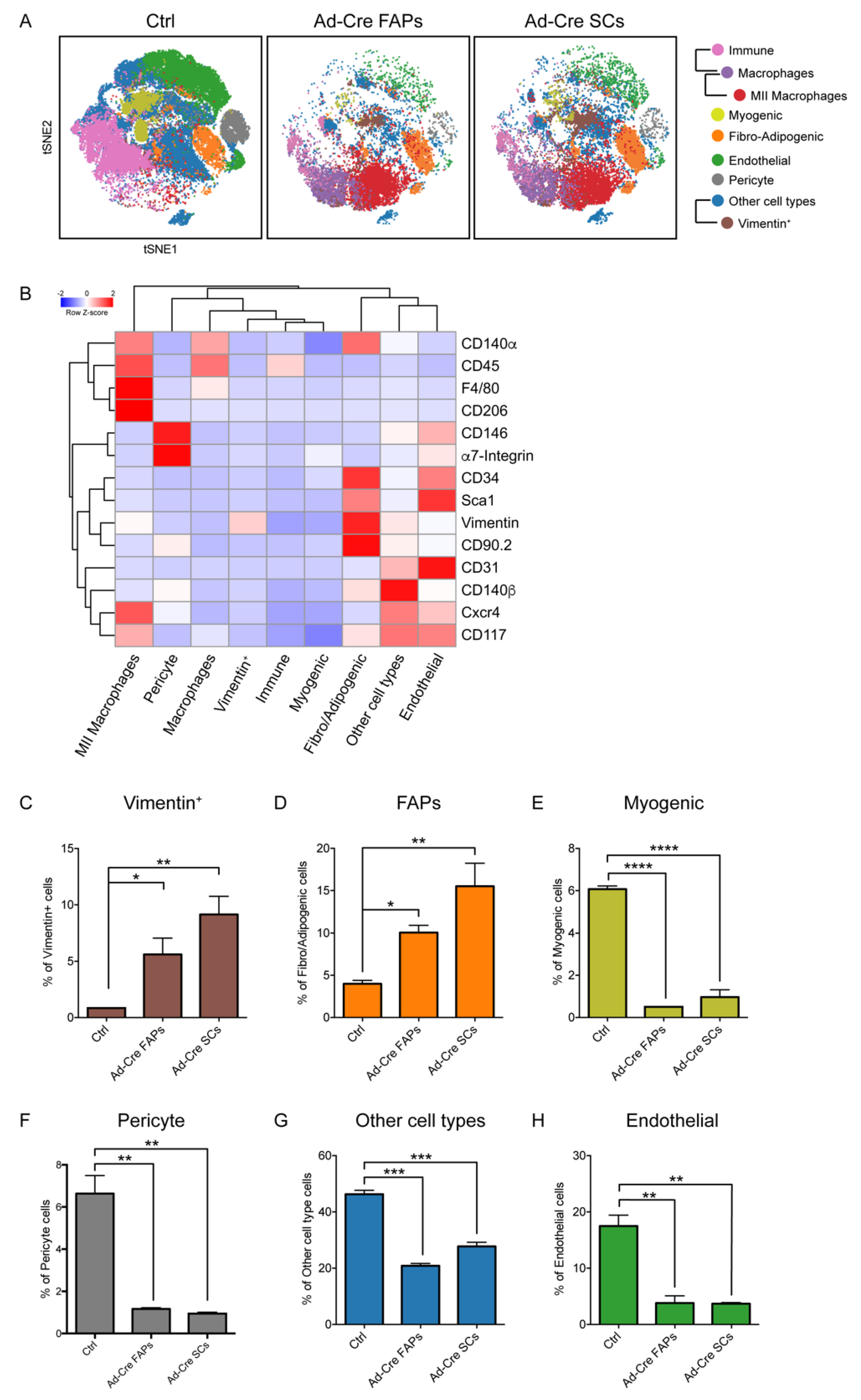
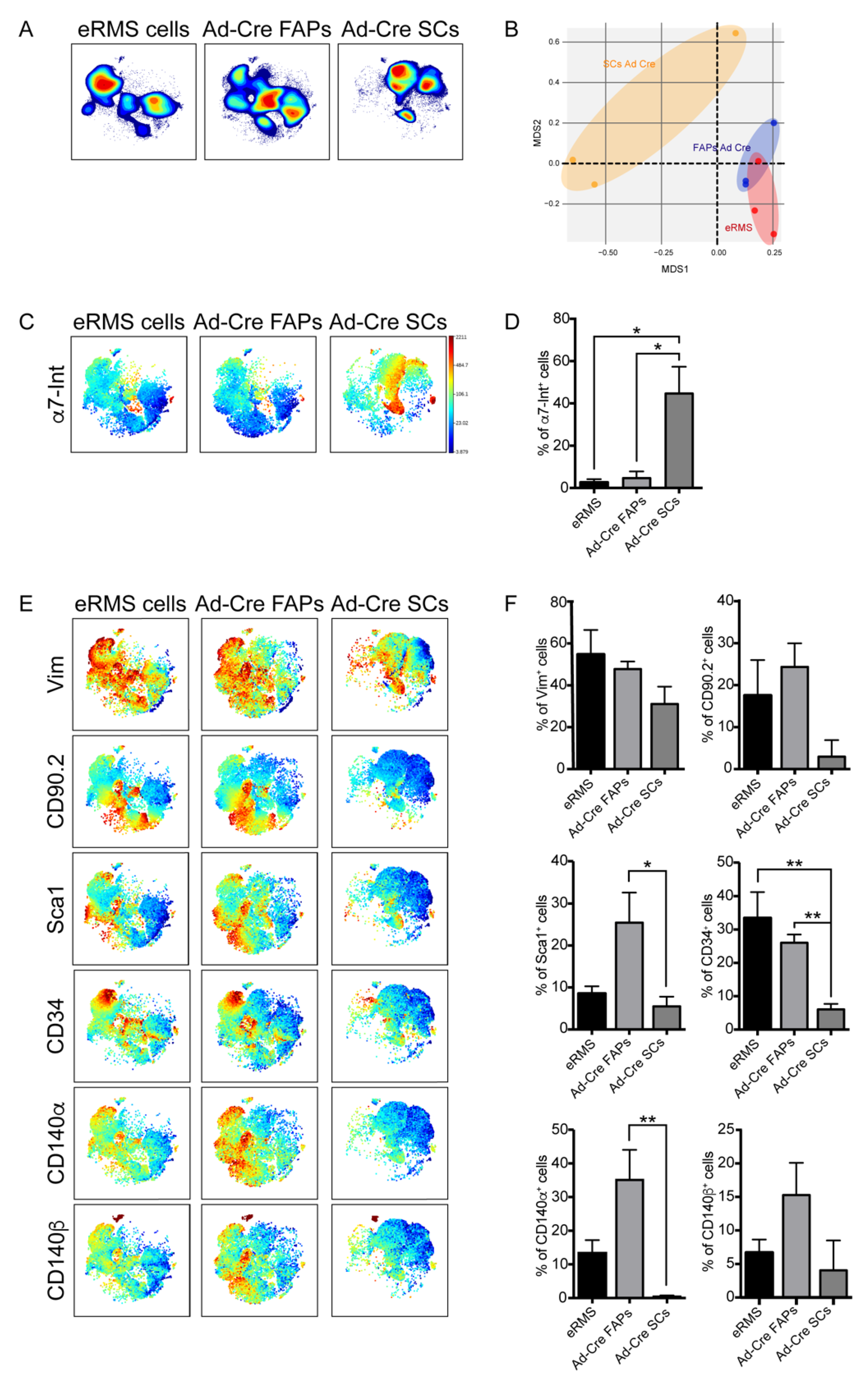
3.4. Immunophenotyping of the In Vitro-Transformed Progenitor Cells Emphasizes FAPs as Candidates eRMS Origin-Cell
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, C. Childhood Rhabdomyosarcoma. J. Dent. Res. 2011, 91, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soleimani, V.D.; Rudnicki, M.A. New Insights into the Origin and the Genetic Basis of Rhabdomyosarcomas. Cancer Cell 2011, 19, 157–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ognjanovic, S.; Linabery, A.M.; Charbonneau, B.; Ross, J.A. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer 2009, 115, 4218–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiffin, N.; Williams, R.D.; Shipley, J.; Pritchard-Jones, K. PAX7 expression in embryonal rhabdomyosarcoma suggests an origin in muscle satellite cells. Br. J. Cancer 2003, 89, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Sebire, N.J. Myogenin and MyoD1 expression in paediatric rhabdomyosarcomas. J. Clin. Pathol. 2003, 56, 412–416. [Google Scholar] [CrossRef]
- Morotti, R.A.; Nicol, K.K.; Parham, D.M.; Teot, L.A.; Moore, J.; Hayes, J.; Meyer, W.; Qualman, S.J. An Immunohistochemical Algorithm to Facilitate Diagnosis and Subtyping of Rhabdomyosarcoma: The Children’s Oncology Group Experience. Am. J. Surg. Pathol. 2006, 30, 962–968. [Google Scholar] [CrossRef]
- Rubin, B.P.; Nishijo, K.; Chen, H.-I.H.; Yi, X.; Schuetze, D.P.; Pal, R.; Prajapati, S.I.; Abraham, J.; Arenkiel, B.R.; Chen, Q.-R.; et al. Evidence for an Unanticipated Relationship between Undifferentiated Pleomorphic Sarcoma and Embryonal Rhabdomyosarcoma. Cancer Cell 2011, 19, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, A.M.; Missiaglia, E.; Galli, G.G.; Hettmer, S.; Urcia, R.; Carrara, M.; Judson, R.N.; Thway, K.; Nadal, G.; Selfe, J.L.; et al. The Hippo Transducer YAP1 Transforms Activated Satellite Cells and Is a Potent Effector of Embryonal Rhabdomyosarcoma Formation. Cancer Cell 2014, 26, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Blum, J.M.; Añó, L.; Li, Z.; Van Mater, D.; Bennett, B.D.; Sachdeva, M.; Lagutina, I.; Zhang, M.; Mito, J.K.; Dodd, L.G.; et al. Distinct and overlapping sarcoma subtypes initiated from muscle stem and progenitor cells. Cell Rep. 2013, 5, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta BBA Bioenerg. 2010, 1805, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asakura, A.; Rudnicki, M.A.; Komaki, M. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation 2001, 68, 245–253. [Google Scholar] [CrossRef]
- De Coppi, P.; Bellini, S.; Conconi, M.T.; Sabatti, M.; Simonato, E.; Gamba, P.G.; Nussdorfer, G.G.; Parnigotto, P.P. Myoblast–Acellular Skeletal Muscle Matrix Constructs Guarantee a Long-Term Repair of Experimental Full-Thickness Abdominal Wall Defects. Tissue Eng. 2006, 12, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Yada, E.; Yamanouchi, K.; Nishihara, M. Adipogenic Potential of Satellite Cells from Distinct Skeletal Muscle Origins in the Rat. J. Veter. Med. Sci. 2006, 68, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Shefer, G.; Wleklinski-Lee, M.; Yablonka-Reuveni, Z. Skeletal muscle satellite cells can spontaneously enter an alternative mesenchymal pathway. J. Cell Sci. 2004, 117, 5393–5404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 Innate Signals Stimulate Fibro/Adipogenic Progenitors to Facilitate Muscle Regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Palacios, D.; Mozzetta, C.; Consalvi, S.; Caretti, G.; Saccone, V.; Proserpio, V.; Marquez, V.E.; Valente, S.; Mai, A.; Forcales, S.V.; et al. TNF/p38α/Polycomb Signaling to Pax7 Locus in Satellite Cells Links Inflammation to the Epigenetic Control of Muscle Regeneration. Cell Stem Cell 2010, 7, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Pannérec, A.; Marazzi, G.; Sassoon, D. Stem cells in the hood: The skeletal muscle niche. Trends Mol. Med. 2012, 18, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-E.; Jin, B.; Li, Y.-P. TNF-α regulates myogenesis and muscle regeneration by activating p38 MAPK. Am. J. Physiol. Physiol. 2007, 292, C1660–C1671. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.N.; Low, M.; Eisner, C.; Rossi, F.M. Isolation, Culture, and Differentiation of Fibro/Adipogenic Progenitors (FAPs) from Skeletal Muscle. Adv. Struct. Saf. Stud. 2017, 1668, 93–103. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.-A.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef] [Green Version]
- Kirsch, D.G.; Dinulescu, D.M.; Miller, J.B.; Grimm, J.; Santiago, P.M.; Young, N.P.; Nielsen, G.P.; Quade, B.J.; Chaber, C.J.; Schultz, C.P.; et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat. Med. 2007, 13, 992–997. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Finck, R.; Simonds, E.F.; Jager, A.; Krishnaswamy, S.; Sachs, K.; Fantl, W.; Pe’Er, D.; Nolan, G.P.; Bendall, S.C. Normalization of mass cytometry data with bead standards. Cytom. Part A 2013, 83, 483–494. [Google Scholar] [CrossRef]
- Kotecha, N.; Krutzik, P.O.; Irish, J.M. Web-Based Analysis and Publication of Flow Cytometry Experiments. Curr. Protoc. Cytom. 2010, 53, 10.17.1–10.17.24. [Google Scholar] [CrossRef]
- Van Der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Amir, E.-A.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’Er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 2015, 87, 636–645. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Grimm, J.; Kirsch, D.G.; Windsor, S.D.; Kim, C.F.B.; Santiago, P.M.; Ntziachristos, V.; Jacks, T.; Weissleder, R. Use of gene expression profiling to direct in vivo molecular imaging of lung cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 14404–14409. [Google Scholar] [CrossRef] [Green Version]
- Riedlinger, W.F.J.; Kozakewich, H.P.W.; Vargas, S.O. Myogenic Markers in the Evaluation of Embryonal Botryoid Rhabdomyosarcoma of the Female Genital Tract. Pediatr. Dev. Pathol. 2005, 8, 355–361. [Google Scholar] [CrossRef]
- Bodenmiller, B.; Zunder, E.R.; Finck, R.; Chen, T.J.; Savig, E.S.; Bruggner, R.V.; Simonds, E.F.; Bendall, S.C.; Sachs, K.; Krutzik, P.O.; et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat. Biotechnol. 2012, 30, 858–867. [Google Scholar] [CrossRef]
- Bjornson, Z.B.; Nolan, G.P.; Fantl, W.J. Single-cell mass cytometry for analysis of immune system functional states. Curr. Opin. Immunol. 2013, 25, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Reggio, A.; Rosina, M.; Palma, A.; Perpetuini, A.C.; Petrilli, L.L.; Gargioli, C.; Fuoco, C.; Micarelli, E.; Giuliani, G.; Cerretani, M.; et al. Adipogenesis of skeletal muscle fibro/adipogenic progenitors is affected by the WNT5a/GSK3/β-catenin axis. Cell Death Differ. 2020, 27, 2921–2941. [Google Scholar] [CrossRef]
- Joe, A.W.B.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M.V. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 2011, 124, 3654–3664. [Google Scholar] [CrossRef] [Green Version]
- Petrilli, L.L.; Spada, F.; Palma, A.; Reggio, A.; Rosina, M.; Gargioli, C.; Castagnoli, L.; Fuoco, C.; Cesareni, G. High-Dimensional Single-Cell Quantitative Profiling of Skeletal Muscle Cell Population Dynamics during Regeneration. Cells 2020, 9, 1723. [Google Scholar] [CrossRef]
- Malecova, B.; Gatto, S.; Etxaniz, U.; Passafaro, M.; Cortez, A.; Nicoletti, C.; Giordani, L.; Torcinaro, A.; De Bardi, M.; Bicciato, S.; et al. Dynamics of cellular states of fibro-adipogenic progenitors during myogenesis and muscular dystrophy. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Giuliani, G.; Vumbaca, S.; Fuoco, C.; Gargioli, C.; Giorda, E.; Massacci, G.; Palma, A.; Reggio, A.; Riccio, F.; Rosina, M.; et al. SCA-1 micro-heterogeneity in the fate decision of dystrophic fibro/adipogenic progenitors. Cell Death Dis. 2021, 12, 1–24. [Google Scholar] [CrossRef]
- The Tabula Muris Consortium; Overall coordination; Logistical coordination; Organ collection and processing; Library preparation and sequencing; Computational data analysis; Cell type annotation; Writing Group; Supplemental text writing group; Principal Investigators. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [CrossRef]
- Marinkovic, M.; Fuoco, C.; Sacco, F.; Perpetuini, A.C.; Giuliani, G.; Micarelli, E.; Pavlidou, T.; Petrilli, L.L.; Reggio, A.; Riccio, F.; et al. Fibro-adipogenic progenitors of dystrophic mice are insensitive to NOTCH regulation of adipogenesis. Life Sci. Alliance 2019, 2, e201900437. [Google Scholar] [CrossRef] [Green Version]
- Uezumi, A.; Fukada, S.-I.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef]
- Burkin, D.J.; Kaufman, S.J. The α7β1 integrin in muscle development and disease. Cell Tissue Res. 1999, 296, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.A.; Apponi, L.H.; Long, K.K.; Pavlath, G.K. Chemokine expression and control of muscle cell migration during myogenesis. J. Cell Sci. 2010, 123, 3052–3060. [Google Scholar] [CrossRef] [Green Version]
- Dellavalle, A.; Sampaolesi, M.; Tonlorenzi, R.; Tagliafico, E.; Sacchetti, B.; Perani, L.; Innocenzi, A.; Gálvez, B.G.; Messina, G.; Morosetti, R.; et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 2007, 9, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Birbrair, A.; Zhang, T.; Wang, Z.-M.; Messi, M.L.; Enikolopov, G.N.; Mintz, A.; Delbono, O. Role of Pericytes in Skeletal Muscle Regeneration and Fat Accumulation. Stem Cells Dev. 2013, 22, 2298–2314. [Google Scholar] [CrossRef] [Green Version]
- Newman, P.J.; Berndt, M.C.; Gorski, J.; White, G.C.; Lyman, S.; Paddock, C.; Muller, W.A. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science 1990, 247, 1219–1222. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Muller, W.A. Fluorescencein SituHybridization Mapping of the Mouse Platelet Endothelial Cell Adhesion Molecule-1 (PECAM1) to Mouse Chromosome 6, Region F3–G1. Genomics 1996, 37, 226–228. [Google Scholar] [CrossRef]
- Li, T.-S.; Hamano, K.; Nishida, M.; Hayashi, M.; Ito, H.; Mikamo, A.; Matsuzaki, M. CD117+ stem cells play a key role in therapeutic angiogenesis induced by bone marrow cell implantation. Am. J. Physiol. Circ. Physiol. 2003, 285, H931–H937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, S.; Wei, J.; Pentinmikko, N.; Leinonen, H.; Salvén, P. Generation of Functional Blood Vessels from a Single c-kit+ Adult Vascular Endothelial Stem Cell. PLoS Biol. 2012, 10, e1001407. [Google Scholar] [CrossRef]
- Avdalyan, A.; Bobrov, I.; Klimachev, V.; Lazarev, A. Prognostic Value of Microvessel Density in Tumor and Peritumoral Area as Evaluated by CD31 Protein Expression and Argyrophilic Nucleolar Organizer Region Count in Endothelial Cells in Uterine Leiomyosarcoma. Sarcoma 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paolo, V.; Russo, I.; Boldrini, R.; Ravà, L.; Pezzullo, M.; Benedetti, M.C.; Galardi, A.; Colletti, M.; Rota, R.; Orlando, D.; et al. Evaluation of Endoglin (CD105) expression in pediatric rhabdomyosarcoma. BMC Cancer 2018, 18, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libura, J.; Drukala, J.; Majka, M.; Tomescu, O.; Navenot, J.M.; Kucia, M.; Marquez, L.; Peiper, S.C.; Barr, F.G.; Janowska-Wieczorek, A.; et al. CXCR4–SDF-1 signaling is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood 2002, 100, 2597–2606. [Google Scholar] [CrossRef] [Green Version]
- Anastasi, S.; Giordano, S.; Sthandier, O.; Gambarotta, G.; Maione, R.; Comoglio, P.; Amati, P. A Natural Hepatocyte Growth Factor/Scatter Factor Autocrine Loop in Myoblast Cells and the Effect of the Constitutive Met Kinase Activation on Myogenic Differentiation. J. Cell Biol. 1997, 137, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, D.; Wold, B.J. Single-Cell Analysis of Regulatory Gene Expression in Quiescent and Activated Mouse Skeletal Muscle Satellite Cells. Dev. Biol. 1997, 191, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, R.; Anderson, J.E.; Nevoret, C.J.; Halevy, O.; Allen, R.E. HGF/SF Is Present in Normal Adult Skeletal Muscle and Is Capable of Activating Satellite Cells. Dev. Biol. 1998, 194, 114–128. [Google Scholar] [CrossRef] [Green Version]
- Hettmer, S.; Liu, J.; Miller, C.M.; Lindsay, M.C.; Sparks, C.A.; Guertin, D.A.; Bronson, R.T.; Langenau, D.M.; Wagers, A.J. Sarcomas induced in discrete subsets of prospectively isolated skeletal muscle cells. Proc. Natl. Acad. Sci. USA 2011, 108, 20002–20007. [Google Scholar] [CrossRef] [Green Version]
- Sesillo, F.B.; Fox, D.; Sacco, A. Muscle Stem Cells Give Rise to Rhabdomyosarcomas in a Severe Mouse Model of Duchenne Muscular Dystrophy. Cell Rep. 2019, 26, 689–701.e6. [Google Scholar] [CrossRef] [Green Version]
- Hatley, M.E.; Tang, W.; Garcia, M.R.; Finkelstein, D.; Millay, D.P.; Liu, N.; Graff, J.; Galindo, R.L.; Olson, E.N. A Mouse Model of Rhabdomyosarcoma Originating from the Adipocyte Lineage. Cancer Cell 2012, 22, 536–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, C.J.; Hanna, J.A.; Garcia, M.R.; Devine, D.J.; Heyrana, A.J.; Finkelstein, D.; Rehg, J.E.; Hatley, M.E. Hedgehog Pathway Drives Fusion-Negative Rhabdomyosarcoma Initiated from Non-myogenic Endothelial Progenitors. Cancer Cell 2018, 33, 108–124.e5. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.-X.; Finckenstein, F.G.; Abdueva, D.A.; Shahbazian, V.; Chung, B.; Weinberg, K.I.; Triche, T.J.; Shimada, H.; Anderson, M.J. Mouse Mesenchymal Stem Cells Expressing PAX-FKHR Form Alveolar Rhabdomyosarcomas by Cooperating with Secondary Mutations. Cancer Res. 2008, 68, 6587–6597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, R.D.; Mito, J.K.; Kirsch, D.G. Animal models of soft-tissue sarcoma. Dis. Models Mech. 2010, 3, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Huh, W.W.; Bejar, D.E. Rhabdomyosarcoma in adolescent and young adult patients: Current perspectives. Adolesc. Health Med. Ther. 2014, 5, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccarelli, C.; Vulcano, F.; Milazzo, L.; Gravina, G.L.; Marampon, F.; Macioce, G.; Giampaolo, A.; Tombolini, V.; Di Paolo, V.; Hassan, H.J.; et al. Key role of MEK/ERK pathway in sustaining tumorigenicity and in vitro radioresistance of embryonal rhabdomyosarcoma stem-like cell population. Mol. Cancer 2016, 15, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Hatina, J.; Kripnerova, M.; Houfkova, K.; Pesta, M.; Kuncova, J.; Sana, J.; Slaby, O.; Rodríguez, R. Sarcoma Stem Cell Heterogeneity. Adv. Exp. Med. Biol. 2019, 1123, 95–118. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Metal |
|---|---|
| Anti-mouse CD146 | 141Pr |
| Anti-mouse CD34 | 144Nd |
| Anti-mouse F4/80 | 146Nd |
| Anti-mouse CD45 | 147Sm |
| Anti-mouse CD140α | 148Nd |
| Anti-mouse CD25 (Il-2R) | 150Nd |
| Anti-mouse CD140β | 151Eu |
| Anti-mouse CD274 | 153 Eu |
| Anti-Vimentin | 154Sm |
| Anti-mouse CD90.2 (Thy-1.2) | 156Gd |
| Anti-mouse Cxcr4 (CD184) | 159Tb |
| Anti-mouse α7-Integrin | 161Dy |
| Anti-mouse Sca1 (Ly-6A/E) | 164Dy |
| Anti-CD31 (Pecam1) | 165Ho |
| Anti-mouse CD117 | 166Er |
| Anti-mouse CD206 | 169Tm |
| Anti-mouse CD4 | 172Yb |
| Anti-Pan-Actin | 175Lu |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrilli, L.L.; Riccio, F.; Giuliani, G.; Palma, A.; Gargioli, C.; Vumbaca, S.; Faron, M.; Palmieri, G.; Pasquini, L.; Sacco, F.; et al. Skeletal Muscle Subpopulation Rearrangements upon Rhabdomyosarcoma Development through Single-Cell Mass Cytometry. J. Clin. Med. 2021, 10, 823. https://doi.org/10.3390/jcm10040823
Petrilli LL, Riccio F, Giuliani G, Palma A, Gargioli C, Vumbaca S, Faron M, Palmieri G, Pasquini L, Sacco F, et al. Skeletal Muscle Subpopulation Rearrangements upon Rhabdomyosarcoma Development through Single-Cell Mass Cytometry. Journal of Clinical Medicine. 2021; 10(4):823. https://doi.org/10.3390/jcm10040823
Chicago/Turabian StylePetrilli, Lucia Lisa, Federica Riccio, Giulio Giuliani, Alessandro Palma, Cesare Gargioli, Simone Vumbaca, Monika Faron, Graziana Palmieri, Luca Pasquini, Francesca Sacco, and et al. 2021. "Skeletal Muscle Subpopulation Rearrangements upon Rhabdomyosarcoma Development through Single-Cell Mass Cytometry" Journal of Clinical Medicine 10, no. 4: 823. https://doi.org/10.3390/jcm10040823
APA StylePetrilli, L. L., Riccio, F., Giuliani, G., Palma, A., Gargioli, C., Vumbaca, S., Faron, M., Palmieri, G., Pasquini, L., Sacco, F., Cesareni, G., Castagnoli, L., & Fuoco, C. (2021). Skeletal Muscle Subpopulation Rearrangements upon Rhabdomyosarcoma Development through Single-Cell Mass Cytometry. Journal of Clinical Medicine, 10(4), 823. https://doi.org/10.3390/jcm10040823