
Cardiac Hypertrophy and Related Dysfunctions in Cushing Syndrome Patients—Literature Review


Abstract
:1. Introduction
2. Physiology of Cortisol
2.1. Glucocorticoid Receptor and Regulating Enzymes
2.2. Physiological Role of Glucocorticoid in Rodent Cardiomyocytes
2.3. Harmful Effects to Cardiomyocytes by Experimental Excessive Glucocorticoid Exposure
2.3.1. Excessive Glucocorticoid and Cardiomyocyte Hypertrophy
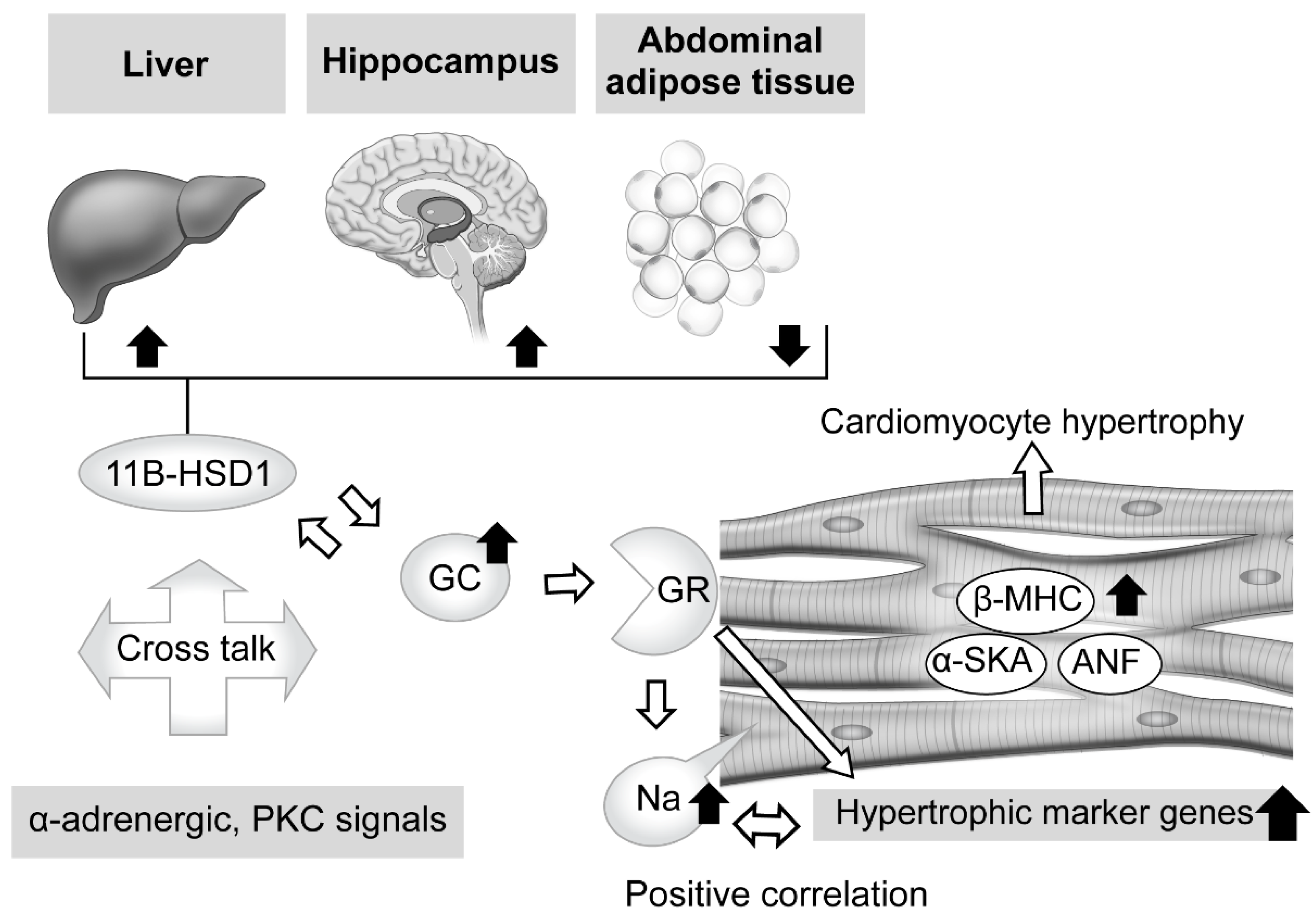
2.3.2. Tissue-Specific Role of 11β-Hydroxysteroid Dehydrogenase Type 1 in Glucocorticoid Excess
3. Effects of Hypercortisolemia on Heart Structure and Function in Clinical Findings
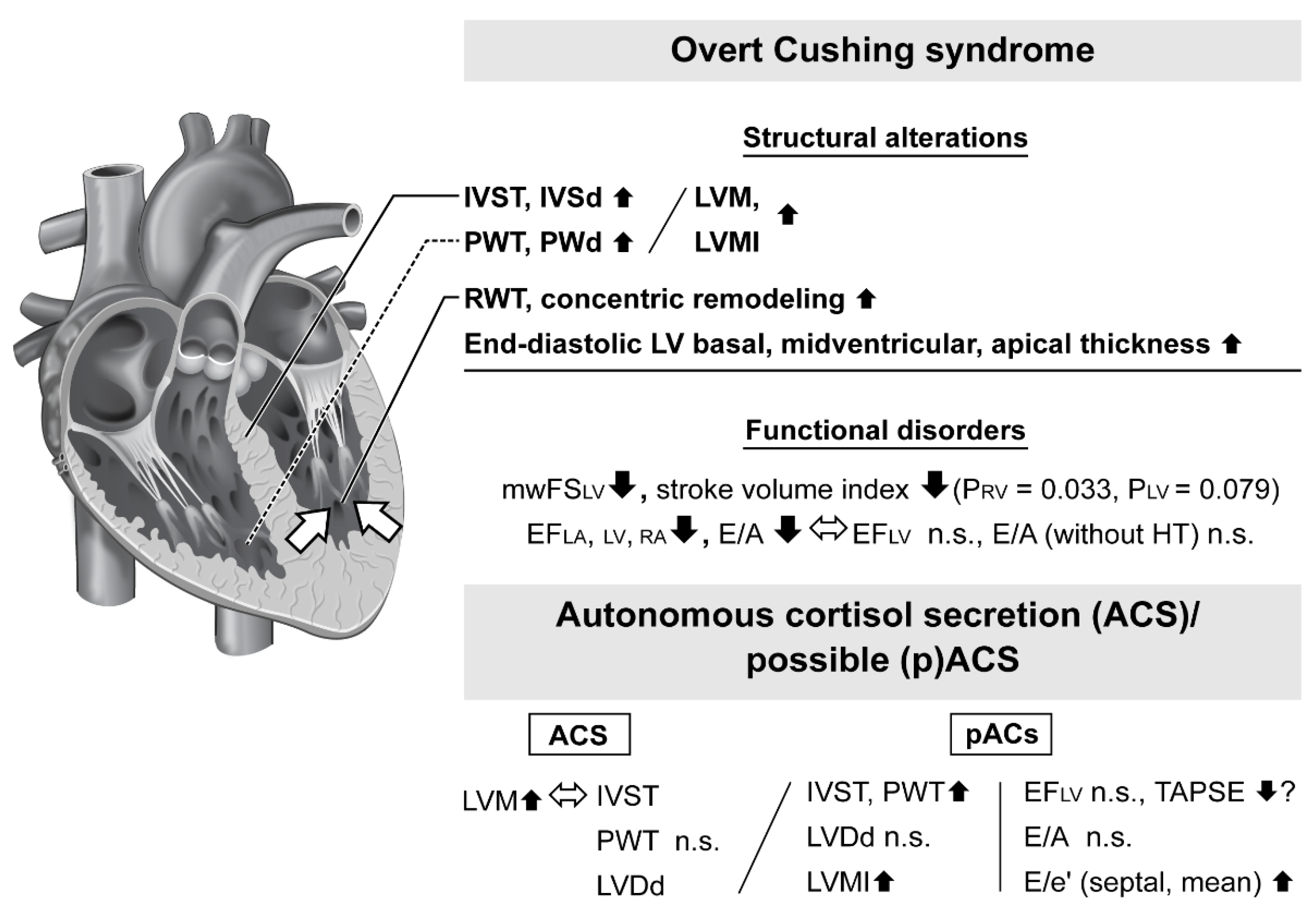
3.1. Heart Structural Alterations in Patients with Overt Cushing Syndrome
3.2. Cardiac Dysfunction in Patients with Overt Cushing Syndrome
3.3. Cardiac Magnetic Resonance Imaging Test for Evaluation of Patients with Overt Cushing Syndrome
3.4. Contributions of Human Serum and Urinary Cortisol Levels to Cardiac Impairment
3.5. Geometric and Functional Impairment in Autonomous Cortisol Secretion Patients
4. Treatment of Adrenal Cushing Syndrome and Effects on Cardiac Impairment
5. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Plotz, C.M.; Knowlton, A.I.; Ragan, C. The natural history of Cushing’s syndrome. Am. J. Med. 1952, 13, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Pikkarainen, L.; Sane, T.; Reunanen, A. The survival and well-being of patients treated for Cushing’s syndrome. J. Intern. Med. 1999, 245, 463–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, J.; Juul, S.; Jørgensen, J.O.; Astrup, J.; Bjerre, P.; Feldt-Rasmussen, U.; Hagen, C.; Jørgensen, J.; Kosteljanetz, M.; Kristensen, L.; et al. Incidence and late prognosis of cushing’s syndrome: A population-based study. J. Clin. Endocrinol. Metab. 2001, 86, 117–123. [Google Scholar] [CrossRef]
- Yaneva, M.; Kalinov, K.; Zacharieva, S. Mortality in Cushing’s syndrome: Data from 386 patients from a single tertiary referral center. Eur. J. Endocrinol. 2013, 169, 621–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntali, G.; Asimakopoulou, A.; Siamatras, T.; Komninos, J.; Vassiliadi, D.; Tzanela, M.; Tsagarakis, S.; Grossman, A.B.; Wass, J.A.; Karavitaki, N. Mortality in Cushing’s syndrome: Systematic analysis of a large series with prolonged follow-up. Eur. J. Endocrinol. 2013, 169, 715–723. [Google Scholar] [CrossRef] [Green Version]
- Favero, V.; Cremaschi, A.; Parazzoli, C.; Falchetti, A.; Gaudio, A.; Gennari, L.; Scillitani, A.; Vescini, F.; Morelli, V.; Aresta, C.; et al. Pathophysiology of Mild Hypercortisolism: From the Bench to the Bedside. Int. J. Mol. Sci. 2022, 23, 673. [Google Scholar] [CrossRef]
- Newell-Price, J.; Bertagna, X.; Grossman, A.B.; Nieman, L.K. Cushing’s syndrome. Lancet 2006, 367, 1605–1617. [Google Scholar] [CrossRef]
- Javanmard, P.; Duan, D.; Geer, E.B. Mortality in Patients with Endogenous Cushing’s Syndrome. Endocrinol. Metab. Clin. North Am. 2018, 47, 313–333. [Google Scholar] [CrossRef]
- Barbot, M.; Zilio, M.; Scaroni, C. Cushing’s syndrome: Overview of clinical presentation, diagnostic tools and complications. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101380. [Google Scholar] [CrossRef]
- Ntali, G.; Hakami, O.; Wattegama, M.; Ahmed, S.; Karavitaki, N. Mortality of Patients with Cushing’s Disease. Exp. Clin. Endocrinol. Diabetes. 2021, 129, 203–207. [Google Scholar] [CrossRef]
- Dekkers, O.M.; Horváth-Puhó, E.; Jørgensen, J.O.; Cannegieter, S.C.; Ehrenstein, V.; Vandenbroucke, J.P.; Pereira, A.M.; Sørensen, H.T. Multisystem morbidity and mortality in Cushing’s syndrome: A cohort study. J. Clin. Endocrinol. Metab. 2013, 98, 2277–2284. [Google Scholar] [CrossRef] [PubMed]
- Ntali, G.; Grossman, A.; Karavitaki, N. Clinical and biochemical manifestations of Cushing’s. Pituitary 2015, 18, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.H.; Kim, J.H.; Park, M.Y.; Kim, S.W. Epidemiology and Comorbidity of Adrenal Cushing Syndrome: A Nationwide Cohort Study. J. Clin. Endocrinol. Metab. 2021, 106, e1362–e1372. [Google Scholar] [CrossRef] [PubMed]
- Reincke, M.; Nieke, J.; Krestin, G.P.; Saeger, W.; Allolio, B.; Winkelmann, W. Preclinical Cushing’s syndrome in adrenal “incidentalomas”: Comparison with adrenal Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1992, 75, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Arlt, W.; Bancos, I.; Dralle, H.; Newell-Price, J.; Sahdev, A.; Tabarin, A.; Terzolo, M.; Tsagarakis, S.; Dekkers, O.M. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 2016, 175, G1–G34. [Google Scholar] [CrossRef] [Green Version]
- Masserini, B.; Morelli, V.; Palmieri, S.; Eller-Vainicher, C.; Zhukouskaya, V.; Cairoli, E.; Orsi, E.; Beck-Peccoz, P.; Spada, A.; Chiodini, I. Lipid abnormalities in patients with adrenal incidentalomas: Role of subclinical hypercortisolism and impaired glucose metabolism. J. Endocrinol. Invest. 2015, 38, 623–628. [Google Scholar] [CrossRef]
- Aresta, C.; Favero, V.; Morelli, V.; Giovanelli, L.; Parazzoli, C.; Falchetti, A.; Pugliese, F.; Gennari, L.; Vescini, F.; Salcuni, A.; et al. Cardiovascular complications of mild autonomous cortisol secretion. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101494. [Google Scholar] [CrossRef]
- Dalmazi, G.D.; Vicennati, V.; Garelli, S.; Casadio, E.; Rinaldi, E.; Giampalma, E.; Mosconi, C.; Golfieri, R.; Paccapelo, A.; Pagotto, U.; et al. Cardiovascular events and mortality in patients with adrenal incidentalomas that are either non-secreting or associated with intermediate phenotype or subclinical Cushing’s syndrome: A 15-year retrospective study. Lancet Diabetes Endocrinol. 2014, 2, 396–405. [Google Scholar] [CrossRef]
- Patrova, J.; Kjellman, M.; Wahrenberg, H.; Falhammar, H. Increased mortality in patients with adrenal incidentalomas and autonomous cortisol secretion: A 13-year retrospective study from one center. Endocrine 2017, 58, 267–275. [Google Scholar] [CrossRef]
- Debono, M.; Bradburn, M.; Bull, M.; Harrison, B.; Ross, R.J.; Newell-Price, J. Cortisol as a marker for increased mortality in patients with incidental adrenocortical adenomas. J. Clin. Endocrinol. Metab. 2014, 99, 4462–4470. [Google Scholar] [CrossRef]
- Barzon, L.; Sonino, N.; Fallo, F.; Palu, G.; Boscaro, M. Prevalence and natural history of adrenal incidentalomas. Eur. J. Endocrinol. 2003, 149, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Maekawa, S.; Ishii, R.; Sanada, M.; Morikawa, T.; Shiraishi, Y.; Yoshida, K.; Nagata, Y.; Sato-Otsubo, A.; Yoshizato, T.; et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 2014, 344, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; Dalmazi, G.D.; Bathon, K.; Ronchi, C.L.; Beuschlein, F. cAMP signaling in cortisol-producing adrenal adenoma. Eur. J. Endocrinol. 2015, 173, M99–M106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Thompson, E.B. Gene regulation by the glucocorticoid receptor: Structure: Function relationship. J. Steroid Biochem. Mol. Biol. 2005, 94, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M. The steroid and thyroid hormone receptor superfamily. Science 1988, 240, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Grad, I.; Picard, D. The glucocorticoid responses are shaped by molecular chaperones. Mol. Cell Endocrinol. 2007, 275, 2–12. [Google Scholar] [CrossRef]
- Funder, J.W. Aldosterone and mineralocorticoid receptors: A personal reflection. Mol. Cell Endocrinol. 2012, 350, 146–150. [Google Scholar] [CrossRef]
- Funder, J.; Myles, K. Exclusion of corticosterone from epithelial mineralocorticoid receptors is insufficient for selectivity of aldosterone action: In Vivo binding studies. Endocrinology 1996, 137, 5264–5268. [Google Scholar] [CrossRef]
- Funder, J.W. Mineralocorticoid receptor antagonists: Emerging roles in cardiovascular medicine. Integr. Blood Press. Control 2013, 6, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Funder, J.W. Mineralocorticoid receptor activation and oxidative stress. Hypertension 2007, 50, 840–841. [Google Scholar] [CrossRef]
- Mihailidou, A.S.; Le, T.Y.L.; Mardini, M.; Funder, J.W. Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension 2009, 54, 1306–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seckl, J.R. 11beta-hydroxysteroid dehydrogenases: Changing glucocorticoid action. Curr. Opin. Pharmacol. 2004, 4, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Hadoke, P.W.; Iqbal, J.; Walker, B.R. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br. J. Pharmacol. 2009, 156, 689–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasov, A.G.; Nashev, L.G.; Gelman, L.; Legeza, B.; Sack, R.; Portmann, R.; Odermatt, A. Direct protein-protein interaction of 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase in the endoplasmic reticulum lumen. Biochim. Biophys. Acta 2008, 1783, 1536–1543. [Google Scholar] [CrossRef] [Green Version]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W.; Lavery, G.G. 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. USA 2014, 111, E2482–E2491. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase type 1, a tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Nashev, L.G.; Schweizer, R.A.; Frick, C.; Odermatt, A. Hexose-6-phosphate dehydrogenase determines the reaction direction of 11beta-hydroxysteroid dehydrogenase type 1 as an oxoreductase. FEBS Lett. 2004, 571, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.R. Glucocorticoids and cardiovascular disease. Eur. J. Endocrinol. 2007, 157, 545–559. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, L. Glucocorticoids and vascular reactivity. Curr. Vasc. Pharmacol. 2004, 2, 1–12. [Google Scholar] [CrossRef]
- Albiston, A.L.; Obeyesekere, V.R.; Smith, R.E.; Krozowski, Z.S. Cloning and tissue distribution of the human 11 beta-hydroxysteroid dehydrogenase type 2 enzyme. Mol. Cell Endocrinol. 1994, 105, R11–R17. [Google Scholar] [CrossRef]
- Brown, R.W.; Chapman, K.E.; Murad, P.; Edwards, C.R.; Seckl, J.R. Purification of 11beta-hydroxysteroid dehydrogenase type 2 from human placenta utilizing a novel affinity labelling technique. Biochem. J. 1996, 313, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.R. Extra-adrenal regeneration of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1, physiological regulator and pharmacological target for energy partitioning. Proc. Nutr. Soc. 2007, 66, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Cain, D.W.; Cidlowski, J.A. Specificity and sensitivity of glucocorticoid signaling in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Rog-Zielinska, E.A.; Thomson, A.; Kenyon, C.J.; Brownstein, D.G.; Moran, C.M.; Szumska, D.; Michailidou, Z.; Richardson, J.; Owen, E.; Watt, A.; et al. Glucocorticoid receptor is required for foetal heart maturation. Hum. Mol. Genet. 2013, 22, 3269–3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, N.; Yang, C.; Xu, A. Dexamethasone treatment improves sarcoplasmic reticulum function and contractile performance in aged myocardium. Mol. Cell. Biochem. 2004, 266, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Cidlowski, J.A. Glucocorticoid signaling in cardiac disease. Horm. Mol. Biol. Clin. Investig. 2010, 4, 559–564. [Google Scholar] [CrossRef]
- Pearl, J.M.; Nelson, D.P.; Schwartz, S.M.; Wagner, C.J.; Bauer, S.M.; Setser, E.A.; Duffy, J.Y. Glucocorticoids reduce ischemia-reperfusion-induced myocardial apoptosis in immature hearts. Ann. Thorac. Surg. 2002, 74, 830–836; discussion 836–837. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Matsui, T.; Novikov, M.; Park, J.; Hemmings, B.; Rosenzweig, A. Serum and glucocorticoid-responsive kinase-1 regulates cardiomyocyte survival and hypertrophic response. Circulation 2005, 111, 1652–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, R.; Oakley, R.H.; Cruz-Topete, D.; Cidlowski, J.A. Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis. Endocrinology 2012, 153, 5346–5360. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Topete, D.; He, B.; Xu, X.; Cidlowski, J.A. Krüppel-like Factor 13 Is a Major Mediator of Glucocorticoid Receptor Signaling in Cardiomyocytes and Protects These Cells from DNA Damage and Death. J. Biol. Chem. 2016, 291, 19374–19386. [Google Scholar] [CrossRef]
- Brereton, P.S.; van Driel, R.R.; Suhaimi, F.B.; Koyama, K.; Dilley, R.; Krozowski, Z. Light and electron microscopy localization of the 11beta-hydroxysteroid dehydrogenase type I enzyme in the rat. Endocrinology 2001, 142, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, E.P.; Romero, D.G.; de Rodriguez, A.F.; Warden, M.P.; Krozowski, Z.; Gomez-Sanchez, C.E. Hexose-6-phosphate dehydrogenase and 11beta-hydroxysteroid dehydrogenase-1 tissue distribution in the rat. Endocrinology 2008, 149, 525–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, W.; Hofland, J.; Jansen, P.M.; Garrelds, I.M.; de Vries, R.; van den Bogaerdt, A.J.; Feelders, R.A.; de Jong, F.H.; Danser, A.H. Steroidogenesis vs. steroid uptake in the heart: Do corticosteroids mediate effects via cardiac mineralocorticoid receptors? J. Hypertens. 2010, 28, 1044–1053. [Google Scholar] [CrossRef]
- Lefer, A.M. Influence of corticosteroids on mechanical performance of isolated rat papillary muscles. Am. J. Physiol. 1968, 214, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.R.; Yau, J.L.; Brett, L.P.; Seckl, J.R.; Monder, C.; Williams, B.C.; Edwards, C.R. 11 beta-hydroxysteroid dehydrogenase in vascular smooth muscle and heart: Implications for cardiovascular responses to glucocorticoids. Endocrinology 1991, 129, 3305–3312. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Andrew, R.; Cruden, N.L.; Kenyon, C.J.; Hughes, K.A.; Newby, D.E.; Hadoke, P.W.; Walker, B.R. Displacement of cortisol from human heart by acute administration of a mineralocorticoid receptor antagonist. J. Clin. Endocrinol. Metab. 2014, 99, 915–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McSweeney, S.J.; Hadoke, P.W.; Kozak, A.M.; Small, G.R.; Khaled, H.; Walker, B.R.; Gray, G.A. Improved heart function follows enhanced inflammatory cell recruitment and angiogenesis in 11betaHSD1-deficient mice post-MI. Cardiovasc. Res. 2010, 88, 159–167. [Google Scholar] [CrossRef] [Green Version]
- White, C.I.; Jansen, M.A.; McGregor, K.; Mylonas, K.J.; Richardson, R.V.; Thomson, A.; Moran, C.M.; Seckl, J.R.; Walker, B.R.; Chapman, K.E.; et al. Cardiomyocyte and Vascular Smooth Muscle-Independent 11β-Hydroxysteroid Dehydrogenase 1 Amplifies Infarct Expansion, Hypertrophy, and the Development of Heart Failure After Myocardial Infarction in Male Mice. Endocrinology 2016, 157, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Rahman, T.J.; Mayosi, B.M.; Hall, D.; Avery, P.J.; Stewart, P.M.; Connell, J.M.; Watkins, H.; Keavney, B. Common variation at the 11-β hydroxysteroid dehydrogenase type 1 gene is associated with left ventricular mass. Circ. Cardiovasc. Genet. 2011, 4, 156–162. [Google Scholar] [CrossRef]
- McGoldrick, E.; Stewart, F.; Parker, R.; Dalziel, S.R. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst. Rev. 2020, 12, CD004454. [Google Scholar] [CrossRef]
- Agnew, E.J.; Ivy, J.R.; Stock, S.J.; Chapman, K.E. Glucocorticoids, antenatal corticosteroid therapy and fetal heart maturation. J. Mol. Endocrinol. 2018, 61, R61–R73. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.J.; Martyn, C.N.; Osmond, C.; Hales, C.N.; Fall, C.H. Growth in utero and serum cholesterol concentrations in adult life. BMJ 1993, 307, 1524–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hales, C.N.; Barker, D.J.; Clark, P.M.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 1991, 303, 1019–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, C.M.; Shiell, A.W. Is blood pressure inversely related to birth weight? The strength of evidence from a systematic review of the literature. J. Hypertens. 1996, 14, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Rich-Edwards, J.W.; Stampfer, M.J.; Manson, J.E.; Rosner, B.; Hankinson, S.E.; Colditz, G.A.; Willett, W.C.; Hennekens, C.H. Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ 1997, 315, 396–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillen, I.C.; Robinson, J.S. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol. Rev. 2005, 85, 571–633. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.G. Foetal experience: Lifelong consequences. J. Neuroendocrinol. 2007, 19, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Lister, K.; Autelitano, D.J.; Jenkins, A.; Hannan, R.D.; Sheppard, K.E. Cross talk between corticosteroids and alpha-adrenergic signaling augments cardiomyocyte hypertrophy: A possible role for SGK1. Cardiovasc. Res. 2006, 70, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002, 105, 2543–2548. [Google Scholar] [CrossRef] [Green Version]
- Baartscheer, A.; Schumacher, C.A.; Belterman, C.N.; Coronel, R.; Fiolet, J.W. [Na+] i and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc. Res. 2003, 57, 986–995. [Google Scholar] [CrossRef]
- Pogwizd, S.M.; Sipido, K.R.; Verdonck, F.; Bers, D.M. Intracellular Na in animal models of hypertrophy and heart failure: Contractile function and arrhythmogenesis. Cardiovasc. Res. 2003, 57, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Pieske, B.; Houser, S.R. [Na+] i handling in the failing human heart. Cardiovasc. Res. 2003, 57, 874–886. [Google Scholar] [CrossRef]
- Murphy, E.; Eisner, D.A. Regulation of intracellular and mitochondrial sodium in health and disease. Circ. Res. 2009, 104, 292–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, D.; Hongo, K.; Ito, K.; Yoshino, T.; Kayama, Y.; Kawai, M.; Date, T.; Yoshimura, M. Corticosteroids increase intracellular free sodium ion concentration via glucocorticoid receptor pathway in cultured neonatal rat cardiomyocytes. Int. J. Cardiol. Heart Vessel. 2014, 3, 49–356. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, M.; Iwasaki, Y.; Nakayama, S.; Okazaki, M.; Taguchi, T.; Tsuda, M.; Makino, S.; Fujimoto, S.; Terada, Y. Tissue-specific regulation of 11β hydroxysteroid dehydrogenase type-1 mRNA expressions in Cushing’s syndrome mouse model. Steroids 2022, 183, 109021. [Google Scholar] [CrossRef] [PubMed]
- Stenzel-Poore, M.P.; Cameron, V.A.; Vaughan, J.; Sawchenko, P.E.; Vale, W. Development of Cushing’s syndrome in corticotropin-releasing factor transgenic mice. Endocrinology 1992, 130, 3378–3386. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Nishiyama, M.; Iwasaki, Y.; Shinahara, M.; Okada, Y.; Tsuda, M.; Okazaki, M.; Tsugita, M.; Taguchi, T.; Makino, S.; et al. Corticotropin-releasing hormone (CRH) transgenic mice display hyperphagia with increased Agouti-related protein mRNA in the hypothalamic arcuate nucleus. Endocr. J. 2011, 58, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Liu, J.; Sheng, Y.; Lv, Y.; Yu, J.; Qi, H.; Di, W.; Lv, S.; Zhou, S.; Ding, G. 11β-hydroxysteroid dehydrogenase type 1 inhibitor attenuates high-fat diet induced cardiomyopathy. J. Mol. Cell. Cardiol. 2018, 125, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Lavall, D.; Schuster, P.; Jacobs, N.; Kazakov, A.; Böhm, M.; Laufs, U. Rac1 GTPase regulates 11β hydroxysteroid dehydrogenase type 2 and fibrotic remodeling. J. Biol. Chem. 2017, 292, 7542–7553. [Google Scholar] [CrossRef] [Green Version]
- Sugihara, N.; Shimizu, M.; Kita, Y.; Shimizu, K.; Ino, H.; Miyamori, I.; Nakabayashi, H.; Takeda, R. Cardiac characteristics and postoperative courses in Cushing’s syndrome. Am. J. Cardiol. 1992, 69, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Muiesan, M.L.; Lupia, M.; Salvetti, M.; Grigoletto, C.; Sonino, N.; Boscaro, M.; Rosei, E.A.; Mantero, F.; Fallo, F. Left ventricular structural and functional characteristics in Cushing’s syndrome. J. Am. Coll. Cardiol. 2003, 41, 2275–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toja, P.M.; Branzi, G.; Ciambellotti, F.; Radaelli, P.; Martin, M.D.; Lonati, L.M.; Scacchi, M.; Parati, G.; Cavagnini, F.; Giraldi, F.P. Clinical relevance of cardiac structure and function abnormalities in patients with Cushing’s syndrome before and after cure. Clin. Endocrinol. 2012, 76, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Tanabe, A.; Tsuiki, M.; Naruse, M.; Takano, K. Hypokalemia, diabetes mellitus, and hypercortisolemia are the major contributing factors to cardiac dysfunction in adrenal Cushing’s syndrome. Endocr. J. 2009, 56, 1009–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamenický, P.; Redheuil, A.; Roux, C.; Salenave, S.; Kachenoura, N.; Raissouni, Z.; Macron, L.; Guignat, L.; Jublanc, C.; Azarine, A.; et al. Cardiac structure and function in Cushing’s syndrome: A cardiac magnetic resonance imaging study. J. Clin. Endocrinol. Metab. 2014, 99, E2144–E2153. [Google Scholar] [CrossRef] [Green Version]
- Avenatti, E.; Rebellato, A.; Iannaccone, A.; Battocchio, M.; Dassie, F.; Veglio, F.; Milan, A.; Fallo, F. Left ventricular geometry and 24-h blood pressure profile in Cushing’s syndrome. Endocrine 2017, 55, 547–554. [Google Scholar] [CrossRef]
- Iacobellis, G.; Petramala, L.; Barbaro, G.; Kargi, A.Y.; Serra, V.; Zinnamosca, L.; Colangelo, L.; Marinelli, C.; Ciardi, A.; De Toma, G.; et al. Epicardial fat thickness and left ventricular mass in subjects with adrenal incidentaloma. Endocrine 2013, 44, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Sbardella, E.; Minnetti, M.; D’Aluisio, D.; Rizza, L.; Di Giorgio, M.R.; Vinci, F.; Pofi, R.; Giannetta, E.; Venneri, M.A.; Vestri, A.; et al. Cardiovascular features of possible autonomous cortisol secretion in patients with adrenal incidentalomas. Eur. J. Endocrinol. 2018, 178, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdecchia, P.; Angeli, F.; Achilli, P.; Castellani, C.; Broccatelli, A.; Gattobigio, R.; Cavallini, C. Echocardiographic left ventricular hypertrophy in hypertension: Marker for future events or mediator of events? Curr. Opin. Cardiol. 2007, 22, 329–334. [Google Scholar] [CrossRef]
- Wicker, P.; Roudaut, R.; Haissaguere, M.; Villega-Arino, P.; Clementy, J.; Dallocchio, M. Prevalence and significance of asymmetric septal hypertrophy in hypertension: An echocardiographic and clinical study. Eur. Heart J. 1983, 4, 1–5. [Google Scholar] [CrossRef]
- Yokota, Y.; Teng, S.S.; Emoto, R.; Miki, T.; Takarada, A.; Seo, T.; Sano, H.; Fukuzaki, H. Mechanism of development of asymmetric septal hypertrophy in patients with essential systemic hypertension. Jpn. Circ. J. 1989, 53, 1173–1184. [Google Scholar] [CrossRef]
- Vensel, L.A.; Devereux, R.B.; Pickering, T.G.; Herrold, E.M.; Borer, J.S.; Laragh, J.H. Cardiac structure and function in renovascular hypertension produced by unilateral and bilateral renal artery stenosis. Am. J. Cardiol. 1986, 58, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Abe, H.; Nagata, S.; Saitoh, F.; Iwata, S.; Ashizawa, A.; Kuramochi, M.; Omae, T. Left ventricular structural characteristics in unilateral renovascular hypertension and primary aldosteronism. Am. J. Cardiol. 1988, 62, 1224–1227. [Google Scholar] [CrossRef] [PubMed]
- Shub, C.; Cueto-Garcia, L.; Sheps, S.G.; Ilstrup, D.M.; Tajik, A.J. Echocardiographic findings in pheochromocytoma. Am. J. Cardiol. 1986, 57, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Heng, M.K.; Janz, R.F.; Jobin, J. Estimation of regional stress in the left ventricular septum and free wall: An echocardiographic study suggesting a mechanism for asymmetric septal hypertrophy. Am. Heart J. 1985, 110, 84–90. [Google Scholar] [CrossRef]
- Barbot, M.; Ceccato, F.; Scaroni, C. The Pathophysiology and Treatment of Hypertension in Patients with Cushing’s Syndrome. Front. Endocrinol. 2019, 10, 321. [Google Scholar] [CrossRef]
- Ganau, A.; Devereux, R.B.; Roman, M.J.; de Simone, G.; Pickering, T.G.; Saba, P.S.; Vargiu, P.; Simongini, I.; Laragh, J.H. Patterns of left ventricular hypertrophy and geometric remodeling in essential hypertension. J. Am. Coll. Cardiol. 1992, 19, 1550–1558. [Google Scholar] [CrossRef] [Green Version]
- Koren, M.J.; Devereux, R.B.; Casale, P.N.; Savage, D.D.; Laragh, J.H. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann. Intern. Med. 1991, 114, 345–352. [Google Scholar] [CrossRef]
- Norton, G.R.; Woodiwiss, A.J.; Gaasch, W.H.; Mela, T.; Chung, E.S.; Aurigemma, G.P.; Meyer, T.E. Heart failure in pressure overload hypertrophy: The relative roles of ventricular remodeling and myocardial dysfunction. J. Am. Coll. Cardiol. 2002, 39, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Suarez, M.G.; Stack, M.; Hinojosa-Amaya, J.M.; Mitchell, M.D.; Varlamov, E.V.; Yedinak, C.G.; Cetas, J.S.; Sheppard, B.; Fleseriu, M. Hypercoagulability in Cushing Syndrome, Prevalence of Thrombotic Events: A Large, Single-Center, Retrospective Study. J. Endocr. Soc. 2019, 4, bvz033. [Google Scholar] [CrossRef] [Green Version]
- Fallo, F.; Budano, S.; Sonino, N.; Muiesan, M.L.; Agabiti-Rosei, E.; Boscaro, M. Left ventricular structural characteristics in Cushing’s syndrome. J. Hum. Hypertens. 1994, 8, 509–513. [Google Scholar] [PubMed]
- Grothues, F.; Smith, G.C.; Moon, J.C.; Bellenger, N.G.; Collins, P.; Klein, H.U.; Pennell, D.J. Comparison of interstudy reproducibility of cardiovascular magnetic resonance with two-dimensional echocardiography in normal subjects and in patients with heart failure or left ventricular hypertrophy. Am. J. Cardiol. 2002, 90, 29–34. [Google Scholar] [CrossRef]
- Codella, N.C.; Lee, H.Y.; Fieno, D.S.; Chen, D.W.; Hurtado-Rua, S.; Kochar, M.; Finn, J.P.; Judd, R.; Goyal, P.; Schenendorf, J.; et al. Improved left ventricular mass quantification with partial voxel interpolation: In vivo and necropsy validation of a novel cardiac MRI segmentation algorithm. Circ. Cardiovasc. Imaging 2012, 5, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.C.; McKenna, W.J.; McCrohon, J.A.; Elliott, P.M.; Smith, G.C.; Pennell, D.J. Toward clinical risk assessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance. J. Am. Coll. Cardiol. 2003, 41, 1561–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirpiris, M.; Sudhir, K.; Yeung, S.; Jennings, G.; Whitworth, J.A. Pressor responsiveness in corticosteroid-induced hypertension in humans. Hypertension 1992, 19, 567–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhir, K.; Jennings, G.L.; Esler, M.D.; Korner, P.I.; Blombery, P.A.; Lambert, G.W.; Scoggins, B.; Whitworth, J.A. Hydrocortisone-induced hypertension in humans: Pressor responsiveness and sympathetic function. Hypertension 1989, 13, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Saruta, T.; Suzuki, H.; Handa, M.; Igarashi, Y.; Kondo, K.; Senba, S. Multiple factors contribute to the pathogenesis of hypertension in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1986, 62, 275–279. [Google Scholar] [CrossRef]
- Giraud, G.D.; Louey, S.; Jonker, S.; Schultz, J.; Thornburg, K.L. Cortisol stimulates cell cycle activity in the cardiomyocyte of the sheep fetus. Endocrinology 2006, 147, 3643–3649. [Google Scholar] [CrossRef] [Green Version]
- Lumbers, E.R.; Boyce, A.C.; Joulianos, G.; Kumarasamy, V.; Barner, E.; Segar, J.L.; Burrell, J.H. Effects of cortisol on cardiac myocytes and on expression of cardiac genes in fetal sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R567–R574. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G.; Corradi, D.; Sharma, A.M. Epicardial adipose tissue: Anatomic, biomolecular and clinical relationships with the heart. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 536–543. [Google Scholar] [CrossRef]
- Iacobellis, G.; Bianco, A.C. Epicardial adipose tissue: Emerging physiological, pathophysiological and clinical features. Trends Endocrinol. Metab. 2011, 22, 450–457. [Google Scholar] [CrossRef]
- Iacobellis, G.; Ribaudo, M.C.; Zappaterreno, A.; Iannucci, C.V.; Leonetti, F. Relation between epicardial adipose tissue and left ventricular mass. Am. J. Cardiol. 2004, 94, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Ribaudo, M.C.; Assael, F.; Vecci, E.; Tiberti, C.; Zappaterreno, A.; Mario, U.D.; Leonetti, F. Echocardiographic epicardial adipose tissue is related to anthropometric and clinical parameters of metabolic syndrome: A new indicator of cardiovascular risk. J. Clin. Endocrinol. Metab. 2003, 88, 5163–5168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobellis, G.; Willens, H.J. Echocardiographic epicardial fat: A review of research and clinical applications. J. Am. Soc. Echocardiogr. 2009, 22, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Cuspidi, C.; Meani, S.; Negri, F.; Giudici, V.; Valerio, C.; Sala, C.; Zanchetti, A.; Mancia, G. Indexation of left ventricular mass to body surface area and height to allometric power of 2.7, is the difference limited to obese hypertensives? J. Hum. Hypertens. 2009, 23, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Evran, M.; Akkuş, G.; Bozdoğan, İ.B.; Gök, M.; Deniz, A.; Sert, M.; Tetiker, T. Carotid Intima-Media Thickness as the Cardiometabolic Risk Indicator in Patients with Nonfunctional Adrenal Mass and Metabolic Syndrome Screening. Med. Sci. Monit. 2016, 22, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Newell-Price, J.; Trainer, P.; Besser, M.; Grossman, A. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr. Rev. 1998, 19, 647–672. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, L.; Akyüz, A.R.; Gurbak, I.; Erkan, H.; Dursun, I.; Celik, S. Presystolic A wave may predict increased arterial stiffness in asymptomatic individuals. Blood Press. Monit. 2016, 21, 144–148. [Google Scholar] [CrossRef]
- El-Aziz, T.A.A. A-wave acceleration: A new Doppler echocardiographic index for evaluation of left ventricular diastolic dysfunction in elderly patients. Angiology 2008, 59, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, A.; Kadoya, M.; Kakutani-Hatayama, M.; Kosaka-Hamamoto, K.; Miyoshi, A.; Shoji, T.; Goda, A.; Asakura, M.; Koyama, H. Subclinical decrease in cardiac autonomic and diastolic function in patients with metabolic disorders: HSCAA study. Metabol. Open 2020, 5, 100025. [Google Scholar] [CrossRef]
- Toniato, A.; Merante-Boschin, I.; Opocher, G.; Pelizzo, M.R.; Schiavi, F.; Ballotta, E. Surgical versus conservative management for subclinical Cushing syndrome in adrenal incidentalomas: A prospective randomized study. Ann. Surg. 2009, 249, 388–391. [Google Scholar] [CrossRef]
- Tsuiki, M.; Tanabe, A.; Takagi, S.; Naruse, M.; Takano, K. Cardiovascular risks and their long-term clinical outcome in patients with subclinical Cushing’s syndrome. Endocr. J. 2008, 55, 737–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiodini, I.; Morelli, V.; Salcuni, A.S.; Eller-Vainicher, C.; Torlontano, M.; Coletti, F.; Iorio, L.; Cuttitta, A.; Ambrosio, A.; Vicentini, L.; et al. Beneficial metabolic effects of prompt surgical treatment in patients with an adrenal incidentaloma causing biochemical hypercortisolism. J. Clin. Endocrinol. Metab. 2010, 95, 2736–2745. [Google Scholar] [CrossRef] [PubMed]
- Iacobone, M.; Citton, M.; Viel, G.; Boetto, R.; Bonadio, I.; Mondi, I.; Tropea, S.; Nitti, D.; Favia, G. Adrenalectomy may improve cardiovascular and metabolic impairment and ameliorate quality of life in patients with adrenal incidentalomas and subclinical Cushing’s syndrome. Surgery 2012, 152, 991–997. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Details and Numbers of Subjects | Age, Years | Sex, F/M | BMI, kg/m2 | Duration to Diagnosis of CS (Months) | Complications (No.) | Assessment | Primary Findings | Reference |
|---|---|---|---|---|---|---|---|---|
| Overt CS (12) | 27–57 | 8/4 | - | - | Diabetes mellitus (7) | Echocardiography |
| [80] |
| CD (2) | Impaired glucose | Electrocardiogram | ||||||
| Adrenal adenoma (9) | tolerance (4) | |||||||
| Adrenal carcinoma (1) | ||||||||
| CS (42) | 39 ± 12 | 30/12 | 28 ± 4.8 | 10.8 ± 14 | Hypertension (32) | Echocardiography |
| [81] |
| CD (36) | Diabetes mellitus (5) | |||||||
| Adrenal adenoma (6) | ||||||||
| Adrenal carcinoma (1) | ||||||||
| Ectopic ACTH production (1) | ||||||||
| CS (71) | 40.1 ± 1.46 | 61/10 | 29.4 ± 1.1 | 39.4 ± 5.49 | Hypertensive | Echocardiography |
| [82] |
| CD (65) | condition (30) | |||||||
| Adrenal adenoma (2) | ||||||||
| Ectopic ACTH production (2) | Diabetes mellitus (8) | |||||||
| ACTH-independent adrenal hyperplasia (2) | ||||||||
| Overt CS (50) | 46.6 ± 14.3 | 44/6 | – | 70.8 ± 69.6 | Hypertensive | Echocardiography |
| [83] |
| Adrenal CS (50) | condition (41) | |||||||
| Diabetes mellitus (25) | ||||||||
| Dyslipidemia (38) | ||||||||
| CS (18) | 35.0 | 16/2 | 27.5 | – | Hypertension (9) | Cardiac magnetic |
| [84] |
| CD (15) | [26.2; 47.2] | [22.2; 33.0] | Diabetes mellitus (5) | resonance imaging | ||||
| Adrenal adenoma (2) | Impaired glucose tolerance (4) | |||||||
| Ectopic ACTH production (1) | Dyslipidemia (6) | |||||||
| CS (25) | 44.6 ± 11.3 | 21/4 | 27.1 | – | Hypertensive | Echocardiography |
| [85] |
| CD (21) | (25.4; 32.4) | condition (16) | ||||||
| Adrenal adenoma (4) | Diabetes mellitus (6) | |||||||
| Incidentaloma (46) | 69.3 ± 12.5 | 24/22 | 30.3 ± 7.5 | – | Mean blood pressure (mmHg); 134 ± 9/82 ± 4 | Echocardiography |
| [86] |
| Mild CS (6) | ||||||||
| NFA (40) | Mean fasting glucose (mg/dL): 128 ± 25.8 | |||||||
| Incidentaloma (71) | 67 | 24/47 | 26.3 | – | Hypertension (22) | Echocardiography, |
| [87] |
| pACS (34) | [59; 72] | [23.5; 29.4] | Diabetes mellitus (7) | brachial oscillometric | ||||
| NFA (37) | Dyslipidemia (24) | blood pressure waves |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanzaki, A.; Kadoya, M.; Katayama, S.; Koyama, H. Cardiac Hypertrophy and Related Dysfunctions in Cushing Syndrome Patients—Literature Review. J. Clin. Med. 2022, 11, 7035. https://doi.org/10.3390/jcm11237035
Kanzaki A, Kadoya M, Katayama S, Koyama H. Cardiac Hypertrophy and Related Dysfunctions in Cushing Syndrome Patients—Literature Review. Journal of Clinical Medicine. 2022; 11(23):7035. https://doi.org/10.3390/jcm11237035
Chicago/Turabian StyleKanzaki, Akinori, Manabu Kadoya, Satoru Katayama, and Hidenori Koyama. 2022. "Cardiac Hypertrophy and Related Dysfunctions in Cushing Syndrome Patients—Literature Review" Journal of Clinical Medicine 11, no. 23: 7035. https://doi.org/10.3390/jcm11237035
APA StyleKanzaki, A., Kadoya, M., Katayama, S., & Koyama, H. (2022). Cardiac Hypertrophy and Related Dysfunctions in Cushing Syndrome Patients—Literature Review. Journal of Clinical Medicine, 11(23), 7035. https://doi.org/10.3390/jcm11237035