
Expression of Iron Metabolism Proteins in Patients with Chronic Heart Failure

 , , ,
, , ,
Abstract
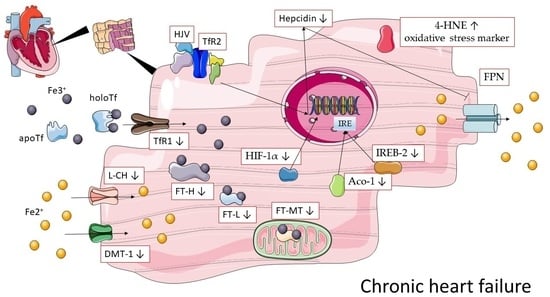
:
1. Introduction
2. Materials and Methods
2.1. Study Population and Protocol
2.2. Study Protocol
Laboratory Test
2.3. Preparation of Cardiac Tissue
2.4. Assessment Iron-Related Proteins
2.5. Statistical Analysis
3. Results
3.1. Clinical Characteristic of the Study Groups (Heart Failure and Non-Failing Subjects)
3.2. Expression of Iron-Handling Proteins, Their Regulatory Factors and an Oxidative Stress Marker in the Non-Failing Compared to the Failing Human Myocardium
Expression of Iron-Handling Proteins in the Non-Failing Compared to the Failing Human Myocardium (Non-Iron Deficient and Iron Deficient)
3.3. Correlations between Iron-Handling Proteins, Regulatory Factors and Oxidative Stress Markers in the Non-Failing Human Myocardium
3.3.1. Relations among Iron Regulatory Proteins
3.3.2. Relations with Regulatory Factors
3.3.3. Relations with Oxidative Stress Marker
3.4. Correlations between Iron-Handling Proteins, Regulatory Factors and an Oxidative Stress Marker in The Failing Human Myocardium
3.4.1. Relations among Iron Regulatory Proteins
3.4.2. Relations with Regulatory Factors
3.4.3. Relations with Oxidative Stress Marker
3.5. Correlations between Iron-Handling Proteins and Clinical Data
4. Discussion
5. Conclusions
6. Limitation of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fitzsimons, S.; Doughty, R.N. Iron deficiency in patients with heart failure. Eur. Heart J. Cardiovasc. Pharmacother. 2015, 1, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Li, J.; Zhang, Y.; Chang, Y.-Z. Cellular iron metabolism and regulation. Adv. Exp. Med. Biol. 2019, 1173, 21–32. [Google Scholar] [CrossRef]
- Mentz, R.J.; Ambrosy, A.P.; Ezekowitz, J.A.; Lewis, G.D.; Butler, J.; Wong, Y.W.; De Pasquale, C.G.; Troughton, R.W.; O’Meara, E.; Rockhold, F.W.; et al. Randomized placebo-controlled trial of ferric carboxymaltose in heart failure with iron deficiency: Rationale and design. Circ. Heart Fail. 2021, 14, e008100. [Google Scholar] [CrossRef]
- Yamani, N.; Ahmed, A.; Gosain, P.; Fatima, K.; Shaikh, A.T.; Qamar, H.; Shahid, I.; Arshad, M.S.; Almas, T.; Figueredo, V. Effect of iron supplementation in patients with heart failure and iron deficiency: A systematic review and meta-analysis. IJC Heart Vasc. 2021, 36, 100871. [Google Scholar] [CrossRef]
- Ponikowski, P.; Kirwan, B.-A.; Anker, S.D.; McDonagh, T.; Dorobantu, M.; Drozdz, J.; Fabien, V.; Filippatos, G.; Göhring, U.M.; Keren, A.; et al. Ferric carboxymaltose for iron deficiency at discharge after acute heart failure: A multicentre, double-blind, randomised, controlled trial. Lancet 2020, 396, 1895–1904. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Das, S.K.; Wang, W.; Zhabyeyev, P.; Basu, R.; McLean, B.; Fan, D.; Parajuli, N.; DesAulniers, J.; Patel, V.B.; Hajjar, R.J.; et al. Iron-overload injury and cardiomyopathy in acquired and genetic models is attenuated by resveratrol therapy. Sci. Rep. 2015, 5, 18132. [Google Scholar] [CrossRef] [Green Version]
- Hodges, Y.K.; Reese, S.M.; Pahl, P.M.B.; Horwitz, L.D. Paradoxical effects of iron chelation on growth of vascular endothelial cells. J. Cardiovasc. Pharmacol. 2005, 45, 539–544. [Google Scholar] [CrossRef]
- Wu, C.; Qiu, H.; Xu, L.; Ye, J.; Yang, Z.; Qian, X.; Meng, X.; Cui, Y.; Song, L.; Gao, R. inhibitory effect of iron on in vitro proliferation of smooth muscle cells. Chin. Med. J. 2013, 126, 3728–3731. [Google Scholar]
- Yoo, J.-H.; Maeng, H.-Y.; Sun, Y.-K.; Kim, Y.-A.; Park, D.-W.; Park, T.S.; Lee, S.T.; Choi, J.-R. Oxidative status in iron-deficiency anemia. J. Clin. Lab. Anal. 2009, 23, 319–323. [Google Scholar] [CrossRef]
- Hassan, T.H.; Badr, M.A.; Karam, N.A.; Zkaria, M.; El Saadany, H.F.; Abdel Rahman, D.M.; Shahbah, D.A.; Al Morshedy, S.M.; Fathy, M.; Esh, A.M.H.; et al. Impact of iron deficiency anemia on the function of the immune system in children. Medicine 2016, 95, e5395. [Google Scholar] [CrossRef]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar]
- Brissot, P.; Bardou-Jacquet, E.; Jouanolle, A.-M.; Loréal, O. Iron Disorders of genetic origin: A changing world. Trends Mol. Med. 2011, 17, 707–713. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Musallam, K.M.; Taher, A.T. Iron deficiency anaemia revisited. J. Intern. Med. 2020, 287, 153–170. [Google Scholar] [CrossRef]
- Anand, I.S.; Gupta, P. Anemia and iron deficiency in heart failure: Current concepts and emerging therapies. Circulation 2018, 138, 80–98. [Google Scholar] [CrossRef]
- Klip, I.T.; Comin-Colet, J.; Voors, A.A.; Ponikowski, P.; Enjuanes, C.; Banasiak, W.; Lok, D.J.; Rosentryt, P.; Torrens, A.; Polonski, L.; et al. Iron deficiency in chronic heart failure: An international pooled analysis. Am. Heart J. 2013, 165, 575–582.e3. [Google Scholar] [CrossRef]
- Rizzo, C.; Carbonara, R.; Ruggieri, R.; Passantino, A.; Scrutinio, D. Iron deficiency: A new target for patients with heart failure. Front. Cardiovasc. Med. 2021, 8, 709872. [Google Scholar] [CrossRef]
- Lewis, G.D.; Malhotra, R.; Hernandez, A.F.; McNulty, S.E.; Smith, A.; Felker, G.M.; Tang, W.H.W.; LaRue, S.J.; Redfield, M.M.; Semigran, M.J.; et al. Effect of oral iron repletion on exercise capacity in patients with heart failure with reduced ejection fraction and iron deficiency: The IRONOUT HF randomized clinical trial. JAMA 2017, 317, 1958–1966. [Google Scholar] [CrossRef]
- Crichton, R.R.; Wilmet, S.; Legssyer, R.; Ward, R.J. Molecular and cellular mechanisms of iron homeostasis and toxicity in mammalian cells. J. Inorg. Biochem. 2002, 91, 9–18. [Google Scholar] [CrossRef]
- Zhang, H.; Zhabyeyev, P.; Wang, S.; Oudit, G.Y. Role of iron metabolism in heart failure: From iron deficiency to iron overload. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 1925–1937. [Google Scholar] [CrossRef]
- Otto-Duessel, M.; Brewer, C.; Wood, J.C. Interdependence of cardiac iron and calcium in a murine model of iron overload. Transl. Res. 2011, 157, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, J.L.; Loggetto, S.R.; Veríssimo, M.P.A.; Fertrin, K.Y.; Baldanzi, G.R.; Fioravante, L.A.B.; Tan, D.M.; Higa, T.; Mashima, D.A.; Piga, A.; et al. A randomized trial of amlodipine in addition to standard chelation therapy in patients with thalassemia major. Blood 2016, 128, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Wijarnpreecha, K.; Kumfu, S.; Chattipakorn, S.C.; Chattipakorn, N. Cardiomyopathy associated with iron overload: How does iron enter myocytes and what are the implications for pharmacological therapy? Hemoglobin 2015, 39, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Crowe, S.; Bartfay, W.J. Amlodipine decreases iron uptake and oxygen free radical production in the heart of chronically iron overloaded mice. Biol. Res. Nurs. 2002, 3, 189–197. [Google Scholar] [CrossRef]
- Maeder, M.T.; Khammy, O.; dos Remedios, C.; Kaye, D.M. Myocardial and systemic iron depletion in heart failure implications for anemia accompanying heart failure. J. Am. Coll. Cardiol. 2011, 58, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Leszek, P.; Sochanowicz, B.; Szperl, M.; Kolsut, P.; Brzóska, K.; Piotrowski, W.; Rywik, T.M.; Danko, B.; Polkowska-Motrenko, H.; Różański, J.M.; et al. Myocardial iron homeostasis in advanced chronic heart failure patients. Int. J. Cardiol. 2012, 159, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Leszek, P.; Sochanowicz, B.; Brzóska, K.; Danko, B.; Kraj, L.; Kuśmierczyk, M.; Piotrowski, W.; Sobieszczańska-Małek, M.; Rywik, T.M.; Polkowska-Motrenko, H.; et al. Does myocardial iron load determine the severity of heart insufficiency? Int. J. Cardiol. 2015, 182, 191–193. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, Y.; Zheng, B.; Han, X.; Ma, D.; Ma, Z.; Guan, S.; Gao, Y.; Li, Z.; Chu, L. Salvia Miltiorrhiza (SM) injection ameliorates iron overload-associated cardiac dysfunction by regulating the expression of DMT1, TfR1, and FP1 in rats. Evid.-Based Complementary Altern. Med. 2021, 2021, e6864723. [Google Scholar] [CrossRef]
- Rhee, J.; Yi, H.; Lam, C.K.; Belbachir, N.; Tian, L.; Thomas, D.; Lau, E.; Paik, D.T.; Lam, M.P.; Sayed, N.; et al. Abstract 15973: Ebselen, a divalent metal transporter 1 inhibitor, effectively blocks iron entry to human Ipsc-derived cardiomyocytes and prevents iron-overload cardiotoxicity. Circulation 2019, 140, A15973. [Google Scholar] [CrossRef]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. Cell Rep. 2015, 13, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Tajes, M.; Díez-López, C.; Enjuanes, C.; Moliner, P.; Ferreiro, J.L.; Garay, A.; Jiménez-Marrero, S.; Yun, S.; Sosa, S.G.; Alcoberro, L.; et al. Neurohormonal activation induces intracellular iron deficiency and mitochondrial dysfunction in cardiac cells. Cell Biosci. 2021, 11, 89. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Wolna, M.; Carr, C.A.; Miller, J.J.J.; Christian, H.C.; Ball, V.; Santos, A.; Diaz, R.; Biggs, D.; Stillion, R.; et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. USA 2015, 112, 3164–3169. [Google Scholar] [CrossRef] [Green Version]
- Worthen, C.; Enns, C. The role of hepatic transferrin receptor 2 in the regulation of iron homeostasis in the body. Front. Pharmacol. 2014, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Bertoli, S.; Paubelle, E.; Bérard, E.; Saland, E.; Thomas, X.; Tavitian, S.; Larcher, M.-V.; Vergez, F.; Delabesse, E.; Sarry, A.; et al. Ferritin heavy/light chain (FTH1/FTL) expression, serum ferritin levels, and their functional as well as prognostic roles in acute myeloid leukemia. Eur. J. Haematol. 2019, 102, 131–142. [Google Scholar] [CrossRef]
- Gao, G.; Chang, Y.-Z. Mitochondrial ferritin in the regulation of brain iron homeostasis and neurodegenerative diseases. Front. Pharmacol. 2014, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Punnonen, K.; Irjala, K.; Rajamäki, A. Serum transferrin receptor and its ratio to serum ferritin in the diagnosis of iron deficiency. Blood 1997, 89, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Silvestre, O.M.; Gonçalves, A.; Junior, W.N.; Claggett, B.; Couper, D.; Eckfeldt, J.H.; Pankow, J.S.; Anker, S.D.; Solomon, S.D. Ferritin levels and risk of heart failure—The atherosclerosis risk in communities study. Eur. J. Heart Fail. 2017, 19, 340–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef]
- Vela, D. Balance of cardiac and systemic hepcidin and its role in heart physiology and pathology. Lab. Investig. 2018, 98, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018, 131, 899–910. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Wolna, M.; Chung, Y.J.; Christian, H.C.; Heather, L.C.; Brescia, M.; Ball, V.; Diaz, R.; Santos, A.; Biggs, D.; et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. eLife 2016, 5, e19804. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, E.A.; Malyszko, J.; Ardehali, H.; Koc-Zorawska, E.; Banasiak, W.; von Haehling, S.; Macdougall, I.C.; Weiss, G.; McMurray, J.J.V.; Anker, S.D.; et al. Iron status in patients with chronic heart failure. Eur. Heart J. 2013, 34, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Ajoolabady, A.; Demillard, L.J.; Yu, W.; Hilaire, M.L.; Zhang, Y.; Ren, J. Dysregulation of iron metabolism in cardiovascular diseases: From iron deficiency to iron overload. Biochem. Pharmacol. 2021, 190, 114661. [Google Scholar] [CrossRef] [PubMed]
- Haddad, S.; Wang, Y.; Galy, B.; Korf-Klingebiel, M.; Hirsch, V.; Baru, A.M.; Rostami, F.; Reboll, M.R.; Heineke, J.; Flögel, U.; et al. Iron-regulatory proteins secure iron availability in cardiomyocytes to prevent heart failure. Eur. Heart J. 2017, 38, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Date, T.; Mochizuki, S.; Belanger, A.J.; Yamakawa, M.; Luo, Z.; Vincent, K.A.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Expression of constitutively stable hybrid hypoxia-inducible factor-1α protects cultured rat cardiomyocytes against simulated ischemia-reperfusion injury. Am. J. Physiol. Cell Physiol. 2005, 288, C314–C320. [Google Scholar] [CrossRef] [Green Version]
- Zolk, O.; Solbach, T.F.; Eschenhagen, T.; Weidemann, A.; Fromm, M.F. Activation of negative regulators of the hypoxia-inducible factor (HIF) pathway in human end-stage heart failure. Biochem. Biophys. Res. Commun. 2008, 376, 315–320. [Google Scholar] [CrossRef]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. P53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef]
- Csala, M.; Kardon, T.; Legeza, B.; Lizák, B.; Mandl, J.; Margittai, É.; Puskás, F.; Száraz, P.; Szelényi, P.; Bánhegyi, G. On the role of 4-hydroxynonenal in health and disease. Biochim. Biophys. Acta 2015, 1852, 826–838. [Google Scholar] [CrossRef] [Green Version]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef]
- Nakamura, K.; Kusano, K.; Nakamura, Y.; Kakishita, M.; Ohta, K.; Nagase, S.; Yamamoto, M.; Miyaji, K.; Saito, H.; Morita, H.; et al. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation 2002, 105, 2867–2871. [Google Scholar] [CrossRef] [Green Version]
- Akoglu, H. User’s guide to correlation coefficients. Turk. J. Emerg. Med. 2018, 18, 91–93. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||||||||
| Unit | Failing Heart Group (n = 58) | Women (n = 13) | Men (n = 45) | p-Value | ||||
| Me | Q | Me | Q | Me | Q | |||
| Demographic Information | ||||||||
| Age | Yrs | 53.5 | 10.5 | 58 | 10 | 53 | 10.5 | 0.4728 |
| Height | Cm | 175.5 | 6.8 | 164 | 3.5 | 178 | 5 | 0 |
| Body weight | Kg | 80 | 10.5 | 61 | 11.5 | 82.5 | 9.5 | 0.0005 |
| BMI- Body-mass index | kg/m2 | 25.6 | 3.3 | 22.8 | 4.1 | 25.9 | 2.6 | 0.1454 |
| Laboratory tests | ||||||||
| NTproBNP | pg/mL | 3955.5 | 2250.5 | 4436 | 1291.5 | 3833 | 2665 | 0.4014 |
| CRP | mg/dL | 0.4 | 0.3 | 0.3 | 0.2 | 0.5 | 0.3 | 0.5101 |
| Creatinine | mg/dL | 1.2 | 0.2 | 1 | 0.1 | 1.2 | 0.2 | 0.0376 |
| Glomerular filtration rate | ml/min/1.73 m2 | 60 | 4.5 | 60 | 5 | 60 | 4.3 | 0.557 |
| Echocardiography parameters | ||||||||
| LVED | Mm | 67 | 8 | 58.5 | 10.5 | 70 | 8.5 | 0.0069 |
| LVSD | Mm | 59 | 13 | 43 | 10.5 | 65 | 12 | 0.0034 |
| LVEF | % | 20 | 7.5 | 20 | 10 | 20 | 5.5 | 0.1812 |
| RV | Mm | 42 | 6.5 | 42 | 11 | 42 | 5.5 | 0.8857 |
| IM | +/++++ | 1.5 | 0.5 | 1.5 | 0.5 | 1.5 | 0.5 | 0.8521 |
| IT | +/++++ | 1.8 | 0.8 | 2.5 | 0.5 | 1.5 | 0.5 | 0.0183 |
| RVSP | mmHg | 47 | 12.5 | 45 | 12.5 | 47 | 11 | 0.6869 |
| TAPSE | Mm | 15 | 2 | 13 | 2 | 16 | 2 | 0.0134 |
| Hemodynamic parameters | ||||||||
| PAPs | mmHg | 42 | 12.5 | 36 | 8.5 | 44 | 12 | 0.1052 |
| PAPm | mmHg | 29 | 7.5 | 26.5 | 5.5 | 29 | 7 | 0.2711 |
| PWPm | mmHg | 21 | 5.5 | 19.5 | 6 | 21 | 7.5 | 0.2624 |
| TCG | mmHg | 8.5 | 3 | 9 | 1.5 | 8 | 3 | 0.4742 |
| CO | l/min | 3.5 | 0.8 | 2.6 | 0.4 | 3.7 | 0.7 | 0.0058 |
| PVR | jWood | 2.2 | 0.9 | 2.7 | 1.4 | 2.2 | 0.7 | 0.2157 |
| SVR | jWood | 13.3 | 2.8 | 22.4 | 10.9 | 12.6 | 2.2 | 0.0969 |
| Iron-related markers | ||||||||
| HT | g/dL | 14.1 | 1.4 | 14.3 | 1.5 | 14 | 1.2 | 0.5261 |
| MCV | Fl | 91 | 3.6 | 90 | 4.8 | 91 | 3.5 | 0.7372 |
| RET % | % | 1.6 | 0.5 | 1.7 | 0.2 | 1.6 | 0.5 | 0.7642 |
| RET-Hb | Pg | 32 | 2 | 26 | 3.1 | 32 | 1.6 | 0.096 |
| Ferritin | ng/mL | 212.1 | 89.1 | 230 | 122.5 | 202.6 | 82.5 | 0.8663 |
| TSAT | % | 21 | 8.6 | 18 | 7.1 | 24.2 | 7.5 | 0.1138 |
| sTfR | mg/L | 3.2 | 1.1 | 3.3 | 1.6 | 3.2 | 1.2 | 0.4381 |
| sTfR1/logFR | - | 0.61 | 0.27 | 0.63 | 0.36 | 0.61 | 0.21 | 0.2327 |
| Erythropoietin | mIU/mL | 18.3 | 11.4 | 28.6 | 16.1 | 16.1 | 10.1 | 0.0386 |
| Aberrations: NTproBNP (N-terminal pro-brain natriuretic peptide), CRP (C-reactive protein), LVED (left ventricle diameters diastolic), LVSD (left ventricle diameters systolic), LVEF(left ventricle ejection fraction), RV (right ventricle diastolic size), IM (mitral insufficiency), IT(tricuspid insufficiency), RVSP (right ventricular systolic pressure), TAPSE (tricuspid annular plane systolic excursion), PAPs (pulmonary artery pressure systolic), PAPm (pulmonary artery pressure mean), PWPm (pulmonary capillary wedge pressure mean), TCG (transpulmonary pressure gradient), CO(cardiac output), PVR (pulmonary vascular resistance), SVR (systemic vascular resistance),HT (hematocrit), MCV (mean corpuscular volume), RET % (reticulocyte %),RET-Hb (reticulocyte hemoglobin), TSAT (transferrin saturation), sTfR (soluble transferrin receptor) sTfR1/logFR (soluble transferrin receptor/logarithm of ferritin) | ||||||||
| (B) | ||||||||
| Heart failure | Women | Men | p-Value | |||||
| Etiology | N | % | N | % | N | % | ||
| Ischemic | 18.0 | 31.0 | 2.0 | 15.4 | 16.0 | 35.6 | ||
| Cardiomyopathy | 36.0 | 62.1 | 9.0 | 69.2 | 27.0 | 60 | 0.2008 | |
| Other | 4.0 | 6.9 | 2 | 15.4 | 2 | 44.4 | ||
| NYHA: 3 | 31 | 53.4 | 6 | 46.2 | 25 | 55.5 | 0.5494 | |
| NYHA: 4 | 27 | 46.6 | 7 | 53.84 | 20 | 44.4 | ||
| Sinus rhythm | 40 | 69 | 7 | 53.84 | 33 | 73.3 | 0.1810 | |
| No Sinus rhythm | 18 | 31 | 6 | 46.2 | 12 | 26.7 | ||
| Aberrations: NYH (New York Heart Association) | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozłowska, B.; Sochanowicz, B.; Kraj, L.; Palusińska, M.; Kołsut, P.; Szymański, Ł.; Lewicki, S.; Śmigielski, W.; Kruszewski, M.; Leszek, P. Expression of Iron Metabolism Proteins in Patients with Chronic Heart Failure. J. Clin. Med. 2022, 11, 837. https://doi.org/10.3390/jcm11030837
Kozłowska B, Sochanowicz B, Kraj L, Palusińska M, Kołsut P, Szymański Ł, Lewicki S, Śmigielski W, Kruszewski M, Leszek P. Expression of Iron Metabolism Proteins in Patients with Chronic Heart Failure. Journal of Clinical Medicine. 2022; 11(3):837. https://doi.org/10.3390/jcm11030837
Chicago/Turabian StyleKozłowska, Bogna, Barbara Sochanowicz, Leszek Kraj, Małgorzata Palusińska, Piotr Kołsut, Łukasz Szymański, Sławomir Lewicki, Witold Śmigielski, Marcin Kruszewski, and Przemysław Leszek. 2022. "Expression of Iron Metabolism Proteins in Patients with Chronic Heart Failure" Journal of Clinical Medicine 11, no. 3: 837. https://doi.org/10.3390/jcm11030837
APA StyleKozłowska, B., Sochanowicz, B., Kraj, L., Palusińska, M., Kołsut, P., Szymański, Ł., Lewicki, S., Śmigielski, W., Kruszewski, M., & Leszek, P. (2022). Expression of Iron Metabolism Proteins in Patients with Chronic Heart Failure. Journal of Clinical Medicine, 11(3), 837. https://doi.org/10.3390/jcm11030837