
Lipid and Lipoprotein Dysregulation in Sepsis: Clinical and Mechanistic Insights into Chronic Critical Illness


 ,
,
Abstract
:1. Introduction
1.1. Sepsis Overview
1.2. Dysregulated Inflammation Driving Organ Failure, Chronic Critical Illness, and Death
2. Anti-Inflammatory and Protective Roles of Lipoproteins and Lipid Mediators in Sepsis
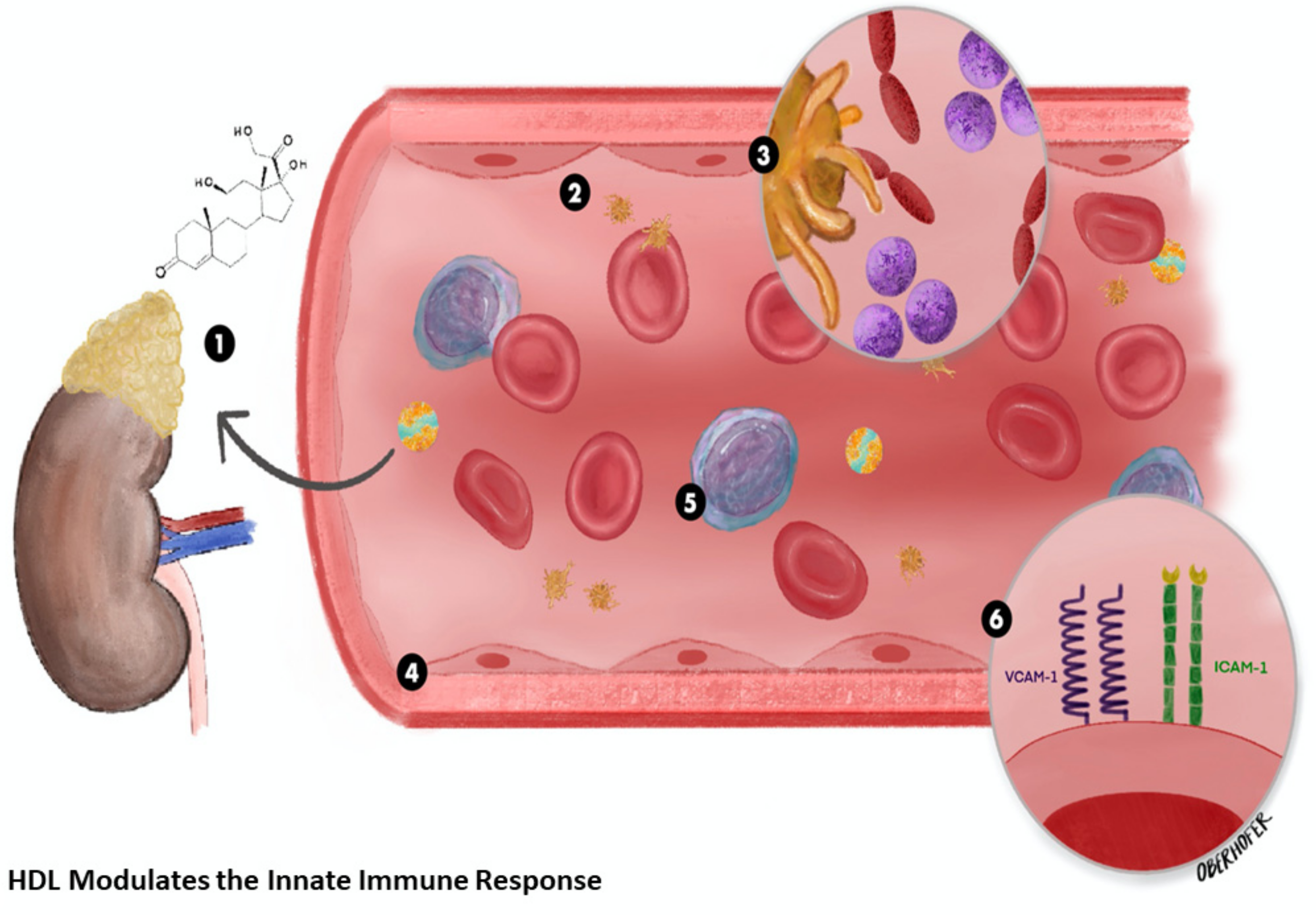
2.1. High-Density Lipoprotein
2.2. Low Density Lipoprotein
2.3. Specialized Pro-Resolving Mediators
3. Alterations of Lipid Metabolism in Sepsis Contribute to Failed Inflammation Resolution
Dysfunctional HDL
4. Lipid-Based Therapies
4.1. Statins
4.2. L-Carnitine
4.3. Lipid Emulsions
4.4. Eritoran
4.5. PCSK9
4.6. Fibrates
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Botero Hernandez, J.S.; Perez Florian, M.C. The history of sepsis from ancient Egypt to the XIX century. In Sepsis—An Ongoing and Significant Challenge; IntechOpen: London, UK, 2012. [Google Scholar]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). J. Am. Med. Assoc. 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.R.; Moldawer, L.L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.I.; Vincent, J.-L. Sepsis and septic shock. Nat. Rev. Dis. Prim. 2016, 2, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention. Sepsis, Clinical Information, Surveillance and Epidemiology. Available online: https://www.cdc.gov/sepsis/datareports/index.html (accessed on 20 November 2020).
- Fleischmann, M.C.; Scherag, A.; Adhikari, N.K.J.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Paoli, C.J.; Reynolds, M.A.; Sinha, M.; Gitlin, M.; Crouser, E. Epidemiology and costs of sepsis in the United States-an analysis based on timing of diagnosis and severity level. Crit. Care Med. 2018, 33, 127–130. [Google Scholar] [CrossRef]
- Martin, G.S.; Mannino, D.M.; Eaton, S.; Moss, M. The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 2003, 348, 1546–1554. [Google Scholar] [CrossRef] [Green Version]
- Kempker, J.A.; Martin, G.S. The changing epidemiology and definitions of sepsis. Clin. Chest. Med. 2016, 37, 165–179. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.; Kumar, N.; Taneja, A.; Kaleekal, T.; Tarima, S.; McGinley, E.; Jimenez, E.; Mohan, A.; Khan, R.A.; Whittle, J.; et al. Nationwide trends of severe sepsis in the 21st century (2000-2007). Chest 2011, 140, 1223–1231. [Google Scholar] [CrossRef]
- Gobatto, A.L.N.; Besen, B.A.M.P.; Azevedo, L.C.P. How can we estimate sepsis incidence and mortality? Shock 2017, 47, 6–11. [Google Scholar] [CrossRef]
- Gohil, S.K.; Cao, C.; Phelan, M.; Tjoa, T.; Rhee, C.; Platt, R.; Huang, S.S. Impact of policies on the rise in sepsis incidence, 2000–2010. Clin. Infect. Dis. 2015, 62, 695–703. [Google Scholar] [CrossRef]
- Rhee, C.; Klompas, M. Sepsis trends: Increasing incidence and decreasing mortality, or changing denominator? J. Thorac. Dis. 2020, 2, S89–S100. [Google Scholar] [CrossRef]
- Buchman, T.G.; Simpson, S.Q.; Sciarretta, K.L.; Finne, K.; Sowers, N.; Collier, M.; Chavan, S.; Oke, I.; Pennini, M.E.; Santhosh, A.; et al. Sepsis among medicare beneficiaries: 2. The trajectories of sepsis, 2012-2018. Crit. Care Med. 2020, 2. [Google Scholar] [CrossRef]
- Hawkins, R.B.; Raymond, S.L.; Stortz, J.A.; Horiguchi, H.; Brakenridge, S.C.; Gardner, A.; Efron, P.A.; Bihorac, A.; Segal, M.; Moore, F.A.; et al. Chronic critical illness and the persistent inflammation, immunosuppression, and catabolism syndrome. Front Immunol. 2018, 9, 1511. [Google Scholar] [CrossRef]
- Rosenthal, M.D.; Moore, F.A. Persistent inflammatory, immunosuppressed, catabolic syndrome (PICS): A new phenotype of multiple organ failure. J. Adv. Nutr. Hum. Metab. 2015, 1, 107–125. [Google Scholar] [CrossRef] [Green Version]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Walton, A.H.; Muenzer, J.T.; Rasche, D.; Boomer, J.S.; Sato, B.; Brownstein, B.H.; Pachot, A.; Brooks, T.L.; Deych, E.; Shannon, W.D.; et al. Reactivation of multiple viruses in patients with sepsis. PLoS ONE 2014, 9, e98819. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Strauss, L. Energy metabolism drives myeloid-derived suppressor cell differentiation and functions in pathology. J. Leukoc. Biol. 2017, 102, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef] [Green Version]
- Plebanek, M.P.; Bhaumik, D.; Bryce, P.J.; Thaxton, C.S. Scavenger receptor type b1 and lipoprotein nanoparticle inhibit myeloid-derived suppressor cells. Mol. Cancer Ther. 2017, 17, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Iwashyna, T.J.; Hodgson, C.L.; Pilcher, D.; Bailey, M.; Van Lint, A.; Chavan, S.; Bellomo, R. Timing of onset and burden of persistent critical illness in Australia and New Zealand: A retrospective, population-based, observational study. Lancet Respir. Med. 2016, 4, 566–573. [Google Scholar] [CrossRef]
- Brakenridge, S.C.; Efron, P.A.; Cox, M.C.; Stortz, J.A.; Hawkins, R.B.; Ghita, G.; Gardner, A.; Mohr, A.M.; Anton, S.D.; Moldawer, L.L.; et al. Current Epidemiology of Surgical Sepsis. Ann. Surg. 2019, 270, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.K.; Ghita, G.L.; Wang, Z.; Ozrazgat-Baslanti, T.; Raymond, S.L.; Mankowski, R.T.; Brumback, B.A.; Efron, P.A.; Bihorac, A.; Moore, F.A.; et al. The development of chronic critical illness determines physical function, quality of life, and long-term survival among early survivors of sepsis in surgical ICUs*. Crit. Care Med. 2019, 47, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef] [Green Version]
- Nofer, J.-R.; Levkau, B.; Wolinska, I.; Junker, R.; Fobker, M.; von Eckardstein, A.; Seedorf, U.; Assmann, G. Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipids. J. Biol. Chem. 2001, 276, 34480–34485. [Google Scholar] [CrossRef] [Green Version]
- Suc, I.; Escargueil-Blanc, I.; Troly, M.; Salvayre, R.; Nègre-Salvayre, A. HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arter. Thromb. Vasc. Biol. 1997, 17, 2158–2166. [Google Scholar] [CrossRef]
- Cirstea, M.; Walley, K.R.; Russell, J.A.; Brunham, L.R.; Genga, K.R.; Boyd, J.H. Decreased high-density lipoprotein cholesterol level is an early prognostic marker for organ dysfunction and death in patients with suspected sepsis. J. Crit. Care 2017, 38, 289–294. [Google Scholar] [CrossRef]
- Lekkou, A.; Mouzaki, A.; Siagris, D.; Ravani, I.; Gogos, C.A. Serum lipid profile, cytokine production, and clinical outcome in patients with severe sepsis. J. Crit. Care 2014, 29, 723–727. [Google Scholar] [CrossRef]
- Levels, J.H.M.; Abraham, P.R.; Ende, A.V.D.; Van Deventer, S.J.H. Distribution and kinetics of lipoprotein-bound endotoxin. Infect. Immun. 2001, 69, 2821–2828. [Google Scholar] [CrossRef] [Green Version]
- Kitchens, R.L.; Thompson, P.A.; Viriyakosol, S.; O’Keefe, G.E.; Munford, R.S. Plasma CD14 decreases monocyte responses to LPS by transferring cell-bound LPS to plasma lipoproteins. J. Clin. Investig. 2001, 108, 485–493. [Google Scholar] [CrossRef]
- Grin, P.M.; Dwivedi, D.J.; Chathely, K.M.; Trigatti, B.L.; Prat, A.; Seidah, N.G.; Liaw, P.C.; Fox-Robichaud, A.E. Low-density lipoprotein (LDL)-dependent uptake of Gram-positive lipoteichoic acid and Gram-negative lipopolysaccharide occurs through LDL receptor. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Guirgis, F.W.; Donnelly, J.P.; Dodani, S.; Howard, G.; Safford, M.M.; Levitan, E.B.; Wang, H.E. Cholesterol levels and long-term rates of community-acquired sepsis. Crit. Care 2016, 20, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N. Novel pro-resolving lipid mediators in inflammation are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalli, J.; Colas, R.A.; Quintana, C.; Barragan-Bradford, D.; Hurwitz, S.; Levy, B.D.; Choi, A.M.; Serhan, C.N.; Baron, R.M. Human sepsis eicosanoid and proresolving lipid mediator temporal profiles. Crit. Care Med. 2017, 45, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Hoda, S.; Hoda, R.S. Robbins and cotran pathologic basis of disease. Am. J. Clin. Pathol. 2020, 154, 869. [Google Scholar] [CrossRef]
- Levy, B.D.; Clish, C.B.; A Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef]
- Godson, C.; Mitchell, S.; Harvey, K.; Petasis, N.A.; Hogg, N.; Brady, H.R. Cutting edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 2000, 164, 1663–1667. [Google Scholar] [CrossRef]
- Chan, M.M.-Y.; Moore, A.R. Resolution of inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J. Immunol. 2010, 184, 6418–6426. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Fredman, G.; Bäckhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nat. Cell Biol. 2012, 484, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Fan, X.H.; Wu, Y.P.; Zhu, J.L.; Wang, F.; Bo, L.L.; Li, J.B.; Bao, R.; Deng, X.M. Resolvin D1 improves survival in experimental sepsis through reducing bacterial load and preventing excessive activation of inflammatory response. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 457–464. [Google Scholar] [CrossRef]
- Levy, B.D.; Kohli, P.; Gotlinger, K.; Haworth, O.; Hong, S.; Kazani, S.; Israel, E.; Haley, K.J.; Serhan, C.N. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J. Immunol. 2007, 178, 496–502. [Google Scholar] [CrossRef] [Green Version]
- Pirault, J.; Bäck, M. Lipoxin and resolvin receptors transducing the resolution of inflammation in cardiovascular disease. Front. Pharmacol. 2018, 9, 1273. [Google Scholar] [CrossRef]
- Banerjee, S.; Bhaduri, J.N. Serum protein-bound carbohydrates and lipids in cholera. Exp. Biol. Med. 1959, 101, 340–341. [Google Scholar] [CrossRef]
- Van Leeuwen, H.J.; Heezius, E.C.J.M.; Dallinga, G.M.; Van Strijp, J.A.G.; Verhoef, J.; Van Kessel, K.P.M. Lipoprotein metabolism in patients with severe sepsis. Crit. Care Med. 2003, 31, 1359–1366. [Google Scholar] [CrossRef]
- Chien, J.-Y.; Jerng, J.-S.; Yu, C.-J.; Yang, P.-C. Low serum level of high-density lipoprotein cholesterol is a poor prognostic factor for severe sepsis*. Crit. Care Med. 2005, 33, 1688–1693. [Google Scholar] [CrossRef]
- Lüthold, S.; Berneis, K.; Bady, P.; Muller, B. Effects of infectious disease on plasma lipids and their diagnostic significance in critical illness. Eur. J. Clin. Investig. 2007, 37, 573–579. [Google Scholar] [CrossRef]
- Barlage, S.; Gnewuch, C.; Liebisch, G.; Wolf, Z.; Audebert, F.-X.; Glück, T.; Fröhlich, D.; Krämer, B.K.; Rothe, G.; Schmitz, G. Changes in HDL-associated apolipoproteins relate to mortality in human sepsis and correlate to monocyte and platelet activation. Intensiv. Care Med. 2009, 35, 1877–1885. [Google Scholar] [CrossRef]
- Ettinger, W.H.; Varma, V.K.; Sorci-Thomas, M.; Parks, J.S.; Sigmon, R.C.; Smith, T.K.; Verdery, R.B. Cytokines decrease apolipoprotein accumulation in medium from Hep G2 cells. Arter. Thromb. Vasc. Biol. 1994, 14, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Feingold, K.R.; Grunfeld, C. Tumor necrosis factor-alpha stimulates hepatic lipogenesis in the rat in vivo. J. Clin. Investig. 1987, 80, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Nonogaki, K.; Fuller, G.M.; Fuentes, N.L.; Moser, A.H.; Staprans, I.; Grunfeld, C.; Feingold, K.R. Interleukin-6 stimulates hepatic triglyceride secretion in rats. Endocrinology 1995, 136, 2143–2149. [Google Scholar] [CrossRef]
- Khovidhunkit, W.; Kim, M.-S.; Memon, R.A.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Thematic review series: The Pathogenesis of Atherosclerosis. Effects of infection and inflammation on lipid and lipoprotein metabolism mechanisms and consequences to the host. J. Lipid Res. 2004, 45, 1169–1196. [Google Scholar] [CrossRef] [Green Version]
- Feingold, K.R.; Serio, M.K.; Adi, S.; Moser, A.H.; Grunfeld, C. Tumor necrosis factor stimulates hepatic lipid synthesis and secretion*. Endocrinology 1989, 124, 2336–2342. [Google Scholar] [CrossRef]
- Beutler, B.; Mahoney, J.; Le Trang, N.; Pekala, P.; Cerami, A. Purification of cachectin, a lipoprotein lipase-suppressing hormone secreted by endotoxin-induced RAW 264.7 cells. J. Exp. Med. 1985, 161, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Robin, A.P.; Askanazi, J.; Greenwood, M.R.; A Carpentier, Y.; E Gump, F.; Kinney, J.M. Lipoprotein lipase activity in surgical patients: Influence of trauma and infection. Surgery 1981, 90, 401–408. [Google Scholar]
- Ilias, I.; Vassiliadi, D.; Theodorakopoulou, M.; Boutati, E.; Maratou, E.; Mitrou, P.; Nikitas, N.; Apollonatou, S.; Dimitriadis, G.; Armaganidis, A.; et al. Adipose tissue lipolysis and circulating lipids in acute and subacute critical illness: Effects of shock and treatment. J. Crit. Care 2014, 29, 1130.e5–1130.e9. [Google Scholar] [CrossRef]
- Rival, T.; Cinq-Frais, C.; Silva-Sifontes, S.; García, J.; Riu, B.; Salvayre, R.; Genestal, M.; Caspar-Bauguil, S. Alteration of plasma phospholipid fatty acid profile in patients with septic shock. Biochimica 2013, 95, 2177–2181. [Google Scholar] [CrossRef]
- Cury-Boaventura, M.F.; Gorjão, R.; De Lima, T.M.; Piva, T.M.; Peres, C.M.; Soriano, F.G.; Curi, R. Toxicity of a Soybean Oil Emulsion on Human Lymphocytes and Neutrophils. J. Parenter. Enter. Nutr. 2006, 30, 115–123. [Google Scholar] [CrossRef]
- Nogueira, A.C.; Kawabata, V.; Biselli, P.; Lins, M.H.; Valeri, C.; Seckler, M.; Hoshino, W.; Júnior, L.G.; Bernik, M.M.S.; Machado, J.B.D.A.; et al. Changes in plasma free fatty acid levels in septic patients are associated with cardiac damage and reduction in heart rate variability. Shock 2008, 29, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Karahan, S.; Cetinkaya, A.; Erden, A.; Avci, D.; Karagoz, H.; Basak, M.; Bulut, K.; Gençer, V.; Mutlu, H. Is hypertriglyceridemia a prognostic factor in sepsis? Ther. Clin. Risk Manag. 2014, 10, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Park, M.S.; Park, B.H.; Jung, W.J.; Lee, I.S.; Kim, S.Y.; Kim, E.Y.; Jung, J.Y.; Kang, Y.A.; Kim, Y.S.; et al. Prognostic implications of serum lipid metabolism over time during sepsis. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Berghe, G.V.D.; Wouters, P.; Weekers, F.; Verwaest, C.; Bruyninckx, F.; Schetz, M.; Vlasselaers, D.; Ferdinande, P.; Lauwers, P.; Bouillon, R. Intensive Insulin Therapy in Critically Ill Patients. N. Engl. J. Med. 2001, 345, 1359–1367. [Google Scholar] [CrossRef]
- Mesotten, D.; Swinnen, J.V.; Vanderhoydonc, F.; Wouters, P.J.; Berghe, G.V.D. Contribution of circulating lipids to the improved outcome of critical illness by glycemic control with intensive insulin therapy. J. Clin. Endocrinol. Metab. 2004, 89, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirasawa, H.; Oda, S.; Nakamura, M. Blood glucose control in patients with severe sepsis and septic shock. World J. Gastroenterol. 2009, 15, 4132–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappi, S.B.; Noritomi, D.T.; Velasco, I.T.; Curi, R.; Loureiro, T.C.A.; Soriano, F.G. Dyslipidemia: A prospective controlled randomized trial of intensive glycemic control in sepsis. Intensiv. Care Med. 2012, 38, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Trinder, M.; Genga, K.R.; Kong, H.J.; Blauw, L.L.; Lo, C.; Li, X.; Cirstea, M.; Wang, Y.; Rensen, P.C.N.; Russell, J.A.; et al. Cholesteryl ester transfer protein influences high-density lipoprotein levels and survival in sepsis. Am. J. Respir. Crit. Care Med. 2019, 199, 854–862. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.P.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Hillenbrand, A.; Knippschild, U.; Weiss, M.; Schrezenmeier, H.; Henne-Bruns, D.; Huber-Lang, M.; Wolf, A.M. Sepsis induced changes of adipokines and cytokines-septic patients compared to morbidly obese patients. BMC Surg. 2010, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Kalani, C.; Venigalla, T.; Bailey, J.; Udeani, G.; Surani, S. Sepsis patients in critical care units with obesity: Is obesity protective? Cureus 2020, 12, e6929. [Google Scholar] [CrossRef] [Green Version]
- Gariballa, S.; Alkaabi, J.; Yasin, J.; Al Essa, A. Total adiponectin in overweight and obese subjects and its response to visceral fat loss. BMC Endocr. Disord. 2019, 19, 1–6. [Google Scholar] [CrossRef]
- Fabiani, H.; Mudjihartini, N.; Lestari, W. Low dietary omega-6 to omega-3 fatty acid intake ratio enhances adiponectin level in obesity. World Nutr. J. 2021, 5, 30–39. [Google Scholar] [CrossRef]
- Mudjihartini, N.; Fabiani, H.; Lestari, W. Dietary omega-6 to omega-3 fatty acids ratio is correlated with high molecular weight adiponectin level in indonesian office workers. Int. J. Nutr. Pharmacol. Neurol. Dis. 2021. [Google Scholar] [CrossRef]
- Guebre-Egziabher, F.; Rabasa-Lhoret, R.; Bonnet, F.; Bastard, J.-P.; Desage, M.; Skilton, M.R.; Vidal, H.; Laville, M. Nutritional intervention to reduce the n−6/n−3 fatty acid ratio increases adiponectin concentration and fatty acid oxidation in healthy subjects. Eur. J. Clin. Nutr. 2007, 62, 1287–1293. [Google Scholar] [CrossRef]
- Simopoulos, A. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Itoh, M.; Suganami, T.; Satoh, N.; Tanimoto-Koyama, K.; Yuan, X.; Tanaka, M.; Kawano, H.; Yano, T.; Aoe, S.; Takeya, M.; et al. Increased adiponectin secretion by highly purified eicosapentaenoic acid in rodent models of obesity and human obese subjects. Arter. Thromb. Vasc. Biol. 2007, 27, 1918–1925. [Google Scholar] [CrossRef] [Green Version]
- Guirgis, F.W.; Dodani, S.; Leeuwenburgh, C.; Moldawer, L.; Bowman, J.; Kalynych, C.; Grijalva, V.; Reddy, S.T.; Jones, A.E.; Moore, F.A. HDL inflammatory index correlates with and predicts severity of organ failure in patients with sepsis and septic shock. PLoS ONE 2018, 13, e0203813. [Google Scholar] [CrossRef]
- Guirgis, F.W.; Dodani, S.; Moldawer, L.; Leeuwenburgh, C.; Bowman, J.; Kalynych, C.; Jones, A.E.; Reddy, S.T.; Moore, F.A. Exploring the predictive ability of dysfunctional high-density lipoprotein for adverse outcomes in emergency department patients with sepsis: A preliminary investigation. Shock 2017, 48, 539–544. [Google Scholar] [CrossRef]
- Shao, B.; Tang, C.; Sinha, A.; Mayer, P.S.; Davenport, G.D.; Brot, N.; Oda, M.N.; Zhao, X.-Q.; Heinecke, J.W. Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ. Res. 2014, 114, 1733–1742. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Aulak, K.; Huang, Y.; Wagner, M.; Gerstenecker, G.; Topbas, C.; Gogonea, V.; DiDonato, A.J.; Tang, W.H.W.; Mehl, R.A.; et al. Site-specific nitration of apolipoprotein A-I at Tyrosine 166 Is both abundant within human atherosclerotic plaque and dysfunctional. J. Biol. Chem. 2014, 289, 10276–10292. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Nukuna, B.; Brennan, M.-L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [Green Version]
- Murch, O.; Collin, M.; Hinds, C.J.; Thiemermann, C. Lipoproteins in inflammation and sepsis. I. Basic science. Intensiv. Care Med. 2007, 33, 13–24. [Google Scholar] [CrossRef]
- Rohrer, L.; Hersberger, M.; Von Eckardstein, A. High density lipoproteins in the intersection of diabetes mellitus, inflammation and cardiovascular disease. Curr. Opin. Lipidol. 2004, 15, 269–278. [Google Scholar] [CrossRef]
- Cabana, V.G.; Lukens, J.R.; Rice, K.S.; Hawkins, T.J.; Getz, G.S. HDL content and composition in acute phase response in three species: Triglyceride enrichment of HDL a factor in its decrease. J. Lipid Res. 1996, 37, 2662–2674. [Google Scholar] [CrossRef]
- Rousset, X.; Vaisman, B.; Amar, M.; Sethi, A.A.; Remaley, A.T. Lecithin: Cholesterol acyltransferase-from biochemistry to role in cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, M.; Korporaal, S.J.A.; van der Sluis, R.J.; Hirsch-Reinshagen, V.; Bochem, A.E.; Wellington, C.L.; Van Berkel, T.J.C.; Kuivenhoven, J.A.; Van Eck, M. LCAT deficiency in mice is associated with a diminished adrenal glucocorticoid function. J. Lipid Res. 2013, 54, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Petropoulou, P.-I.; Berbée, J.F.; Theodoropoulos, V.; Hatziri, A.; Stamou, P.; Karavia, E.A.; Spyridonidis, A.; Karagiannides, I.; Kypreos, K.E. Lack of LCAT reduces the LPS-neutralizing capacity of HDL and enhances LPS-induced inflammation in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2106–2115. [Google Scholar] [CrossRef] [Green Version]
- Guirgis, F.W.; Leeuwenburgh, C.; Grijalva, V.; Bowman, J.; Kalynych, C.; Moldawer, L.; Moore, F.A.; Reddy, S.T. HDL cholesterol efflux is impaired in older patients with early sepsis: A subanalysis of a prospective pilot study. Shock 2018, 50, 66–70. [Google Scholar] [CrossRef]
- Jones, T.K.; Wong, H.R.; Meyer, N.J. HDL Cholesterol: A “Pathogen Lipid Sink” for Sepsis? Am. J. Respir. Crit. Care Med. 2019, 199, 812–814. [Google Scholar] [CrossRef]
- Rao, R.; Albers, J.J.; Wolfbauer, G.; Pownall, H.J. Molecular and macromolecular specificity of human plasma phospholipid transfer protein†. Biochemistry 1997, 36, 3645–3653. [Google Scholar] [CrossRef] [PubMed]
- Settasatian, N.; Duong, M.; Curtiss, L.K.; Ehnholm, C.; Jauhiainen, M.; Huuskonen, J.; Rye, K.-A. The mechanism of the remodeling of high density lipoproteins by phospholipid transfer protein. J. Biol. Chem. 2001, 276, 26898–26905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deckert, V.; Lemaire, S.; Ripoll, P.-J.; De Barros, J.-P.P.; Labbé, J.; Borgne, C.C.-L.; Turquois, V.; Maquart, G.; LaRose, D.; Desroche, N.; et al. Recombinant human plasma phospholipid transfer protein (PLTP) to prevent bacterial growth and to treat sepsis. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Lenten, B.J.; Van Hama, S.Y.; Beer, F.C.; Stafforini, D.M.; McIntyre, T.M.; Prescott, S.M.; La Du, B.N.; Fogelman, A.M.; Navab, M. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. J. Clin. Investig. 1995, 96, 2758–2767. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, Y.; Toh, R.; Hasokawa, M.; Nakajima, H.; Honjo, T.; Otsui, K.; Mori, K.; Miyamoto-Sasaki, M.; Shinohara, M.; Nishimura, K.; et al. Serum myeloperoxidase/paraoxonase 1 ratio as potential indicator of dysfunctional high-density lipoprotein and risk stratification in coronary artery disease. Atherosclerosis 2014, 234, 288–294. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.D.; Berliner, J.A.; Hama, S.Y.; La Du, B.N.; Faull, K.F.; Fogelman, A.M.; Navab, M. Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J. Clin. Investig. 1995, 96, 2882–2891. [Google Scholar] [CrossRef]
- Prescott, S.M.; McIntyre, T.M.; Zimmerman, A. Minireview platelet-activating. J. Biol. Chem. 1990, 265, 17381–17384. [Google Scholar] [CrossRef]
- Opal, S.; Laterre, P.-F.; Abraham, E.; Francois, B.; Wittebole, X.; Lowry, S.; Dhainaut, J.-F.; Warren, B.; Dugernier, T.; Lopez, A.; et al. Recombinant human platelet-activating factor acetylhydrolase for treatment of severe sepsis: Results of a phase III, multicenter, randomized, double-blind, placebo-controlled, clinical trial. Crit. Care Med. 2004, 32, 332–341. [Google Scholar] [CrossRef]
- E Yu, J.; Han, S.-Y.; Wolfson, B.; Zhou, Q. The role of endothelial lipase in lipid metabolism, inflammation, and cancer. Histol. Histopathol. 2017, 33, 1–10. [Google Scholar] [CrossRef]
- Guirgis, F.W.; Black, L.P.; Rosenthal, M.D.; Henson, M.; Ferreira, J.; Leeuwenburgh, C.; Kalynych, C.; Moldawer, L.L.; Miller, T.; Jones, L.; et al. LIPid Intensive Drug therapy for Sepsis Pilot (LIPIDS-P): Phase I/II clinical trial protocol of lipid emulsion therapy for stabilising cholesterol levels in sepsis and septic shock. BMJ Open 2019, 9, e029348. [Google Scholar] [CrossRef]
- Jin, W.; Sun, G.-S.; Marchadier, D.; Octtaviani, E.; Glick, J.M.; Rader, D.J. Endothelial cells secrete triglyceride lipase and phospholipase activities in response to cytokines as a result of endothelial lipase. Circ. Res. 2003, 92, 644–650. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Ishida, T.; Kojima, Y.; Tanaka, H.; Yasuda, T.; Shinohara, M.; Toh, R.; Hirata, K.-I. Targeted deletion of endothelial lipase increases HDL particles with anti-inflammatory properties both in vitro and in vivo. J. Lipid Res. 2011, 52, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Jr, G.H.S. Serum amyloid A—A review. Mol. Med. 2018, 24, 1–27. [Google Scholar] [CrossRef]
- Pepys, M.; Baltz, M.L. Acute Phase Proteins with Special Reference to C-Reactive Protein and Related Proteins (Pentaxins) and Serum Amyloid A Protein. Adv. Immunol. 1983, 34, 141–212. [Google Scholar] [CrossRef]
- A Coetzee, G.; Strachan, A.F.; van der Westhuyzen, D.R.; Hoppe, H.C.; Jeenah, M.S.; de Beer, F.C. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J. Biol. Chem. 1986, 261, 9644–9651. [Google Scholar] [CrossRef]
- Cabana, V.G.; Siegel, J.N.; Sabesin, S.M. Effects of the acute phase response on the concentration and density distribution of plasma lipids and apolipoproteins. J. Lipid Res. 1989, 30, 39–49. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in Innate Immunity and Inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Jiang, H.; Cui, X.; Liang, G.; Chen, Y.; Wang, T.; Sun, Z.; Qi, L. Elevated serum levels of lipoprotein-associated phospholipase�A2 predict mortality rates in patients with sepsis. Mol. Med. Rep. 2017, 17, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Balsinde, J.; Winstead, M.V.; A Dennis, E. Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6. [Google Scholar] [CrossRef]
- Ansell, B.J.; Navab, M.; Hama, S.; Kamranpour, N.; Fonarow, G.; Hough, G.; Rahmani, S.; Mottahedeh, R.; Dave, R.; Reddy, S.T.; et al. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation 2003, 108, 2751–2756. [Google Scholar] [CrossRef]
- Cavigiolio, G.; Geier, E.G.; Shao, B.; Heinecke, J.W.; Oda, M.N. Exchange of apolipoprotein A-I between lipid-associated and lipid-free states. J. Biol. Chem. 2010, 285, 18847–18857. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S.; Jr, H.B.B.; Ansell, B.J.; Barter, P.J.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Al-Banna, N.; Lehmann, C. Oxidized LDL and LOX-1 in experimental sepsis. Mediat. Inflamm. 2013, 2013, 761789. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Masaki, T.; Sawamura, T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef]
- Wu, Z.; Sawamura, T.; Kurdowska, A.K.; Ji, H.-L.; Idell, S.; Fu, J. LOX-1 deletion improves neutrophil responses, enhances bacterial clearance, and reduces lung injury in a murine polymicrobial sepsis model. Infect. Immun. 2011, 79, 2865–2870. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yin, Y.; Zhou, Z.; He, M.; Dai, Y. OxLDL-induced IL-1beta secretion promoting foam cells formation was mainly via CD36 mediated ROS production leading to NLRP3 inflammasome activation. Inflamm. Res. 2014, 63, 33–43. [Google Scholar] [CrossRef]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; I Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nat. Cell Biol. 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Danielski, L.G.; Della Giustina, A.; Bonfante, S.; Barichello, T.; Petronilho, F. The NLRP3 inflammasome and its role in sepsis development. Inflammation 2020, 43, 24–31. [Google Scholar] [CrossRef]
- Lopategi, A.; Flores-Costa, R.; Rius, B.; López-Vicario, C.; Alcaraz-Quiles, J.; Titos, E.; Clària, J. Frontline Science: Specialized proresolving lipid mediators inhibit the priming and activation of the macrophage NLRP3 inflammasome. J. Leukoc. Biol. 2018, 105, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Zhang, Y.; Zhang, R.; Qiao, S.; Fan, J. Resolvin D2 recovers neural injury by suppressing inflammatory mediators expression in lipopolysaccharide-induced Parkinson’s disease rat model. Biochem. Biophys. Res. Commun. 2015, 460, 799–805. [Google Scholar] [CrossRef]
- Bento, A.F.; Claudino, R.F.; Dutra, R.C.; Marcon, R.; Calixto, J.B. Omega-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J. Immunol. 2011, 187, 1957–1969. [Google Scholar] [CrossRef] [Green Version]
- Spite, M.; Norling, L.V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N.A.; Flower, R.J.; Perretti, M.; Serhan, C.N. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nat. Cell Biol. 2009, 461, 1287–1291. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Qiu, C.; Yang, L.; Zhang, Z.; Zhang, Q.; Wang, B.; Wang, X. GPR18 expression on PMNs as biomarker for outcome in patient with sepsis. Life Sci. 2019, 217, 49–56. [Google Scholar] [CrossRef]
- Kurihara, T.; Jones, C.N.; Yu, Y.; Fischman, A.J.; Watada, S.; Tompkins, R.G.; Fagan, S.P.; Irimia, D. Resolvin D2 restores neutrophil directionality and improves survival after burns. FASEB J. 2013, 27, 2270–2281. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, S.; Shinohara, M.; Oshita, T.; Nagao, M.; Tanaka, N.; Mori, T.; Hara, T.; Irino, Y.; Toh, R.; Ishida, T.; et al. Novel mechanism of regulation of the 5-lipoxygenase/leukotriene B4 pathway by high-density lipoprotein in macrophages. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Moya, M.D.L.L.; McGillicuddy, F.C.; Hinkle, C.C.; Byrne, M.; Joshi, M.R.; Nguyen, V.; Tabita-Martinez, J.; Wolfe, M.L.; Badellino, K.; Pruscino, L.; et al. Inflammation modulates human HDL composition and function in vivo. Atherosclerosis 2012, 222, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Cavaillon, J.; Singer, M.; Skirecki, T. Sepsis therapies: Learning from 30 years of failure of translational research to propose new leads. EMBO Mol. Med. 2020, 12, e10128. [Google Scholar] [CrossRef]
- Panacek, E.A.; Marshall, J.C.; Albertson, T.E.; Johnson, D.H.; Johnson, S.; MacArthur, R.D.; Miller, M.; Barchuk, W.T.; Fischkoff, S.; Kaul, M.; et al. Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab′)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels*. Crit. Care Med. 2004, 32, 2173–2182. [Google Scholar] [CrossRef]
- Meisel, C.; Schefold, J.C.; Pschowski, R.; Baumann, T.; Hetzger, K.; Gregor, J.; Weber-Carstens, S.; Hasper, D.; Keh, D.; Zuckermann, H.; et al. Granulocyte–Macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression. Am. J. Respir. Crit. Care Med. 2009, 180, 640–648. [Google Scholar] [CrossRef]
- Shakoory, B.; Carcillo, J.A.; Chatham, W.W.; Amdur, R.L.; Zhao, H.; Dinarello, C.A.; Cron, R.Q.; Opal, S.M. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome. Crit. Care Med. 2016, 44, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welte, T.; Dellinger, R.P.; Ebelt, H.; Ferrer, M.; Opal, S.M.; Singer, M.; Vincent, J.-L.; Werdan, K.; Martin-Loeches, I.; Almirall, J.; et al. Efficacy and safety of trimodulin, a novel polyclonal antibody preparation, in patients with severe community-acquired pneumonia: A randomized, placebo-controlled, double-blind, multicenter, phase II trial (CIGMA study). Intensiv. Care Med. 2018, 44, 438–448. [Google Scholar] [CrossRef] [Green Version]
- Liappis, A.P.; Kan, V.L.; Rochester, C.; Simon, G. The Effect of statins on mortality in patients with bacteremia. Clin. Infect. Dis. 2001, 33, 1352–1357. [Google Scholar] [CrossRef]
- Goodin, J.; Manrique, C.; Dulohery, M.; Sampson, J.; Saettele, M.; Dabbagh, O. effect of statins on the clinical outcomes of patients with sepsis. Anaesth. Intensiv. Care 2011, 39, 1051–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Ji, M.; Si, X. The effects of statin therapy on mortality in patients with sepsis. Medicine 2018, 97, e11578. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.S.; Venkatesh, B. Are there any benefits from statin treatment for the septic patient? Curr. Atheroscler. Rep. 2013, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.M.; Snaith, C.; Thickett, D.R.; Linhartova, L.; Melody, T.; Hawkey, P.; Barnett, A.H.; Jones, A.; Hong, T.; Cooke, M.W.; et al. Randomized double-blind placebo-controlled trial of 40 mg/day of atorvastatin in reducing the severity of sepsis in ward patients (ASEPSIS Trial). Crit. Care 2012, 16, R231. [Google Scholar] [CrossRef] [Green Version]
- Craig, T.R.; Duffy, M.J.; Shyamsundar, M.; McDowell, C.; O’Kane, C.M.; Elborn, J.S.; McAuley, D.F. A Randomized Clinical Trial of Hydroxymethylglutaryl-Coenzyme A Reductase Inhibition for Acute Lung Injury (The HARP Study). Am. J. Respir. Crit. Care Med. 2011, 183, 620–626. [Google Scholar] [CrossRef]
- Ko, H.H.; Lareu, R.R.; Dix, B.R.; Hughes, J.D. Statins: Antimicrobial resistance breakers or makers? PeerJ 2017, 5, e3952. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-C.; Lee, M.-T.G.; Hsu, T.-C.; Porta, L.; Chang, S.-S.; Yo, C.-H.; Tsai, K.-C.; Lee, M. A Population-based cohort study on the drug-specific effect of statins on sepsis outcome. Chest 2018, 153, 805–815. [Google Scholar] [CrossRef]
- Arenas, J.; Huertas, R.; Campos, Y.; Díaz, A.; Villalón, J.M.; Vilas, E. Effects of l-carnitine on the pyruvate dehydrogenase complex and carnitine palmitoyl transferase activities in muscle of endurance athletes. FEBS Lett. 1994, 341, 91–93. [Google Scholar] [CrossRef] [Green Version]
- Takeyama, N.; Takagi, D.; Matsuo, N.; Kitazawa, Y.; Tanaka, T. Altered hepatic fatty acid metabolism in endotoxicosis: Effect of L-carnitine on survival. Am. J. Physiol. Metab. 1989, 256, E31–E38. [Google Scholar] [CrossRef]
- Takeyama, N.; Itoh, Y.; Kitazawa, Y.; Tanaka, T. Altered hepatic mitochondrial fatty acid oxidation and ketogenesis in endotoxic rats. Am. J. Physiol. Metab. 1990, 259, E498–E505. [Google Scholar] [CrossRef]
- Jones, A.E.; Puskarich, M.A.; Shapiro, N.I.; Guirgis, F.W.; Runyon, M.; Adams, J.Y.; Sherwin, R.; Arnold, R.; Roberts, B.W.; Kurz, M.C.; et al. Effect of levocarnitine vs placebo as an adjunctive treatment for septic shock. JAMA Netw. Open 2018, 1, e186076. [Google Scholar] [CrossRef]
- Jennaro, T.S.; Puskarich, M.A.; McCann, M.R.; Gillies, C.E.; Pai, M.P.; Karnovsky, A.; Evans, C.R.; Jones, A.E.; Stringer, K.A. Using l-carnitine as a pharmacologic probe of the interpatient and metabolic variability of sepsis. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 913–923. [Google Scholar] [CrossRef]
- Serhan, C.N.; Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [Green Version]
- Tjonahen, E.; Oh, S.F.; Siegelman, J.; Elangovan, S.; Percarpio, K.B.; Hong, S.; Arita, M.; Serhan, C.N. Resolvin E2: Identification and Anti-Inflammatory Actions: Pivotal Role of Human 5-Lipoxygenase in Resolvin E Series Biosynthesis. Chem. Biol. 2006, 13, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Singer, P.; Shapiro, H.; Theilla, M.; Anbar, R.; Singer, J.; Cohen, J. Anti-inflammatory properties of omega-3 fatty acids in critical illness: Novel mechanisms and an integrative perspective. Intensiv. Care Med. 2008, 34, 1580–1592. [Google Scholar] [CrossRef]
- Pizzini, A.; Lunger, L.; Demetz, E.; Hilbe, R.; Weiss, G.; Ebenbichler, C.; Tancevski, I. The role of omega-3 fatty acids in reverse cholesterol transport: A review. Nutrition 2017, 9, 1099. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Ishida, T.; Nagao, M.; Mori, T.; Monguchi, T.; Sasaki, M.; Mori, K.; Kondo, K.; Nakajima, H.; Honjo, T.; et al. Administration of high dose eicosapentaenoic acid enhances anti-inflammatory properties of high-density lipoprotein in Japanese patients with dyslipidemia. Atherosclerosis 2014, 237, 577–583. [Google Scholar] [CrossRef]
- Burillo, E.; Mateo-Gallego, R.; Cenarro, A.; Fiddyment, S.; Bea, A.M.; Jorge, I.; Vázquez, J.; Civeira, F. Beneficial effects of omega-3 fatty acids in the proteome of high-density lipoprotein proteome. Lipids Heal. Dis. 2012, 11, 116. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wang, S.; Zhao, Y.; Luo, Y.; Tong, H.; Su, L. Correlation analysis of omega-3 fatty acids and mortality of sepsis and sepsis-induced ARDS in adults: Data from previous randomized controlled trials. Nutr. J. 2018, 17, 57. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Sharma, S.; McIntyre, L.; Rhodes, A.; Evans, L.; Almenawer, S.; LeDuc, L.; Angus, D.C.; Alhazzani, W. Omega-3 supplementation in patients with sepsis: A systematic review and meta-analysis of randomized trials. Ann. Intensiv. Care 2017, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.C.; Bilku, D.K.; Al-Leswas, D.; Neal, C.P.; Horst, C.; Cooke, J.; Metcalfe, M.S.; Dennison, A.R. A randomized controlled trial investigating the effects of parenteral fish oil on survival outcomes in critically ill patients with sepsis. J. Parenter. Enter. Nutr. 2014, 39, 301–312. [Google Scholar] [CrossRef]
- Das, U.N. Polyunsaturated fatty acids and sepsis. Nutrients 2019, 65, 39–43. [Google Scholar] [CrossRef]
- Dellinger, R.P.; Tomayko, J.F.; Angus, D.C.; Opal, S.; Cupo, M.A.; McDermott, S.; Ducher, A.; Calandra, T.; Cohen, J. Efficacy and safety of a phospholipid emulsion (GR270773) in Gram-negative severe sepsis: Results of a phase II multicenter, randomized, placebo-controlled, dose-finding clinical trial. Crit. Care Med. 2009, 37, 2929–2938. [Google Scholar] [CrossRef]
- Parker, T.; Levine, D.; Gordon, B.; Saal, S. Subgroup analysis of the lipid infusion and patient outcomes in sepsis trial (LIPOS) reveals benefit in a subgroup not treated with stress replacement doses of corticosteroids. Crit. Care 2014, 18, P62. [Google Scholar] [CrossRef] [Green Version]
- Guirgis, F.W.; Black, L.P.; Devos, E.; Henson, M.; Ferreira, J.; Miller, T.; Rosenthal, M.; Leeuwenburgh, C.; Kalynych, C.; Moldawer, L.; et al. Lipid intensive drug therapy for sepsis pilot: A bayesian phase I clinical trial. J. Am. Coll. Emerg. Physicians Open 2020, 1, 1332–1340. [Google Scholar] [CrossRef]
- Shirey, K.A.; Lai, W.; Scott, A.J.; Lipsky, M.; Mistry, P.; Pletneva, L.M.; Karp, C.L.; McAlees, J.W.; Gioannini, T.L.; Weiss, J.; et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nat. Cell Biol. 2013, 497, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Barochia, A.; Solomon, S.; Cui, X.; Natanson, C.; Eichacker, P.Q. Eritoran tetrasodium (E5564) treatment for sepsis: Review of preclinical and clinical studies. Expert Opin. Drug Metab. Toxicol. 2011, 7, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Misspelled Affiliation Name. JAMA 2013, 310, 324. [CrossRef] [Green Version]
- Azzam, K.M.; Fessler, M.B. Crosstalk between reverse cholesterol transport and innate immunity. Trends Endocrinol. Metab. 2012, 23, 169–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNutt, M.C.; Kwon, H.J.; Chen, C.; Chen, J.R.; Horton, J.D.; Lagace, T.A. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 Cells. J. Biol. Chem. 2009, 284, 10561–10570. [Google Scholar] [CrossRef] [Green Version]
- Tveten, K.; Str⊘m, T.B.; Berge, K.E.; Leren, T.P. PCSK9-mediated degradation of the LDL receptor generates a 17 kDa C-terminal LDL receptor fragment. J. Lipid Res. 2013, 54, 1560–1566. [Google Scholar] [CrossRef] [Green Version]
- Topchiy, E.; Cirstea, M.; Kong, H.J.; Boyd, J.H.; Wang, Y.; Russell, J.A.; Walley, K.R. Lipopolysaccharide is cleared from the circulation by hepatocytes via the low density lipoprotein receptor. PLoS ONE 2016, 11, e0155030. [Google Scholar] [CrossRef] [Green Version]
- Feingold, K.R.; Moser, A.H.; Shigenaga, J.K.; Patzek, S.M.; Grunfeld, C. Inflammation stimulates the expression of PCSK9. Biochem. Biophys. Res. Commun. 2008, 374, 341–344. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Ishibashi, M.; A Seimon, T.; Lee, M.; Sharma, S.M.; Fitzgerald, K.A.; Samokhin, A.O.; Wang, Y.; Sayers, S.; Aikawa, M.; et al. Free cholesterol accumulation in macrophage membranes activates toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ. Res. 2009, 104, 455–465. [Google Scholar] [CrossRef]
- Li, Y.; Schwabe, R.F.; DeVries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.-C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef] [Green Version]
- Vecchié, A.; Bonaventura, A.; Meessen, J.; Novelli, D.; Minetti, S.; Elia, E.; Ferrara, D.; Ansaldo, A.M.; Scaravilli, V.; Villa, S.; et al. PCSK9 is associated with mortality in patients with septic shock: Data from the ALBIOS study. J. Intern. Med. 2021, 289, 179–192. [Google Scholar] [CrossRef]
- Walley, K.R.; Thain, K.R.; Russell, J.A.; Reilly, M.P.; Meyer, N.J.; Ferguson, J.F.; Christie, J.D.; Nakada, T.-A.; Fjell, C.D.; Thair, S.A.; et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci. Transl. Med. 2014, 6, 258ra143. [Google Scholar] [CrossRef] [Green Version]
- Boyd, J.H.; Fjell, C.D.; Russell, J.A.; Sirounis, D.; Cirstea, M.S.; Walley, K.R. Increased plasma PCSK9 levels are associated with reduced endotoxin clearance and the development of acute organ failures during sepsis. J. Innate Immun. 2016, 8, 211–220. [Google Scholar] [CrossRef]
- Mitchell, K.A.; Moore, J.X.; Rosenson, R.S.; Irvin, R.; Guirgis, F.W.; Shapiro, N.; Safford, M.; Wang, H.E. PCSK9 loss-of-function variants and risk of infection and sepsis in the reasons for geographic and racial differences in stroke (REGARDS) cohort. PLoS ONE 2019, 14, e0210808. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.-M.; Valdes, A.L.; Gromada, J.; Anderson, N.; Horton, J.D. Inhibition of PCSK9 does not improve lipopolysaccharide-induced mortality in mice. J. Lipid Res. 2017, 58, 1661–1669. [Google Scholar] [CrossRef] [Green Version]
- Desvergne, B. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef]
- Standage, S.W.; Caldwell, C.C.; Zingarelli, B.; Wong, H.R. Reduced peroxisome proliferator-activated receptor α expression is associated with decreased survival and increased tissue bacterial load in sepsis. Shock 2012, 37, 164–169. [Google Scholar] [CrossRef] [Green Version]
- Cámara-Lemarroy, C.R.; La Garza, F.J.G.-D.; Cordero-Perez, P.; Ibarra-Hernandez, J.M.; Muñoz-Espinosa, L.E.; Fernandez-Garza, N.E. Gemfibrozil attenuates the inflammatory response and protects rats from abdominal sepsis. Exp. Ther. Med. 2015, 9, 1018–1022. [Google Scholar] [CrossRef] [Green Version]
- Ann, S.-J.; Chung, J.H.; Park, B.H.; Kim, S.H.; Jang, J.; Park, S.; Kang, S.-M.; Lee, S.-H. PPARα agonists inhibit inflammatory activation of macrophages through upregulation of β-defensin 1. Atheroscler. 2015, 240, 389–397. [Google Scholar] [CrossRef]
- Paumelle, R.; Haas, J.T.; Hennuyer, N.; Baugé, E.; Deleye, Y.; Mesotten, D.; Langouche, L.; Vanhoutte, J.; Cudejko, C.; Wouters, K.; et al. Hepatic PPARα is critical in the metabolic adaptation to sepsis. J. Hepatol. 2019, 70, 963–973. [Google Scholar] [CrossRef]
- Hecker, M.; Behnk, A.; Morty, R.E.; Sommer, N.; Vadász, I.; Herold, S.; Seeger, W.; Mayer, K. PPAR-α activation reduced LPS-induced inflammation in alveolar epithelial cells. Exp. Lung Res. 2015, 41, 393–403. [Google Scholar] [CrossRef]
- Tancevski, I.; Nairz, M.; Duwensee, K.; Auer, K.; Schroll, A.; Heim, C.; Feistritzer, C.; Hoefer, J.; Gerner, R.R.; Moschen, A.R.; et al. Fibrates ameliorate the course of bacterial sepsis by promoting neutrophil recruitment via CXCR 2. EMBO Mol. Med. 2014, 6, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jozefowicz, E.; Brisson, H.; Rozenberg, S.; Mebazaa, A.; Gelé, P.; Callebert, J.; Lebuffe, G.; Vallet, B.; Bordet, R.; Tavernier, B. Activation of peroxisome proliferator-activated receptor-α by fenofibrate prevents myocardial dysfunction during endotoxemia in rats. Crit. Care Med. 2007, 35, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Wiel, E.; Lebuffe, G.; Robin, E.; Gasan, G.; Corseaux, D.; Tavernier, B.; Jude, B.; Bordet, R.; Vallet, B. Pretreatment with peroxysome proliferator-activated receptor α agonist fenofibrate protects endothelium in rabbit Escherichia coli endotoxin-induced shock. Intensiv. Care Med. 2005, 31, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.E.; Ousman, S.S.; Sobel, R.A.; Zuniga, L.; Baranzini, S.E.; Youssef, S.; Crowell, A.; Loh, J.; Oksenberg, J.R.; Steinman, L. Peroxisome proliferator–activated receptor (PPAR)α expression in T cells mediates gender differences in development of T cell–mediated autoimmunity. J. Exp. Med. 2007, 204, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Seymour, C.W.; Kennedy, J.N.; Wang, S.; Chang, C.-C.H.; Elliott, C.F.; Xu, Z.; Berry, S.; Clermont, G.; Cooper, G.; Gomez, H.; et al. Derivation, Validation, and Potential Treatment Implications of Novel Clinical Phenotypes for Sepsis. JAMA 2019, 321, 2003–2017. [Google Scholar] [CrossRef]
- The ProCESS Investigators. A randomized trial of protocol-based care for early septic shock. N. Engl. J. Med. 2014, 370, 1683–1693. [Google Scholar] [CrossRef] [Green Version]
- Rivers, E.; Nguyen, B.; Havstad, S.; Ressler, J.; Muzzin, A.; Knoblich, B.; Peterson, E.; Tomlanovich, M. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N. Engl. J. Med. 2001, 345, 1368–1377. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, T.E.; Azad, T.D.; Donato, M.; Haynes, W.A.; Perumal, T.M.; Henao, R.; Bermejo-Martin, J.F.; Almansa, R.; Tamayo, E.; Howrylak, J.A.; et al. Unsupervised analysis of transcriptomics in bacterial sepsis across multiple datasets reveals three robust clusters. Crit. Care Med. 2018, 46, 915–925. [Google Scholar] [CrossRef]
- Scicluna, B.P.; A van Vught, L.; Zwinderman, A.H.; A Wiewel, M.; E Davenport, E.; Burnham, K.L.; Nürnberg, P.; Schultz, M.J.; Horn, J.; Cremer, O.L.; et al. Classification of patients with sepsis according to blood genomic endotype: A prospective cohort study. Lancet Respir. Med. 2017, 5, 816–826. [Google Scholar] [CrossRef]
- E Davenport, E.; Burnham, K.L.; Radhakrishnan, J.; Humburg, P.; Hutton, P.; Mills, T.C.; Rautanen, A.; Gordon, A.C.; Garrard, C.; Hill, A.V.S.; et al. Genomic landscape of the individual host response and outcomes in sepsis: A prospective cohort study. Lancet Respir. Med. 2016, 4, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Buras, J.A.; Holzmann, B.; Sitkovsky, M. Animal Models of sepsis: Setting the stage. Nat. Rev. Drug Discov. 2005, 4, 854–865. [Google Scholar] [CrossRef]
- Efron, P.A.; Mohr, A.M.; Moore, F.A.; Moldawer, L.L. The future of murine sepsis and trauma research models. J. Leukoc. Biol. 2015, 98, 945–952. [Google Scholar] [CrossRef]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [Green Version]
- Zolfaghari, P.S.; Pinto, B.B.; Dyson, A.; Singer, M. The metabolic phenotype of rodent sepsis: Cause for concern? Intensiv. Care Med. Exp. 2013, 1, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sari, G.; Meester, E.J.; van der Zee, L.C.; Wouters, K.; van Lennep, J.R.; Peppelenbosch, M.; Boonstra, A.; Van der Heiden, K.; Mulder, M.M.; Vanwolleghem, T. A mouse model of humanized liver shows a human-like lipid profile, but does not form atherosclerotic plaque after western type diet. Biochem. Biophys. Res. Commun. 2020, 524, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Brewer, H.B.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl ester transfer protein. Arter. Thromb. Vasc. Biol. 2003, 23, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Oppi, S.; Lüscher, T.F.; Stein, S. Mouse Models for Atherosclerosis Research—Which Is My Line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agellon, L.; Walsh, A.; Hayek, T.; Moulin, P.; Jiang, X.; Shelanski, S.; Breslow, J.; Tall, A. Reduced high density lipoprotein cholesterol in human cholesteryl ester transfer protein transgenic mice. J. Biol. Chem. 1991, 266, 10796–10801. [Google Scholar] [CrossRef]
- Jiang, X.C.; Masucci-Magoulas, L.; Mar, J.; Lin, M.; Walsh, A.; Breslow, J.L.; Tall, A. Down-regulation of mRNA for the low density lipoprotein receptor in transgenic mice containing the gene for human cholesteryl ester transfer protein. Mechanism to explain accumulation of lipoprotein B particles. J. Biol. Chem. 1993, 268, 27406–27412. [Google Scholar] [CrossRef]
- Breslow, J.L. Mouse Models of Atherosclerosis. Sciences 1996, 272, 685–688. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Enzyme | Level | Function | Pathology during Sepsis |
|---|---|---|---|
| LCAT | ↓ | Promotes cholesterol efflux from cells to nascent HDL [84] | Diminished adrenal glucocorticoid function [85] Reduced LPS-neutralizing ability of HDL [86] |
| CETP | ↓ | Exchange of CE & TG between HDL and Apo-B-containing lipoproteins; promotes HDL maturation [87] | Missense variant in sepsis patients associated with HDL reduction, decreased survival, and increased organ failure [88] |
| PLTP | ↑ | Transfer of amphipathic molecules including phospholipids [89] | Regulates HDL size and composition [90] Recombinant PLTP in mice decreases bacterial growth and accelerates LPS detoxification [91] |
| PON | ↓ | Hydrolyzes lipid peroxides | Declines 71% in sepsis day 1–3 [92] Fails to inhibit oxLDL [93,94] |
| PAF-AH | ↓ | Hydrolyzes PAF | Declines 90% in sepsis day 1–3 [92] Failure to hydrolyze PAF, leading to immune cell activation, platelet activation, vascular permeability, and hypotension [95] Recombinant PAF-AH had no mortality benefit when used in septic patients [96] |
| EL | ↑ | Hydrolyzes HDL particles to liberate FFAs [97] | Upregulation leads to reduced HDL levels [98] Upregulation in inflammatory states may play a role in the resulting low-HDL state [99] EL knockout mice had increased survival to LPS-induced inflammation [100] |
| SAA | ↑ | Cytokine-like, propagates APR, modifies HDL transport [101] | >1000-fold increase during APR, displacing Apo-A-I [102] Comprises up to 80% of the proteins in higher-density HDL molecules [103] Increased HDL catabolism [104] Enhanced MDSC survival [105] |
| PLA2 | ↑ | Hydrolyzes phospholipids to generate an FFA and lysophospholipid | Elevated lipoprotein-associated levels independent predictor of mortality in sepsis [106] Mainly mobilizes AA [107] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barker, G.; Leeuwenburgh, C.; Brusko, T.; Moldawer, L.; Reddy, S.T.; Guirgis, F.W. Lipid and Lipoprotein Dysregulation in Sepsis: Clinical and Mechanistic Insights into Chronic Critical Illness. J. Clin. Med. 2021, 10, 1693. https://doi.org/10.3390/jcm10081693
Barker G, Leeuwenburgh C, Brusko T, Moldawer L, Reddy ST, Guirgis FW. Lipid and Lipoprotein Dysregulation in Sepsis: Clinical and Mechanistic Insights into Chronic Critical Illness. Journal of Clinical Medicine. 2021; 10(8):1693. https://doi.org/10.3390/jcm10081693
Chicago/Turabian StyleBarker, Grant, Christiaan Leeuwenburgh, Todd Brusko, Lyle Moldawer, Srinivasa T. Reddy, and Faheem W. Guirgis. 2021. "Lipid and Lipoprotein Dysregulation in Sepsis: Clinical and Mechanistic Insights into Chronic Critical Illness" Journal of Clinical Medicine 10, no. 8: 1693. https://doi.org/10.3390/jcm10081693
APA StyleBarker, G., Leeuwenburgh, C., Brusko, T., Moldawer, L., Reddy, S. T., & Guirgis, F. W. (2021). Lipid and Lipoprotein Dysregulation in Sepsis: Clinical and Mechanistic Insights into Chronic Critical Illness. Journal of Clinical Medicine, 10(8), 1693. https://doi.org/10.3390/jcm10081693