
Chordoma—Current Understanding and Modern Treatment Paradigms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Embryology and Pathophysiology
2.1. Embryology of the Notochord
2.2. Chordomagenesis
3. Epidemiology
4. Clinical Presentation
5. Diagnosis
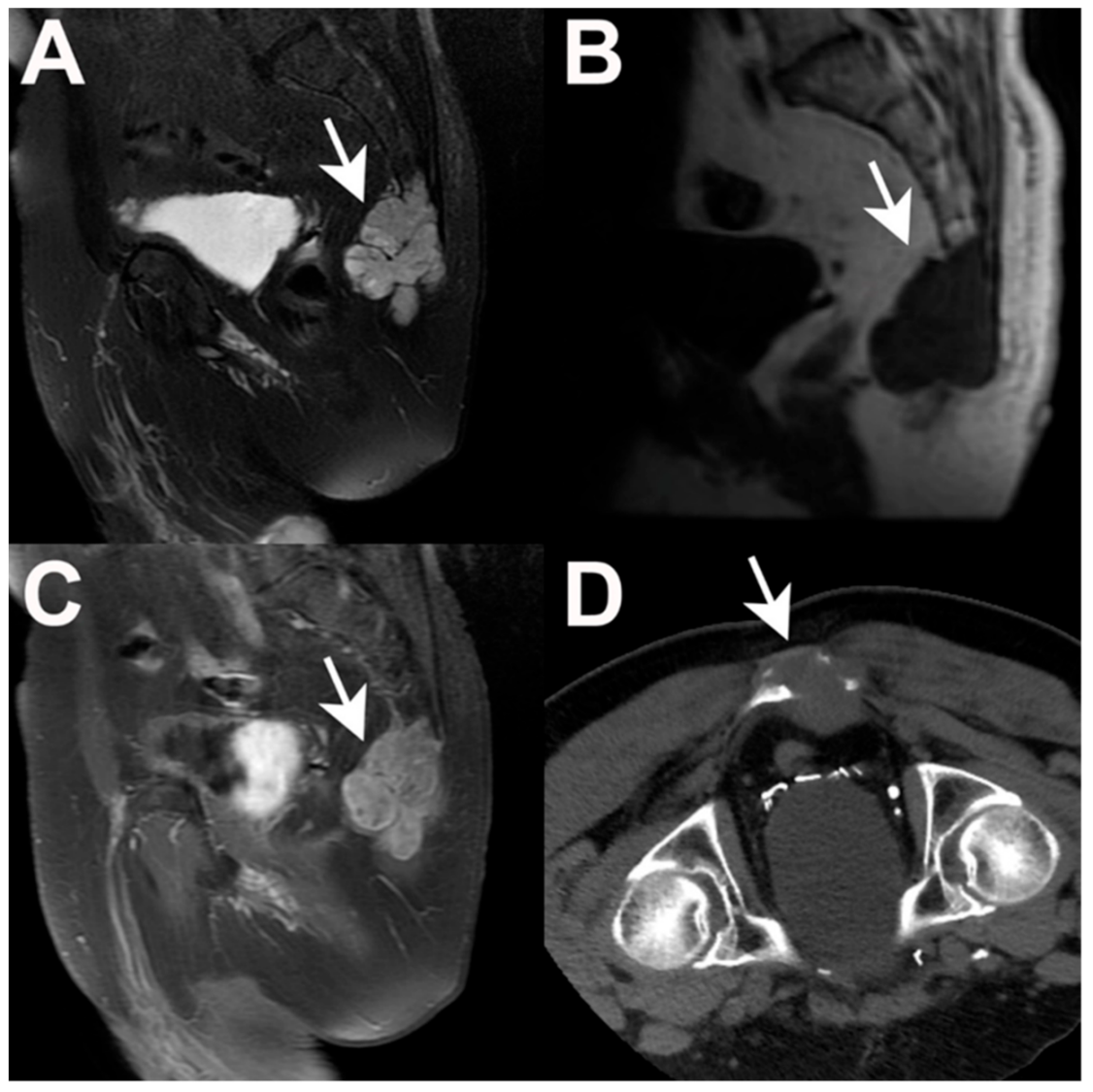
5.1. Imaging
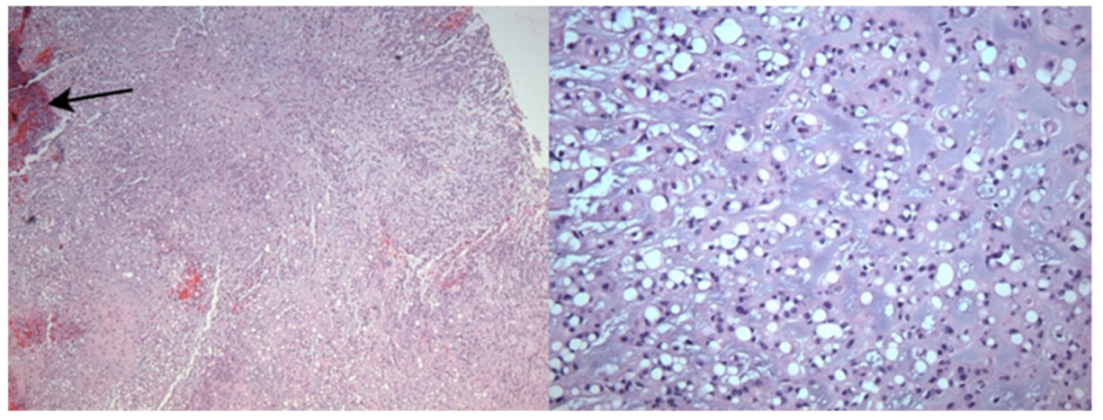
5.2. Histopathology
6. Treatment
6.1. Surgery
6.1.1. Biopsy and Preoperative Workup
6.1.2. Skull Base Chordomas
6.1.3. Mobile Spine Chordomas
6.1.4. Sacral Chordomas
6.2. Radiotherapy
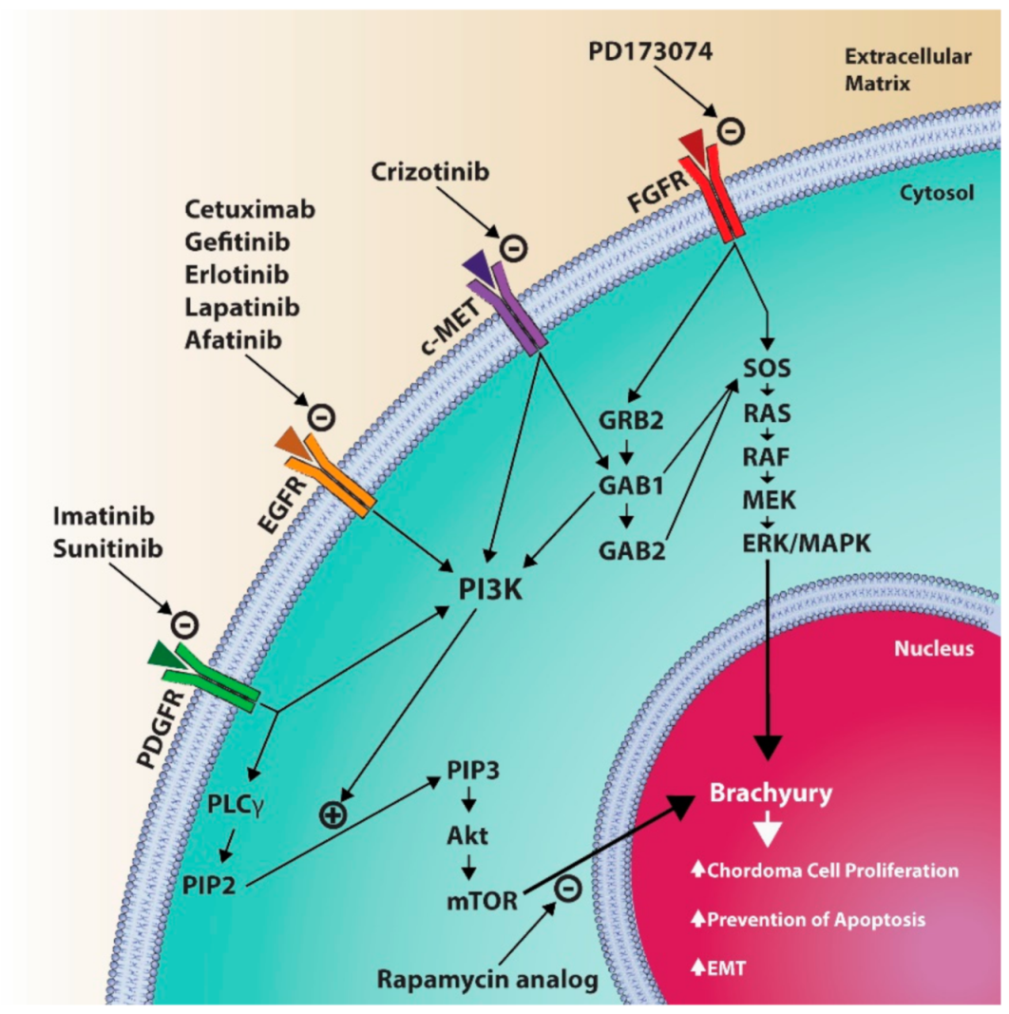
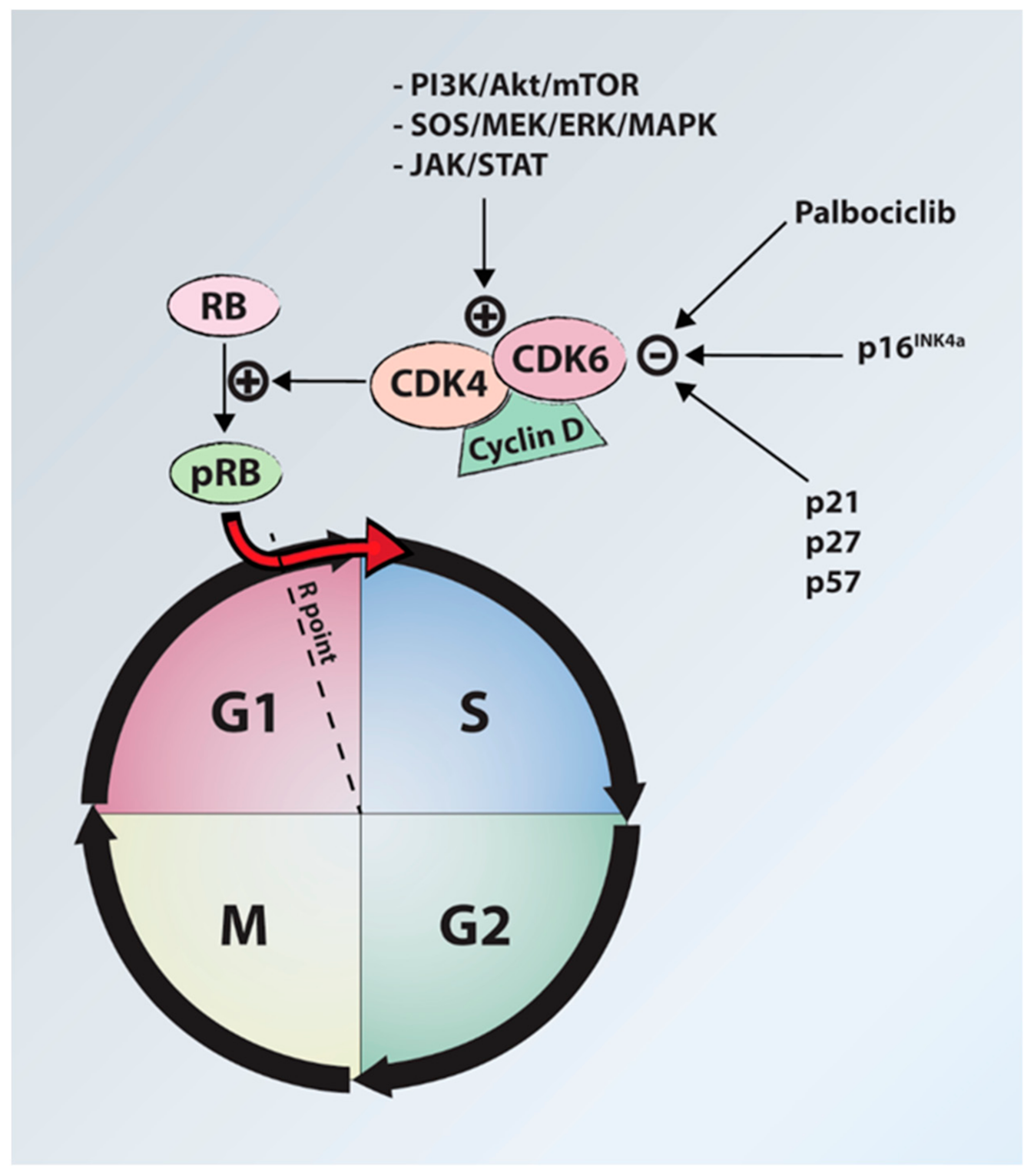
6.3. Medical Treatments
7. Prognosis
8. Expert Opinion
Author Contributions
Funding
Conflicts of Interest
References
- Virchow, R. Untersuchungen Ueber Die Entwicklung Des Schaedelgrundes; Druck und Verlag von Georg Reamer: Berlin, Germany, 1857. [Google Scholar]
- Fletcher, C.D.M.; Unni, K.K.; Mertens, F. (Eds.) World Health Organization Classification of Tumours. In Pathology and Genetics of Tumours of Soft Tissue and Bone, 4th ed.; IARC Press: Lyon, France, 2002. [Google Scholar]
- McMaster, M.L.; Goldstein, A.M.; Bromley, C.M.; Ishibe, N.; Parry, D.M. Chordoma: Incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control 2001, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Z.; Tian, K.; Wang, K.; Li, D.; Ma, J.; Jia, G.; Zhang, L.; Zhang, J. Clinical features and surgical outcomes of patients with skull base chordoma: A retrospective analysis of 238 patients. J. Neurosurg. 2017, 127, 1257–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, P.; Szoverfi, Z.; Fisher, C.G.; Boriani, S.; Gokaslan, Z.L.; Dekutoski, M.B.; Chou, D.; Quraishi, N.A.; Reynolds, J.J.; Luzzati, A.; et al. Surgical treatment of sacral chordoma: Prognostic variables for local recurrence and overall survival. Eur. Spine J. 2014, 24, 1092–1101. [Google Scholar] [CrossRef]
- Gokaslan, Z.L.; Zadnik, P.L.; Sciubba, D.M.; Germscheid, N.M.; Goodwin, C.R.; Wolinsky, J.-P.; Bettegowda, C.; Groves, M.L.; Luzzati, A.; Rhines, L.D.; et al. Mobile spine chordoma: Results of 166 patients from the AOSpine Knowledge Forum Tumor database. J. Neurosurg. Spine 2015, 24, 644–651. [Google Scholar] [CrossRef] [Green Version]
- Ailon, T.; Torabi, R.; Fisher, C.G.; Rhines, L.D.; Clarke, M.J.; Bettegowda, C.; Boriani, S.; Yamada, Y.J.; Kawahara, N.; Varga, P.P.; et al. Management of locally recurrent chordoma of the mobile spine and sacrum: A systematic review. Spine 2016, 41, S193–S198. [Google Scholar] [CrossRef] [PubMed]
- York, J.E.; Kaczaraj, A.; Abi-Said, D.; Fuller, G.N.; Skibber, J.M.; Janjan, N.A.; Gokaslan, Z.L. Sacral chordoma: 40-Year experience at a major cancer center. Neurosurgery 1999, 44, 74–79. [Google Scholar] [CrossRef]
- Boriani, S.; Chevalley, F.; Weinstein, J.N.; Biagini, R.; Campanacci, L.; De Iure, F.; Piccill, P. Chordoma of the spine above the sacrum. Treatment and outcome in 21 cases. Spine 1996, 21, 1569–1577. [Google Scholar] [CrossRef]
- Koga, T.; Shin, M.; Saito, N. Treatment with high marginal dose is mandatory to achieve long-term control of skull base chordomas and chondrosarcomas by means of stereotactic radiosurgery. J. Neuro-Oncol. 2010, 98, 233–238. [Google Scholar] [CrossRef]
- George, B.; Bresson, D.; Herman, P.; Froelich, S. Chordomas: A Review. Neurosurg. Clin. N. Am. 2015, 26, 437–452. [Google Scholar] [CrossRef]
- Bergh, P.; Kindblom, L.G.; Gunterberg, B.; Remotti, F.; Ryd, W.; Meis-Kindblom, J.M. Prognostic factors in chordoma of the sacrum and mobile spine: A study of 39 patients. Cancer 2000, 88, 2122–2134. [Google Scholar] [CrossRef]
- Ruggieri, P.; Angelini, A.; Ussia, G.; Montalti, M.; Mercuri, M. Surgical margins and local control in resection of sacral chordomas. Clin. Orthop. Relat. Res. 2010, 468, 2939–2947. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, B.; Dickey, I.D.; Yaszemski, M.J.; Inwards, C.Y.; Sim, F.H. Operative management of sacral chordoma. J. Bone Jt. Surgery-American Vol. 2005, 87, 2211. [Google Scholar] [CrossRef]
- George, B.; Bresson, D.; Bouazza, S.; Froelich, S.; Mandonnet, E.; Hamdi, S.; Orabi, M.; Polivka, M.; Cazorla, A.; Adle-Biassette, H.; et al. Les chordomes. Neurochirurgie 2014, 60, 63–140. [Google Scholar] [CrossRef]
- Hsieh, P.C.; Xu, R.; Sciubba, D.M.; McGirt, M.J.; Nelson, C.; Witham, T.F.; Wolinksy, J.-P.; Gokaslan, Z.L. Long-term clinical outcomes following en bloc resections for sacral chordomas and chondrosarcomas: A series of twenty consecutive patients. Spine 2009, 34, 2233–2239. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Casali, P.G.; Vullo, S.L.; Mariani, L.; Palassini, E.; Mercuri, M.; Alberghini, M.; Pilotti, S.; Zanella, L.; Gronchi, A.; et al. Chordoma of the mobile spine and sacrum: A retrospective analysis of a series of patients surgically treated at two referral centers. Ann. Surg. Oncol. 2009, 17, 211–219. [Google Scholar] [CrossRef]
- Tzortzidis, F.; Elahi, F.; Wright, D.; Natarajan, S.K.; Sekhar, L.N. Patient outcome at long-term follow-up after aggressive microsurgical resection of cranial base chordomas. Neurosurgery 2006, 59, 230–237. [Google Scholar] [CrossRef]
- Sahgal, A.; Chan, M.W.; Atenafu, E.G.; Masson-Cote, L.; Bahl, G.; Yu, E.; Millar, B.-A.; Chung, C.; Catton, C.; O’Sullivan, B.; et al. Image-guided, intensity-modulated radiation therapy (IG-IMRT) for skull base chordoma and chondrosarcoma: Preliminary outcomes. Neuro Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Delaney, T.F.; Liebsch, N.J.; Pedlow, F.X.; Adams, J.; Ba, E.A.W.; Yeap, B.Y.; Ms, N.D.; Nielsen, G.P.; Harmon, D.C.; Yoon, S.S.; et al. Long-term results of Phase II study of high dose photon/proton radiotherapy in the management of spine chordomas, chondrosarcomas, and other sarcomas. J. Surg. Oncol. 2014, 110, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, R.L.; Folkert, W.; Liebsch, N.J.; Chen, Y.-L.E.; Pedlow, F.X.; Schwab, J.H.; Rosenberg, A.E.; Nielsen, G.P.; Szymonifka, J.; Ferreira, A.E.; et al. High-dose proton-based radiation therapy in the management of spine chordomas: Outcomes and clinicopathological prognostic factors. J. Neurosurg. Spine 2015, 23, 788–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillay, N.; Plagnol, V.; Tarpey, P.S.; Lobo, S.B.; Presneau, N.; Szuhai, K.; Halai, D.; Berisha, F.; Cannon, S.R.; Mead, S.; et al. A common single-nucleotide variant in T is strongly associated with chordoma. Nat. Genet. 2012, 44, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Longhi, A.; Ferraresi, V.; Grignani, G.; Comandone, A.; Stupp, R.; Bertuzzi, A.; Tamborini, E.; Pilotti, S.; Messina, A.; et al. Phase II study of imatinib in advanced chordoma. J. Clin. Oncol. 2012, 30, 914–920. [Google Scholar] [CrossRef]
- Yang, C.; Schwab, J.H.; Schoenfeld, A.J.; Hornicek, F.J.; Wood, K.B.; Nielsen, G.P.; Choy, E.; Mankin, H.; Duan, Z. A novel target for treatment of chordoma: Signal transducers and activators of transcription 3. Mol. Cancer Ther. 2009, 8, 2597–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salisbury, J.; Deverell, M.H.; Cookson, M.J.; Whimster, W.F. Three-dimensional reconstruction of human embryonic notochords: Clue to the pathogenesis of chordoma. J. Pathol. 1993, 171, 59–62. [Google Scholar] [CrossRef]
- Walcott, B.P.; Nahed, B.V.; Mohyeldin, A.; Coumans, J.-V.; Kahle, K.T.; Ferreira, M.J. Chordoma: Current concepts, management, and future directions. Lancet Oncol. 2012, 13, e69–e76. [Google Scholar] [CrossRef]
- Vujovic, S.; Henderson, S.; Presneau, N.; Odell, E.; Jacques, T.S.; Tirabosco, R.; Boshoff, C.; Flanagan, A.M. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J. Pathol. 2006, 209, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.R.; Ng, D.; Alcorta, D.A.; Liebsch, N.J.; Sheridan, E.; Li, S.; Goldstein, A.M.; Parry, D.M.; Kelley, M.J. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat. Genet. 2009, 41, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Stemple, D.L. Structure and function of the notochord: An essential organ for chordate development. Development 2005, 132, 2503–2512. [Google Scholar] [CrossRef] [Green Version]
- Mehnert, F.; Beschorner, R.; Küker, W.; Hahn, U.; Nägele, T. Retroclival ecchordosis physaliphora: MR imaging and review of the literature. Am. J. Neuroradiol. 2004, 25, 1851–1855. [Google Scholar] [CrossRef]
- Kreshak, J.; Larousserie, F.; Picci, P.; Boriani, S.; Mirra, J.; Merlino, B.; Brunocilla, E.; Vanel, D. Difficulty distinguishing benign notochordal cell tumor from chordoma further suggests a link between them. Cancer Imaging 2014, 14, 4. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.-S.; Cohn, M.J.; Harfe, B.D. Identification of nucleus pulposus precursor cells and notochordal remnants in the mouse: Implications for disk degeneration and chordoma formation. Dev. Dyn. 2008, 237, 3953–3958. [Google Scholar] [CrossRef] [Green Version]
- Yakkioui, Y.; Temel, Y.; Creytens, D.; Jahanshahi, A.; Fleischeuer, R.; Santegoeds, R.G.C.; van Overbeeke, J.J. A Comparison of Cell-Cycle Markers in Skull Base and Sacral Chordomas. World Neurosurg. 2014, 82, e311–e318. [Google Scholar] [CrossRef]
- Horbinski, C.; Oakley, G.J.; Cieply, K.; Mantha, G.S.; Nikiforova, M.N.; Dacic, S.; Seethala, R.R. The prognostic value of Ki-67, p53, epidermal growth factor receptor, 1p36, 9p21, 10q23, and 17p13 in skull base chordomas. Arch. Pathol. Lab. Med. 2010, 134, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.; Mohyeldin, A.; Shah, S.R.; Ap Rhys, C.M.; Johnson, L.F.; Sedora-Roman, N.I.; Kosztowski, T.A.; Awad, O.A.; McCarthy, E.F.; Loeb, D.M.; et al. Generation of chordoma cell line JHC7 and the identification of Brachyury as a novel molecular target. J. Neurosurg. 2011, 115, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Le, L.P.; Nielsen, G.P.; Rosenberg, A.E.; Thomas, D.; Batten, J.M.; Deshpande, V.; Schwab, J.; Duan, Z.; Xavier, R.J.; Hornicek, F.J.; et al. Recurrent chromosomal copy number alterations in sporadic chordomas. PLoS ONE 2011, 6, e18846. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Sasaki, H.; Kimura, T.; Miwa, T.; Takahashi, S.; Kawase, T.; Yoshida, K. Molecular and clinical risk factors for recurrence of skull base chordomas: Gain on chromosome 2p, expression of brachyury, and lack of irradiation negatively correlate with patient prognosis. J. Neurosurg. 2018, 128, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Otani, R.; Mukasa, A.; Shin, M.; Omata, M.; Takayanagi, S.; Tanaka, S.; Ueki, K.; Saito, N. Brachyury gene copy number gain and activation of the PI3K/Akt pathway: Association with upregulation of oncogenic Brachyury expression in skull base chordoma. J. Neurosurg. 2018, 128, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, S.; Schwab, J.H.; Nielsen, G.P.; Choy, E.; Ye, S.; Zhang, Z.; Mankin, H.; Hornicek, F.J.; Duan, Z. Tissue Microarray Immunohistochemical Detection of Brachyury Is Not a Prognostic Indicator in Chordoma. PLoS ONE 2013, 8, e75851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Mintz, A.; Shah, S.R.; Quinones-Hinojosa, A.; Hsu, W. The FGFR/MEK/ERK/brachyury pathway is critical for chordoma cell growth and survival. Carcinogenesis 2014, 35, 1491–1499. [Google Scholar] [CrossRef] [Green Version]
- Tamborini, E.; Miselli, F.; Negri, T.; Lagonigro, M.S.; Staurengo, S.; Dagrada, G.P.; Stacchiotti, S.; Pastore, E.; Gronchi, A.; Perrone, F.; et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin. Cancer Res. 2006, 12, 6920–6928. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, P.M.; Yu, Z.; Kowalski, D.; Joe, J.; Manger, P.; Psyrri, A.; Sasaki, C. Differential expression of epidermal growth factor receptor, c-Met, and HER2/neu in chordoma compared with 17 other malignancies. Arch. Otolaryngol. Head Neck Surg. 2005, 131, 707. [Google Scholar] [CrossRef] [Green Version]
- Presneau, N.; Shalaby, A.; Idowu, B.; Gikas, P.D.; Cannon, S.R.; Gout, I.; Diss, T.C.; Tirabosco, R.; Flanagan, A.M. Potential therapeutic targets for chordoma: PI3K/AKT/TSC1/TSC2/mTOR pathway. Br. J. Cancer 2009, 100, 1406–1414. [Google Scholar] [CrossRef]
- Cheng, E.Y.; Özerdemoglu, R.A.; Transfeldt, E.E.; Thompson, R.C. Lumbosacral chordoma: Prognostic factors and treatment. Spine 1999, 24, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Fourney, D.R.; Gokaslan, Z.L. Current management of sacral chordoma. Neurosurg. Focus 2003, 15, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Boriani, S.; Bandiera, S.; Biagini, R.; Bacchini, P.; Boriani, L.; Cappuccio, M.; Chevalley, F.; Gasbarrini, A.; Picci, P.; Weinstein, J.N. Chordoma of the mobile spine: Fifty years of experience. Spine 2006, 31, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Disler, D.G.; Miklic, D. Imaging findings in tumors of the sacrum. Am. J. Roentgenol. 1999, 173, 1699–1706. [Google Scholar] [CrossRef]
- Kendall, B.E.; Lee, B.C.P. Cranial chordomas. Br. J. Radiol. 1977, 50, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Yui, T.; Hayashi, T.; Honma, H.; Suzuki, T.; Takemura, H.; Suzaki, H. A Case of Extraosseous Chordoma of the Nasopharynx. Pract. Otorhinolaryngol. 2012, 105, 1165–1170. [Google Scholar] [CrossRef]
- Antonelli, M.; Raso, A.; Mascelli, S.; Gessi, M.; Nozza, P.; Coli, A.; Gardiman, M.P.; Arcella, A.; Massimino, M.; Buttarelli, F.R.; et al. SMARCB1/INI1 Involvement in Pediatric Chordoma: A Mutational and Immunohistochemical Analysis. Am. J. Surg. Pathol. 2017, 41, 56–61. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Vanel, D.; Righi, A.; Donati, D.M.; Errani, C. Parosteal extra-axial chordoma of the second metacarpal bone: A case report with literature review. Skelet. Radiol. 2018, 47, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.; Cote, G.; Chebib, I.; Choy, E.; DeLaney, T.; Deshpande, V.; Hornicek, F.; Miao, R.; Schwab, J.; Nielsen, G.; et al. Clinicopathologic characteristics of poorly differentiated chordoma. Mod. Pathol. 2018, 31, 1237–1245. [Google Scholar] [CrossRef]
- Hasselblatt, M.; Thomas, C.; Hovestadt, V.; Schrimpf, D.; Johann, P.; Bens, S.; Oyen, F.; Peetz-Dienhart, S.; Crede, Y.; Wefers, A.; et al. Poorly differentiated chordoma with SMARCB1/INI1 loss: A distinct molecular entity with dismal prognosis. Acta Neuropathol. 2016, 132, 149–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMaster, M.L.; Goldstein, A.M.; Parry, D.M. Clinical features distinguish childhood chordoma associated with tuberous sclerosis complex (TSC) from chordoma in the general paediatric population. J. Med. Genet. 2011, 48, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Ranger, A.; Szymczak, A. DO intracranial neoplasms differ in ollier disease and maffucci syndrome? An in-depth analysis of the literature. Neurosurgery 2009, 65, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Erdem, E.; Angtuaco, E.C.; Van Hemert, R.; Park, J.S.; Al-Mefty, O. Comprehensive Review of Intracranial Chordoma. Radio Graph. 2003, 23, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Farsad, K.; Kattapuram, S.V.; Sacknoff, R.; Ono, J.; Nielsen, G.P. Sacral Chordoma. RadioGraphics 2009, 29, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.; Scognamiglio, T.; Zhao, Y.; Schwartz, T.H.; Phillips, C. Prognostic Implications of Gadolinium Enhancement of Skull Base Chordomas. Am. J. Neuroradiol. 2018, 39, 1509–1514. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.; Lv, G.; Zhang, Q.; Wang, S.; Li, J.; Wang, X. Prognostic Factors in Skull Base Chordoma: A Systematic Literature Review and Meta-Analysis. World Neurosurg. 2018, 109, 307–327. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, J.; Zhang, L.; Jia, G.; Tang, J.; Wang, L.; Wang, Z. Prognostic factors for long-term outcome of patients with surgical resection of skull base chordomas-106 cases review in one institution. Neurosurg. Rev. 2010, 33, 451–456. [Google Scholar] [CrossRef]
- Oakley, G.J.; Fuhrer, K.; Seethala, R.R. Brachyury, SOX-9, and podoplanin, new markers in the skull base chordoma vs. chondrosarcoma differential: A tissue microarray-based comparative analysis. Mod. Pathol. 2008, 21, 1461–1469. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Yang, H.-L.; Lü, J.; Liu, J.Y.; Chen, X.Q. Prognostic factors of sacral chordoma after surgical therapy: A study of 36 patients. Spinal Cord 2009, 48, 166–171. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Sun, J.; Bai, H.X.; Huang, X.; Zou, Y.; Tan, X.; Zhang, Z.; Tang, X.; Tao, Y.; Xiao, B.; et al. Prognostic factors in patients with spinal chordoma: An integrative analysis of 682 patients. Clin. Neurosurg. 2017, 81, 812–823. [Google Scholar] [CrossRef]
- Colli, B.O.; Al-Mefty, O. Chordomas of the craniocervical junction: Follow-up review and prognostic factors. J. Neurosurg. 2001, 95, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Sheybani, A.; Menezes, A.H.; Buatti, J.M.; Hitchon, P.W. Disease outcomes for skull base and spinal chordomas: A single center experience. Clin. Neurol. Neurosurg. 2015, 130, 67–73. [Google Scholar] [CrossRef]
- Samii, A.; Gerganov, V.M.; Herold, C.; Hayashi, N.; Naka, T.; Mirzayan, M.J.; Ostertag, H.; Samii, M. Chordomas of the skull base: Surgical management and outcome. J. Neurosurg. 2007, 107, 319–324. [Google Scholar] [CrossRef]
- Hulen, C.A.; Temple, H.T.; Fox, W.P.; Sama, A.A.; Green, B.A.; Eismont, F.J. Oncologic and functional outcome following sacrectomy for sacral chordoma. J. Bone Joint. Surg. Am. Vol. 2006, 88, 1532–1539. [Google Scholar] [CrossRef]
- Kaiser, T.E.; Pritchard, D.J.; Unni, K.K. Clinicopathologic study of sacrococcygeal chordoma. Cancer 1984, 53, 2574–2578. [Google Scholar] [CrossRef]
- Arnautović, K.I.; Al-Mefty, O. Surgical seeding of chordomas. J. Neurosurg. 2001, 95, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.F.; Nyquist, G.G.; Moore, N.; Anand, V.K.; Schwartz, T.H. Endoscopic endonasal transclival resection of chordomas: Operative technique, clinical outcome, and review of the literature. J. Neurosurg. 2010, 112, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Koutourousiou, M.; Gardner, P.A.; Tormenti, M.J.; Henry, S.L.; Stefko, S.T.; Kassam, A.; Fernandez-Miranda, J.C.; Snyderman, C.H. Endoscopic endonasal approach for resection of cranial base chordomas: Outcomes and learning curve. Neurosurgery 2012, 71, 614–625. [Google Scholar] [CrossRef]
- Society, M.T.; Enneking, W.F. Staging of musculoskeletal neoplasms. Skelet. Radiol. 1985, 13, 183–194. [Google Scholar] [CrossRef]
- Dea, N.; Gokaslan, Z.; Choi, D.; Fisher, C. Spine Oncology—Primary Spine Tumors. Neurosurgery 2017, 80, S124–S130. [Google Scholar] [CrossRef] [Green Version]
- Boriani, S.; Weinstein, J.N.; Biagini, R. Primary bone tumors of the spine. Terminology and surgical staging. Spine 1997, 22, 1036–1044. [Google Scholar] [CrossRef]
- Chan, P.; Boriani, S.; Fourney, D.R.; Biagini, R.; Dekutoski, M.B.; Fehlings, M.G.; Ryken, T.C.; Gokaslan, Z.L.; Vrionis, F.D.; Harrop, J.S.; et al. An assessment of the reliability of the Enneking and Weinstein-Boriani-Biagini classifications for staging of primary spinal tumors by the Spine Oncology Study Group. Spine 2009, 34, 384–391. [Google Scholar] [CrossRef]
- Clarke, M.J.; Dasenbrock, B.H.; Bydon, A.; Sciubba, D.M.; McGirt, M.J.; Hsieh, P.C.; Yassari, R.; Gokaslan, Z.L.; Wolinsky, J.-P. Posterior-only approach for en bloc sacrectomy: Clinical outcomes in 36 consecutive patients. Neurosurgery 2012, 71, 357–364. [Google Scholar] [CrossRef]
- Sciubba, D.M.; Chi, J.H.; Rhines, L.D.; Gokaslan, Z.L. Chordoma of the Spinal Column. Neurosurg. Clin. N. Am. 2008, 19, 5–15. [Google Scholar] [CrossRef]
- Fourney, D.R.; Rhines, L.D.; Hentschel, S.J.; Skibber, J.M.; Wolinsky, J.-P.; Weber, K.L.; Suki, D.; Gallia, G.L.; Garonzik, I.; Gokaslan, Z.L. En bloc resection of primary sacral tumors: Classification of surgical approaches and outcome. J. Neurosurg. Spine 2005, 3, 111–122. [Google Scholar] [CrossRef]
- Kayani, B.; Hanna, S.; Sewell, M.D.; Saifuddin, A.; Molloy, S.; Briggs, T.W.R. A review of the surgical management of sacral chordoma. Eur. J. Surg. Oncol. 2014, 40, 1412–1420. [Google Scholar] [CrossRef]
- Mindea, S.A.; Chinthakunta, S.; Moldavsky, M.; Gudipally, M.; Khalil, S. Biomechanical comparison of spinopelvic reconstruction techniques in the setting of total sacrectomy. Spine 2012, 37, E1622–E1627. [Google Scholar] [CrossRef] [PubMed]
- Bederman, S.S.; Shah, K.N.; Hassan, J.M.; Hoang, B.H.; Kiester, P.D.; Bhatia, N.N. Surgical techniques for spinopelvic reconstruction following total sacrectomy: A systematic review. Eur. Spine J. 2013, 23, 305–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, J.H.; Healey, J.H.; Rose, P.; Casas-Ganem, J.; Boland, P.J. The surgical management of sacral chordomas. Spine 2009, 34, 2700–2704. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.S.; Aghi, M.K.; Muzikansky, A.; Shih, H.A.; Barker, F.G.; Curry, W.T. Outcomes and patterns of care in adult skull base chordomas from the Surveillance, Epidemiology, and End Results (SEER) database. J. Clin. Neurosci. 2014, 21, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Bohman, L.-E.; Koch, M.; Bailey, R.L.; Alonso-Basanta, M.; Lee, J.Y. Skull base chordoma and chondrosarcoma: Influence of clinical and demographic factors on prognosis: A SEER analysis. World Neurosurg. 2014, 82, 806–814. [Google Scholar] [CrossRef] [PubMed]
- MoojenMark, W.A.; Vleggeert-Lankamp, C.L.A.; Krol, A.D.G.; Dijkstra, S.P.D. Long-term results: Adjuvant radiotherapy in en bloc resection of sacrococcygeal chordoma is advisable. Spine 2011, 36, E656–E661. [Google Scholar] [CrossRef]
- Kano, H.; Iqbal, F.O.; Sheehan, J.P.; Mathieu, D.; Seymour, Z.A.; Niranjan, A.; Flickinger, J.C.; Kondziolka, D.; Pollock, B.E.; Rosseau, G.; et al. Stereotactic radiosurgery for chordoma: A report from the North American Gamma knife consortium. Neurosurgery 2011, 68, 379–389. [Google Scholar] [CrossRef]
- Jin, C.J.; Berry-Candelario, J.; Reiner, A.S.; Laufer, I.; Higginson, D.S.; Schmitt, A.M.; Lis, E.; Barzilai, O.; Boland, P.; Yamada, Y.; et al. Long-term outcomes of high-dose single-fraction radiosurgery for chordomas of the spine and sacrum. J. Neurosurgery: Spine 2020, 32, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Levin, W.P.; Kooy, H.; Loeffler, J.S.; Delaney, T.F. Proton beam therapy. Br. J. Cancer 2005, 93, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Paganetti, H. (Ed.) Proton Therapy Physics; Taylor & Francis Group, LLC: Abingdon, UK, 2012. [Google Scholar]
- Di Maio, S.; Temkin, N.; Ramanathan, D.; Sekhar, L.N. Current comprehensive management of cranial base chordomas: 10-yearmeta-analysis of observational studies. J. Neurosurg. 2011, 115, 1094–1105. [Google Scholar] [CrossRef]
- Park, L.; Delaney, T.F.; Liebsch, N.J.; Hornicek, F.J.; Goldberg, S.; Mankin, H.; Rosenberg, A.E.; Rosenthal, D.I.; Suit, H.D. Sacral chordomas: Impact of high-dose proton/photon-beam radiation therapy combined with or without surgery for primary versus recurrent tumor. Int. J. Radiat. Oncol. 2006, 65, 1514–1521. [Google Scholar] [CrossRef]
- Folkert, M.R.; Bilsky, M.H.; Cohen, M.G.N.; Zaider, M.; Dauer, L.T.; Cox, B.W.; Boland, P.J.; Laufer, I.; Yamada, Y. Intraoperative32P high-dose rate brachytherapy of the dura for recurrent primary and metastatic intracranial and spinal tumors. Neurosurgery 2012, 71, 1003–1011. [Google Scholar] [CrossRef]
- Bilsky, M.H.; Gerszten, P.; Laufer, I.; Yamada, Y. Radiation for Primary Spine Tumors. Neurosurg. Clin. N. Am. 2008, 19, 119–123. [Google Scholar] [CrossRef]
- Barry, J.J.; Jian, B.J.; Sughrue, M.E.; Kane, A.J.; Mills, S.A.; Tihan, T.; Parsa, A.T. The next step: Innovative molecular targeted therapies for treatment of intracranial chordoma patients. Neurosurgery 2011, 68, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, S.; Merriam, P.; Maki, R.G.; Abbeele, A.D.V.D.; Yap, J.T.; Akhurst, T.; Harmon, D.C.; Bhuchar, G.; O’Mara, M.M.; D’Adamo, D.R.; et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J. Clin. Oncol. 2009, 27, 3154–3160. [Google Scholar] [CrossRef] [Green Version]
- Houessinon, A.; Boone, M.; Constans, J.-M.; Toussaint, P.; Chauffert, B. Sustained response of a clivus chordoma to erlotinib after imatinib failure. Case Rep. Oncol. 2015, 8, 25–29. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Tamborini, E.; Vullo, S.L.; Bozzi, F.; Messina, A.; Morosi, C.; Casale, A.; Crippa, F.; Conca, E.; Negri, T.; et al. Phase ii study on lapatinib in advanced egfr-positive chordoma. Ann. Oncol. 2013, 24, 1931–1936. [Google Scholar] [CrossRef]
- Magnaghi, P.; Salom, B.; Cozzi, L.; Amboldi, N.; Ballinari, D.; Tamborini, E.; Gasparri, F.; Montagnoli, A.; Raddrizzani, L.; Somaschini, A.; et al. Afatinib is a new therapeutic approach in chordoma with a unique ability to target EGFR and Brachyury. Mol. Cancer Ther. 2017, 17, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Shen, J.K.; Choy, E.; Zhang, Y.; Mankin, H.J.; Hornicek, F.J.; Duan, Z. CDK4 expression in chordoma: A potential therapeutic target. J. Orthop. Res. 2018, 36, 1581–1589. [Google Scholar] [CrossRef] [Green Version]
- Iannalfi, A.; D’Ippolito, E.; Riva, G.; Molinelli, S.; Gandini, S.; Viselner, G.; Fiore, M.R.; Vischioni, B.; Vitolo, V.; Bonora, M.; et al. Proton and carbon ion radiotherapy in skull base chordomas: A prospective study based on a dual particle and a patient-customized treatment strategy. Neuro Oncol. 2020, 22, 1348–1358. [Google Scholar] [CrossRef]
- Von Witzleben, A.; Goerttler, L.T.; Marienfeld, R.; Barth, H.; Lechel, A.; Mellert, K.; Bohm, M.; Kornmann, M.; Mayer-Steinacker, R.; Von Baer, A.; et al. Preclinical characterization of novel Chordoma cell systems and their targeting by pharmocological inhibitors of the CDK4/6 cell-cycle pathway. Cancer Res. 2015, 75, 3823–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.-X.; Guo, K.-M.; Lv, G.-H.; Huang, W.; Li, J.; Wang, X.-B.; Jiang, Y.; She, X.-L. Clinicopathologic implications of CD8+/Foxp3+ratio and miR-574-3p/PD-L1 axis in spinal chordoma patients. Cancer Immunol. Immunother. 2018, 67, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R.; Friedman, E.R.; Richards, J.; Tsang, K.Y.; Heery, C.R.; Schlom, J.; Hodge, J.W. Enhanced killing of chordoma cells by antibody-dependent cell-mediated cytotoxicity employing the novel anti-PD-L1 antibody avelumab. Oncotarget 2016, 7, 33498–33511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliorini, D.; Mach, N.; Aguiar, D.; Vernet, R.; Landis, B.N.; Becker, M.; McKee, T.; Dutoit, V.; Dietrich, P.-Y. First report of clinical responses to immunotherapy in 3 relapsing cases of chordoma after failure of standard therapies. OncoImmunology 2017, 6, e1338235. [Google Scholar] [CrossRef] [Green Version]
- Fernando, R.; Litzinger, M.; Trono, P.; Hamilton, D.H.; Schlom, J.; Palena, C. The T-box transcription factor Brachyury promotes epithelial-mesenchymal transition in human tumor cells. J. Clin. Investig. 2010, 120, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, D.H.; David, J.M.; Dominguez, C.; Palena, C. Development of Cancer Vaccines Targeting Brachyury, a Transcription Factor Associated with Tumor Epithelial-Mesenchymal Transition. Cells Tissues Organs. 2017, 203, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Palena, C.; Fernando, R.I.; Hamilton, D.H. An immunotherapeutic intervention against tumor progression: Targeting a driver of the epithelial-to-mesenchymal transition. Oncoimmunology 2014, 3, e27220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottone, L.; Cribbs, A.P.; Khandelwal, G.; Wells, G.; Ligammari, L.; Philpott, M.; Tumber, A.; Lombard, P.; Hookway, E.S.; Szommer, T.; et al. Inhibition of Histone H3K27 Demethylases Inactivates Brachyury (TBXT) and Promotes Chordoma Cell Death. Cancer Res. 2020, 80, 4540–4551. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yan, Y.-G.; Wang, W.-J.; Ouyang, Z.-H.; Li, X.-L.; Zhang, T.-L.; Wang, X.-B.; Wang, B.; Lv, G.-H.; Li, J.; et al. Development and Validation of a 6-miRNA Prognostic Signature in Spinal Chordoma. Front. Oncol. 2020, 10, 556902. [Google Scholar] [CrossRef]
- La Corte, E.; Cas, M.D.; Raggi, A.; Patanè, M.; Broggi, M.; Schiavolin, S.; Calatozzolo, C.; Pollo, B.; Pipolo, C.; Bruzzone, M.G.; et al. Long and Very-Long-Chain Ceramides Correlate with A More Aggressive Behavior in Skull Base Chordoma Patients. Int. J. Mol. Sci. 2019, 20, 4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenonos, G.A.; Fernandez-Miranda, J.C.; Mukherjee, D.; Chang, Y.-F.; Panayidou, K.; Snyderman, C.H.; Wang, E.W.; Seethala, R.R.; Gardner, P.A. Prospective validation of a molecular prognostication panel for clival chordoma. J. Neurosurg. 2019, 130, 1528–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barber, S.M.; Sadrameli, S.S.; Lee, J.J.; Fridley, J.S.; Teh, B.S.; Oyelese, A.A.; Telfeian, A.E.; Gokaslan, Z.L. Chordoma—Current Understanding and Modern Treatment Paradigms. J. Clin. Med. 2021, 10, 1054. https://doi.org/10.3390/jcm10051054
Barber SM, Sadrameli SS, Lee JJ, Fridley JS, Teh BS, Oyelese AA, Telfeian AE, Gokaslan ZL. Chordoma—Current Understanding and Modern Treatment Paradigms. Journal of Clinical Medicine. 2021; 10(5):1054. https://doi.org/10.3390/jcm10051054
Chicago/Turabian StyleBarber, Sean M., Saeed S. Sadrameli, Jonathan J. Lee, Jared S. Fridley, Bin S. Teh, Adetokunbo A. Oyelese, Albert E. Telfeian, and Ziya L. Gokaslan. 2021. "Chordoma—Current Understanding and Modern Treatment Paradigms" Journal of Clinical Medicine 10, no. 5: 1054. https://doi.org/10.3390/jcm10051054
APA StyleBarber, S. M., Sadrameli, S. S., Lee, J. J., Fridley, J. S., Teh, B. S., Oyelese, A. A., Telfeian, A. E., & Gokaslan, Z. L. (2021). Chordoma—Current Understanding and Modern Treatment Paradigms. Journal of Clinical Medicine, 10(5), 1054. https://doi.org/10.3390/jcm10051054