
Cervical Paraspinal Chordoma: A Literature Review with a Novel Case Report

 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
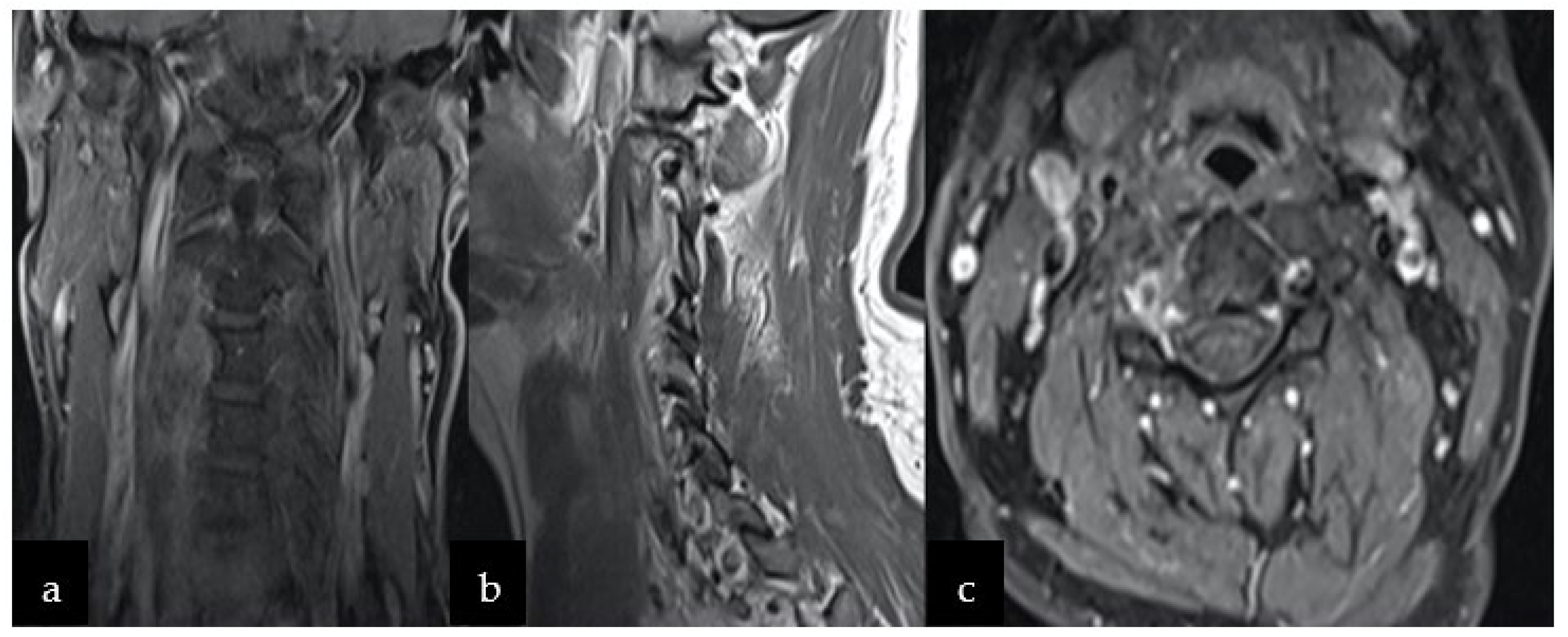
2. Case Report
3. Discussion
3.1. Epidemiological Data
3.2. Diagnostic Process in the COVID-19 Pandemic
3.3. Radiological Features
3.4. Chordoma Classification
3.5. Therapeutic Options
3.5.1. Radiotherapeutic Approach
3.5.2. Current Research and Targeted Therapy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Noor, A.; Bindal, P.; Ramirez, M.; Vredenburgh, J. Chordoma: A case report and review of literature. Am. J. Case Rep. 2020, 21, e918927. [Google Scholar] [CrossRef] [PubMed]
- Awuor, V.; Stewart, C.; Camma, A.; Renner, J.; Tongson, J. Rare case of an extraosseous cervical chordoma with both intradural and extensive extraspinal involvement. Surg. Neurol. Int. 2017, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Alan, O.; Akin Telli, T.; Ercelep, O.; Tanrikulu Simsek, E.; Basoglu Tuylu, T.; Mutis, A.; Hasanov, R.; Kaya, S.; Akgül Babacan, N.; Dane, F.; et al. Chordoma: A case series and review of the literature. J. Med. Case Rep. 2018, 12, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anagiotos, A.; Preuss, S.F.; Drebber, U.; Jumah, M.D. Multiple craniocervical chordomas presenting as a parapharyngeal mass. Head Neck 2013, 35, E325–E327. [Google Scholar] [CrossRef]
- Teles, A.R.; Mattei, T.A.; Goulart, C.R.; Mendel, E. Chordomas and chondrosarcomas of the spine: Preoperative planning, surgical strategies, and complications avoidance. In Chordomas and Chondrosarcomas of the Skull Base and Spine; Academic Press: Cambridge, MA, USA, 2018; ISBN 9780128043332. [Google Scholar]
- Denaro, L.; Berton, A.; Ciuffreda, M.; Loppini, M.; Candela, V.; Brandi, M.L.; Longo, U.G. Surgical management of chordoma: A systematic review. J. Spinal Cord Med. 2020, 43, 797–812. [Google Scholar] [CrossRef]
- Perez-Roman, R.; Burks, S.; Debs, L.; Cajigas, I.; Levi, A. The Risk of Peripheral Nerve Tumor Biopsy in Suspected Benign Etiologies. Neurosurgery 2020, 86, E326–E332. [Google Scholar] [CrossRef]
- Pendleton, C.; Spinner, R. Image-guided percutaneous biopsy of peripheral nerve tumors of indeterminate nature: Risks and benefits. Acta Neurochir. 2020, 162, 1425–1429. [Google Scholar] [CrossRef]
- Healey, J.H.; Lane, J.M. Chordoma: A critical review of diagnosis and treatment. Orthop. Clin. N. Am. 1989, 20, 417–426. [Google Scholar]
- Anonymous. Report of Brain Tumor Registry of Japan (1984–2000). Neurol. Med. Chir. 2009, 49, PS1–PS96. [Google Scholar]
- Varlet, P.; Nielsen, G.P.; Righi, A.; Tanaka, S.; Tirabosco, R. Central Nervous System Tumours: WHO Classification of Tumours, 5th ed.; WHO: Geneva, Switzerland, 2021; Volume 6. [Google Scholar]
- McMaster, M.L.; Goldstein, A.M.; Bromley, C.M.; Ishibe, N.; Parry, D.M. Chordoma: Incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control 2001, 12, 1–11. [Google Scholar] [CrossRef]
- Radaelli, S.; Stacchiotti, S.; Ruggieri, P.; Donati, D.; Casali, P.G.; Palmerini, E.; Collini, P.; Gambarotti, M.; Porcu, L.; Boriani, S.; et al. Sacral Chordoma: Long-term outcome of a large series of patients surgically treated at two reference centers. Spine 2016, 41, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.D.M.; Unni, K.K.; Mertens, F. Pathology and Genetics of Tumours of Soft Tissue and Bone. WHO Classification of Tumours, 3rd ed.; WHO: Geneva, Switzerland, 2002; Volume 5. [Google Scholar]
- Sciubba, D.M.; Chi, J.H.; Rhines, L.D.; Gokaslan, Z.L. Chordoma of the Spinal Column. Neurosurg. Clin. N. Am. 2008, 19, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, N. Letter to the Editor Telemedicine: Could it represent a new problem for spine surgeons to solve? Glob. Spine J. 2022, 12, 1306–1307. [Google Scholar] [CrossRef] [PubMed]
- Onor, M.L.; Misan, S. The clinical interview and the doctor-patient relationship in telemedicine. Telemed. J. e-Health 2005, 11, 102–105. [Google Scholar] [CrossRef]
- Barber, S.M.; Sadrameli, S.S.; Lee, J.J.; Fridley, J.S.; Teh, B.S.; Oyelese, A.A.; Telfeian, A.E.; Gokaslan, Z.L. Chordoma—Current understanding and modern treatment paradigms. J. Clin. Med. 2021, 10, 1054. [Google Scholar] [CrossRef]
- Elefante, A.; Caranci, F.; Del Basso De Caro, M.L.; Peca, C.; Guadagno, E.; Severino, R.; Mariniello, G.; Maiuri, F. Paravertebral high cervical chordoma a case report. Neuroradiol. J. 2013, 26, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Firooznia, H.; Pinto, R.S.; Lin, J.P.; Baruch, H.H.; Zausner, J. Chordoma: Radiologic evaluation of 20 cases. Am. J. Roentgenol. 1976, 127, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Moore, K.R. Diagnostic Imaging: Spine, 3rd ed.; Elsevier Health Sciences: Philadelphia, PA, USA, 2015. [Google Scholar]
- Murpbey, M.D.; Andrews, C.L.; Flemming, D.J.; Temple, H.T.; Smith, W.S.; Smirniotopoulos, J.G. From the Archives of the AFIP: Primary Tumors of the Spine: Radiologic-Pathologic Correlation. Radiographics 1996, 16, 1131–1158. [Google Scholar] [CrossRef]
- Atlas, S.W. (Ed.) Magnetic Resonance Imaging of the Brain and Spine, 4th ed., Vol. 1 and 2. Am. J. Neuroradiol. 2009, 30, e76–e77. [Google Scholar] [CrossRef] [Green Version]
- Caranci, F.; Brunese, L.; Reginelli, A.; Napoli, M.; Fonio, P.; Briganti, F. Neck Neoplastic Conditions in the Emergency Setting: Role of Multidetector Computed Tomography. Semin. Ultrasound CT MRI 2012, 33, 443–448. [Google Scholar] [CrossRef]
- Bannur, U.; Chawda, S.J.; O’Donovan, D.G.; Kaddour, H.; David, K.M. Paravertebral cervical chordoma—A case report. Br. J. Neurosurg. 2011, 25, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Murphey, M.D.; Smith, W.S.; Smith, S.E.; Kransdorf, M.J.; Temple, H.T. From the archives of the AFIP: Imaging of musculoskeletal neurogenic tumors: Radiologic-pathologic correlation. Radiographics 1999, 19, 1253–1280. [Google Scholar] [CrossRef] [Green Version]
- Kleihues, P.; Cavenee, W.K. WHO Classification of Tumors: Pathology and Genetics of Tumours of the Nervous System; WHO: Geneva, Switzerland, 2000. [Google Scholar]
- Abul-Kasim, K.; Thurnher, M.M.; McKeever, P.; Sundgren, P.C. Intradural spinal tumors: Current classification and MRI features. Neuroradiology 2008, 50, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.D.M.; Bridge, J.A.; Hogendoorn, P.C.W.; Mertens, F. WHO Classification of Tumours of Soft Tissue and Bone, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020; ISBN 9789283224341. [Google Scholar]
- Elefante, A.; Peca, C.; Del Basso De Caro, M.L.; Russo, C.; Formicola, F.; Mariniello, G.; Brunetti, A.; Maiuri, F. Symptomatic spinal cord metastasis from cerebral oligodendroglioma. Neurol. Sci. 2012, 33, 609–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoun, S.G.; Elguindy, M.; Barrie, U.; El Ahmadieh, T.Y.; Plitt, A.; Moreno, J.R.; Truelson, J.M.; Bagley, C.A. Four-Level Vertebrectomy for En Bloc Resection of a Cervical Chordoma. World Neurosurg. 2018, 118, 316–323. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, Z.J.; Liu, X.G.; Ma, Q.J.; Wei, F.; Lv, Y.; Dang, G.T. Upper cervical spine chordoma of C2–C3. Eur. Spine J. 2009, 18, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Sommer, J. Building a global consensus approach to chordoma: A position paper from the medical and patient community. Lancet Oncol. 2015, 16, e71–e83. [Google Scholar]
- D’Haen, B.; De Jaegere, T.; Goffin, J.; Dom, R.; Demaerel, P.; Plets, C. Chordoma of the lower cervical spine. Clin. Neurol. Neurosurg. 1995, 97, 245–248. [Google Scholar] [CrossRef]
- Goes, R.; Van Overbeeke, J.J. A vertebral extra dural chordoma at C5, possibly deriving from a clival chordoma. Surg. Neurol. Int. 2015, 6, 94. [Google Scholar] [CrossRef]
- Rhines, L.D.; Fourney, D.R.; Siadati, A.; Suk, I.; Gokaslan, Z.L. En bloc resection of multilevel cervical chordoma with C-2 involvement. Case report and description of operative technique. J. Neurosurg. Spine 2005, 2, 199–205. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Z.; Tian, K.; Wang, K.; Li, D.; Ma, J.; Jia, G.; Zhang, L.; Zhang, J. Clinical features and surgical outcomes of patients with skull base chordoma: A retrospective analysis of 238 patients. J. Neurosurg. 2017, 127, 1257–1267. [Google Scholar] [CrossRef] [Green Version]
- Gokaslan, Z.L.; Zadnik, P.L.; Sciubba, D.M.; Germscheid, N.; Goodwin, C.R.; Wolinsky, J.P.; Bettegowda, C.; Groves, M.L.; Luzzati, A.; Rhines, L.D.; et al. Mobile spine chordoma: Results of 166 patients from the AOSpine Knowledge Forum Tumor database. J. Neurosurg. Spine 2016, 24, 644–651. [Google Scholar] [CrossRef] [Green Version]
- Hulen, C.A.; Temple, H.T.; Fox, W.P.; Sama, A.A.; Green, B.A.; Eismont, F.J. Oncologic And Functional Outcome Following Sacrectomy For Sacral Chordoma. J. Bone Jt. Surgery-American Vol. 2006, 88, 1532–1539. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, P.; Angelini, A.; Ussia, G.; Montalti, M.; Mercuri, M. Surgical margins and local control in resection of sacral chordomas. Clin. Orthop. Relat. Res. 2010, 468, 2939–2947. [Google Scholar] [CrossRef] [Green Version]
- Boriani, S.; Bandiera, S.; Biagini, R.; Bacchini, P.; Boriani, L.; Cappuccio, M.; Chevalley, F.; Gasbarrini, A.; Picci, P.; Weinstein, J.N. Chordoma of the mobile spine: Fifty years of experience. Spine 2006, 31, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, J.; Zhang, L.; Jia, G.; Tang, J.; Wang, L.; Wang, Z. Prognostic factors for long-term outcome of patients with surgical resection of skull base chordomas-106 cases review in one institution. Neurosurg. Rev. 2010, 33, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Yang, H.L.; Lu, J.; Liu, J.Y.; Chen, X.Q. Prognostic factors of sacral chordoma after surgical therapy: A study of 36 patients. Spinal Cord 2010, 48, 166–171. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Sun, J.; Bai, H.X.; Huang, X.; Zou, Y.; Tan, X.; Zhang, Z.; Tang, X.; Tao, Y.; Xiao, B.; et al. Prognostic factors in patients with spinal chordoma: An integrative analysis of 682 patients. Clin. Neurosurg. 2017, 81, 812–823. [Google Scholar] [CrossRef]
- Colli, B.O.; Al-Mefty, O. Chordomas of the skull base: Follow-up review and prognostic factors. Neurosurg. Focus 2001, 10, 1–11. [Google Scholar] [CrossRef]
- Ahmed, R.; Sheybani, A.; Menezes, A.H.; Buatti, J.M.; Hitchon, P.W. Disease outcomes for skull base and spinal chordomas: A single center experience. Clin. Neurol. Neurosurg. 2015, 130, 67–73. [Google Scholar] [CrossRef]
- Samii, A.; Gerganov, V.M.; Herold, C.; Hayashi, N.; Naka, T.; Mirzayan, M.J.; Ostertag, H.; Samii, M. Chordomas of the skull base: Surgical management and outcome. J. Neurosurg. 2007, 107, 319–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, T.E.; Pritchard, D.J.; Unni, K.K. Clinicopathologic study of sacrococcygeal chordoma. Cancer 1984, 53, 2574–2578. [Google Scholar] [CrossRef]
- Arnautović, K.I.; Al-Mefty, O. Surgical seeding of chordomas. Neurosurg. Focus 2001, 10, 1–6. [Google Scholar] [CrossRef]
- DeLaney, T.F.; Liebsch, N.J.; Pedlow, F.X.; Adams, J.; Dean, S.; Yeap, B.Y.; McManus, P.; Rosenberg, A.E.; Nielsen, G.P.; Harmon, D.C.; et al. Phase II Study of High-Dose Photon/Proton Radiotherapy in the Management of Spine Sarcomas. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 732–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moojen, W.A.; Vleggeert-Lankamp, C.L.A.; Krol, A.D.G.; Dijkstra, S.P.D. Long-term results: Adjuvant radiotherapy in en bloc resection of sacrococcygeal chordoma is advisable. Spine 2011, 36, E656–E661. [Google Scholar] [CrossRef]
- Nishida, Y.; Kamada, T.; Imai, R.; Tsukushi, S.; Yamada, Y.; Sugiura, H.; Shido, Y.; Wasa, J.; Ishiguro, N. Clinical outcome of sacral chordoma with carbon ion radiotherapy compared with surgery. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Matloob, S.A.; Nasir, H.A.; Choi, D. Proton beam therapy in the management of skull base chordomas: Systematic review of indications, outcomes, and implications for neurosurgeons. Br. J. Neurosurg. 2016, 30, 382–387. [Google Scholar] [CrossRef]
- Dassoulas, K.; Schlesinger, D.; Yen, C.P.; Sheehan, J. The role of Gamma Knife surgery in the treatment of skull base chordomas. J. Neurooncol. 2009, 94, 243–248. [Google Scholar] [CrossRef]
- Hug, E.B.; Loredo, L.N.; Slater, J.D.; DeVries, A.; Grove, R.I.; Schaefer, R.A.; Rosenberg, A.E.; Slater, J.M. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J. Neurosurg. 1999, 91, 432–439. [Google Scholar] [CrossRef]
- Indelicato, D.J.; Rotondo, R.L.; Begosh-Mayne, D.; Scarborough, M.T.; Gibbs, C.P.; Morris, C.G.; Mendenhall, W.M. A Prospective Outcomes Study of Proton Therapy for Chordomas and Chondrosarcomas of the Spine. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 297–303. [Google Scholar] [CrossRef]
- Tamborini, E.; Miselli, F.; Negri, T.; Lagonigro, M.S.; Staurengo, S.; Dagrada, G.P.; Stacchiotti, S.; Pastore, E.; Gronchi, A.; Perrone, F.; et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin. Cancer Res. 2006, 12, 6920–6928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberger, P.M.; Yu, Z.; Kowalski, D.; Joe, J.; Manger, P.; Psyrri, A.; Sasaki, C.T. Differential expression of epidermal growth factor receptor, c-Met, and HER2/neu in chordoma compared with 17 other malignancies. Arch. Otolaryngol. Head Neck Surg. 2005, 131, 707–711. [Google Scholar] [CrossRef] [Green Version]
- Stacchiotti, S.; Longhi, A.; Ferraresi, V.; Grignani, G.; Comandone, A.; Stupp, R.; Bertuzzi, A.; Tamborini, E.; Pilotti, S.; Messina, A.; et al. Phase II study of imatinib in advanced chordoma. J. Clin. Oncol. 2012, 30, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Marrari, A.; Tamborini, E.; Palassini, E.; Virdis, E.; Messina, A.; Crippa, F.; Morosi, C.; Gronchi, A.; Pilotti, S.; et al. Response to imatinib plus sirolimus in advanced chordoma. Ann. Oncol. 2009, 20, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- Hindi, N.; Casali, P.G.; Morosi, C.; Messina, A.; Palassini, E.; Pilotti, S.; Tamborini, E.; Radaelli, S.; Gronchi, A.; Stacchiotti, S. Imatinib in advanced chordoma: A retrospective case series analysis. Eur. J. Cancer 2015, 51, 2609–2614. [Google Scholar] [CrossRef]
- Bompas, E.; Le Cesne, A.; Tresch-Bruneel, E.; Lebellec, L.; Laurence, V.; Collard, O.; Saada-Bouzid, E.; Isambert, N.; Blay, J.Y.; Amela, E.Y.; et al. Sorafenib in patients with locally advanced and metastatic chordomas: A phase II trial of the French Sarcoma Group (GSF/GETO). Ann. Oncol. 2015, 26, 2168–2173. [Google Scholar] [CrossRef]
- Magnaghi, P.; Salom, B.; Cozzi, L.; Amboldi, N.; Ballinari, D.; Tamborini, E.; Gasparri, F.; Montagnoli, A.; Raddrizzani, L.; Somaschini, A.; et al. Afatinib is a new therapeutic approach in chordoma with a unique ability to target EGFR and brachyury. Mol. Cancer Ther. 2018, 17, 603–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacchiotti, S.; Tamborini, E.; Lo Vullo, S.; Bozzi, F.; Messina, A.; Morosi, C.; Casale, A.; Crippa, F.; Conca, E.; Negri, T.; et al. Phase ii study on lapatinib in advanced egfr-positive chordoma. Ann. Oncol. 2013, 24, 1931–1936. [Google Scholar] [CrossRef]
- Kelley, M.J.; Shi, J.; Ballew, B.; Hyland, P.L.; Li, W.Q.; Rotunno, M.; Alcorta, D.A.; Liebsch, N.J.; Mitchell, J.; Bass, S.; et al. Characterization of T gene sequence variants and germline duplications in familial and sporadic chordoma. Hum. Genet. 2014, 133, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Vujovic, S.; Henderson, S.; Presneau, N.; Odell, E.; Jacques, T.S.; Tirabosco, R.; Boshoff, C.; Flanagan, A.M. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J. Pathol. 2006, 209, 157–165. [Google Scholar] [CrossRef]
- Yang, X.R.; Ng, D.; Alcorta, D.A.; Liebsch, N.J.; Sheridan, E.; Li, S.; Goldstein, A.M.; Parry, D.M.; Kelley, M.J. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat. Genet. 2009, 41, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Von Witzleben, A.; Goerttler, L.T.; Marienfeld, R.; Barth, H.; Lechel, A.; Mellert, K.; Böhm, M.; Kornmann, M.; Mayer-Steinacker, R.; Von Baer, A.; et al. Preclinical characterization of novel Chordoma cell systems and their targeting by pharmocological inhibitors of the CDK4/6 cell-cycle pathway. Cancer Res. 2015, 75, 3823–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traylor, J.I.; Pernik, M.N.; Plitt, A.R.; Lim, M.; Garzon-Muvdi, T. Immunotherapy for chordoma and chondrosarcoma: Current evidence. Cancers 2021, 13, 2408. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R.; Schlom, J.; Hodge, J.W. A potential therapy for chordoma via antibody-dependent cell-mediated cytotoxicity employing NK or high-affinity NK cells in combination with cetuximab. J. Neurosurg. 2018, 128, 1419–1427. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Wang, X.; Wang, Y.; Hornicek, F.J.; Nielsen, G.P.; Duan, Z.; Ferrone, S.; Schwab, J.H. CSPG4 as a prognostic biomarker in chordoma. Spine J. 2016, 16, 722–727. [Google Scholar] [CrossRef]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-Daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014, 2, 25. [Google Scholar] [CrossRef] [Green Version]
- Heery, C.R.; Singh, B.H.; Rauckhorst, M.; Marté, J.L.; Donahue, R.N.; Grenga, I.; Rodell, T.C.; Dahut, W.; Arlen, P.M.; Madan, R.A.; et al. Phase i Trial of a Yeast-Based Therapeutic Cancer Vaccine (GI-6301) Targeting the Transcription Factor Brachyury. Cancer Immunol. Res. 2015, 3, 1248–1256. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabbar, R.; Jankowski, J.; Pawełczyk, A.; Szmyd, B.; Solek, J.; Pierzak, O.; Wojdyn, M.; Radek, M. Cervical Paraspinal Chordoma: A Literature Review with a Novel Case Report. J. Clin. Med. 2022, 11, 4117. https://doi.org/10.3390/jcm11144117
Jabbar R, Jankowski J, Pawełczyk A, Szmyd B, Solek J, Pierzak O, Wojdyn M, Radek M. Cervical Paraspinal Chordoma: A Literature Review with a Novel Case Report. Journal of Clinical Medicine. 2022; 11(14):4117. https://doi.org/10.3390/jcm11144117
Chicago/Turabian StyleJabbar, Redwan, Jakub Jankowski, Agnieszka Pawełczyk, Bartosz Szmyd, Julia Solek, Olaf Pierzak, Maciej Wojdyn, and Maciej Radek. 2022. "Cervical Paraspinal Chordoma: A Literature Review with a Novel Case Report" Journal of Clinical Medicine 11, no. 14: 4117. https://doi.org/10.3390/jcm11144117
APA StyleJabbar, R., Jankowski, J., Pawełczyk, A., Szmyd, B., Solek, J., Pierzak, O., Wojdyn, M., & Radek, M. (2022). Cervical Paraspinal Chordoma: A Literature Review with a Novel Case Report. Journal of Clinical Medicine, 11(14), 4117. https://doi.org/10.3390/jcm11144117