
Epigenetic Alterations in Inborn Errors of Immunity

 ,
,  , and
, and
Abstract
:1. Introduction
2. Physiologic Roles of Epigenetics
3. Epigenetics in the Immune System
4. Epigenetic Alterations in Inborn Errors of Immunity
4.1. Inborn Errors of Humoral Immunity
4.2. Inborn Errors of Adaptive Immunity
4.3. Inborn Errors of Innate Immunity
4.4. Inborn Errors of Immunity with Immune Dysregulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ikegami, K.; Ohgane, J.; Tanaka, S.; Yagi, S.; Shiota, K. Interplay between DNA methylation, histone modification and chromatin remodeling in stem cells and during development. Int. J. Dev. Biol. 2009, 53, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, K. DNA methylation profiles of CpG islands for cellular differentiation and development in mammals. Cytogenet. Genome Res. 2004, 105, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Hawkins, R.D. Three-dimensional genome architecture and emerging technologies: Looping in disease. Genome Med. 2017, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012, 13, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic mechanisms in mammals. Cell Mol. Life Sci. 2009, 66, 596–612. [Google Scholar] [CrossRef] [Green Version]
- Cerrato, F.; Sparago, A.; Ariani, F.; Brugnoletti, F.; Calzari, L.; Coppedè, F.; De Luca, A.; Gervasini, C.; Giardina, E.; Gurrieri, F.; et al. DNA Methylation in the Diagnosis of Monogenic Diseases. Genes 2020, 11, 355. [Google Scholar] [CrossRef] [Green Version]
- Argelaguet, R.; Clark, S.J.; Mohammed, H.; Stapel, L.C.; Krueger, C.; Kapourani, C.A.; Imaz-Rosshandler, I.; Lohoff, T.; Xiang, Y.; Hanna, C.W.; et al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature 2019, 576, 487–491. [Google Scholar] [CrossRef]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Barr, C.L.; Kim, A.; Yue, F.; Lee, A.Y.; Eubanks, J.; Dempster, E.L.; Ren, B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012, 148, 816–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuso, A.; Lucarelli, M. CpG and Non-CpG Methylation in the Diet-Epigenetics-Neurodegeneration Connection. Curr. Nutr. Rep. 2019, 8, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, D.; Rao, A.K.D.M.; Rajkumar, T.; Mani, S. Non-CpG methylation-a key epigenetic modification in cancer. Brief Funct. Genom. 2021, 20, 304–311. [Google Scholar] [CrossRef]
- Joe, S.; Nam, H. Prediction model construction of mouse stem cell pluripotency using CpG and non-CpG DNA methylation markers. BMC Bioinform. 2020, 21, 175. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Velasco, G.; Francastel, C. Genetics meets DNA methylation in rare diseases. Clin. Genet. 2019, 95, 210–220. [Google Scholar] [CrossRef]
- Monk, D.; Mackay, D.J.G.; Eggermann, T.; Maher, E.R.; Riccio, A. Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019, 20, 235–248. [Google Scholar] [CrossRef]
- Pignata, L.; Palumbo, O.; Cerrato, F.; Acurzio, B.; de Álava, E.; Roma, J.; Gallego, S.; Mora, J.; Carella, M.; Riccio, A.; et al. Both Epimutations and Chromosome Aberrations Affect Multiple Imprinted Loci in Aggressive Wilms Tumors. Cancers 2020, 12, 3411. [Google Scholar] [CrossRef]
- Sadikovic, B.; Aref-Eshghi, E.; Levy, M.A.; Rodenhiser, D. DNA methylation signatures in mendelian developmental disorders as a diagnostic bridge between genotype and phenotype. Epigenomics 2019, 11, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Panni, S.; Lovering, R.C.; Porras, P.; Orchard, S. Non-coding RNA regulatory networks. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194417. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Zouali, M. DNA methylation signatures of autoimmune diseases in human B lymphocytes. Clin. Immunol. 2021, 222, 108622. [Google Scholar] [CrossRef]
- Ji, H.; Ehrlich, L.I.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Kulis, M.; Merkel, A.; Heath, S.; Queirós, A.C.; Schuyler, R.P.; Castellano, G.; Beekman, R.; Raineri, E.; Esteve, A.; Clot, G.; et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat. Genet. 2015, 47, 746–756. [Google Scholar] [CrossRef]
- Selimyan, R.; Gerstein, R.M.; Ivanova, I.; Precht, P.; Subrahmanyam, R.; Perlot, T.; Alt, F.W.; Sen, R. Localized DNA demethylation at recombination intermediates during immunoglobulin heavy chain gene assembly. PLoS Biol. 2013, 11, e1001475. [Google Scholar] [CrossRef] [Green Version]
- Sellars, M.; Huh, J.R.; Day, K.; Issuree, P.D.; Galan, C.; Gobeil, S.; Absher, D.; Green, M.R.; Littman, D.R. Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T cell lineages. Nat. Immunol. 2015, 16, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, M.; Gliwiński, M.; Trzonkowski, P.; Iwaszkiewicz-Grzes, D. Regulatory T Cells-Related Genes Are under DNA Methylation Influence. Int. J. Mol. Sci. 2021, 22, 7144. [Google Scholar] [CrossRef]
- Gamper, C.J.; Agoston, A.T.; Nelson, W.G.; Powell, J.D. Identification of DNA methyltransferase 3a as a T cell receptor-induced regulator of Th1 and Th2 differentiation. J. Immunol. 2009, 183, 2267–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Arvey, A.; Chinen, T.; van der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014, 158, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Correa, L.O.; Jordan, M.S.; Carty, S.A. DNA Methylation in T-Cell Development and Differentiation. Crit. Rev. Immunol. 2020, 40, 135–156. [Google Scholar] [CrossRef]
- Luetke-Eversloh, M.; Cicek, B.B.; Siracusa, F.; Thom, J.T.; Hamann, A.; Frischbutter, S.; Baumgrass, R.; Chang, H.D.; Thiel, A.; Dong, J.; et al. NK cells gain higher IFN-γ competence during terminal differentiation. Eur. J. Immunol. 2014, 44, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petriv, O.I.; Kuchenbauer, F.; Delaney, A.D.; Lecault, V.; White, A.; Kent, D.; Marmolejo, L.; Heuser, M.; Berg, T.; Copley, M.; et al. Comprehensive microRNA expression profiling of the hematopoietic hierarchy. Proc. Natl. Acad. Sci. USA 2010, 107, 15443–15448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanson, T.M.; Skinner, J.P.; Kumar, A.; Zhan, Y.; Lew, A.M.; Chong, M.M. The role of microRNAs in lymphopoiesis. Int. J. Hematol. 2014, 100, 246–253. [Google Scholar] [CrossRef]
- Tian, X.; Tian, J.; Tang, X.; Ma, J.; Wang, S. Long non-coding RNAs in the regulation of myeloid cells. J. Hematol. Oncol. 2016, 9, 99. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Weissman, S.M.; Newburger, P.E. Long intergenic non-coding RNA HOTAIRM1 regulates cell cycle progression during myeloid maturation in NB4 human promyelocytic leukemia cells. RNA Biol. 2014, 11, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Dahariya, S.; Paddibhatla, I.; Kumar, S.; Raghuwanshi, S.; Pallepati, A.; Gutti, R.K. Long non-coding RNA: Classification, biogenesis and functions in blood cells. Mol. Immunol. 2019, 112, 82–92. [Google Scholar] [CrossRef]
- Giardino, G.; Gallo, V.; Prencipe, R.; Gaudino, G.; Romano, R.; De Cataldis, M.; Lorello, P.; Palamaro, L.; Di Giacomo, C.; Capalbo, D.; et al. Unbalanced Immune System: Immunodeficiencies and Autoimmunity. Front. Pediatr. 2016, 4, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmonte, O.M.; Castagnoli, R.; Calzoni, E.; Notarangelo, L.D. Inborn Errors of Immunity with Immune Dysregulation: From Bench to Bedside. Front. Pediatr. 2019, 7, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020, 40, 24–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousfiha, A.; Jeddane, L.; Picard, C.; Al-Herz, W.; Ailal, F.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J. Clin. Immunol. 2020, 40, 66–81. [Google Scholar] [CrossRef] [Green Version]
- van Schouwenburg, P.A.; Davenport, E.E.; Kienzler, A.K.; Marwah, I.; Wright, B.; Lucas, M.; Malinauskas, T.; Martin, H.C.; Lockstone, H.E.; Cazier, J.B.; et al. Application of whole genome and RNA sequencing to investigate the genomic landscape of common variable immunodeficiency disorders. Clin. Immunol. 2015, 160, 301–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abolhassani, H.; Hammarström, L.; Cunningham-Rundles, C. Current genetic landscape in common variable immune deficiency. Blood 2020, 135, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Almamun, M.; Levinson, B.T.; Gater, S.T.; Schnabel, R.D.; Arthur, G.L.; Davis, J.W.; Taylor, K.H. Genome-wide DNA methylation analysis in precursor B-cells. Epigenetics 2014, 9, 1588–1595. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.T.; Xiao, Y.; Muench, M.O.; Xiao, J.; Fomin, M.E.; Wiencke, J.K.; Zheng, S.; Dou, X.; de Smith, A.; Chokkalingam, A.; et al. A global DNA methylation and gene expression analysis of early human B-cell development reveals a demethylation signature and transcription factor network. Nucleic Acids Res. 2012, 40, 11339–11351. [Google Scholar] [CrossRef]
- Tallmadge, R.L.; Shen, L.; Tseng, C.T.; Miller, S.C.; Barry, J.; Felippe, M.J. Bone marrow transcriptome and epigenome profiles of equine common variable immunodeficiency patients unveil block of B lymphocyte differentiation. Clin. Immunol. 2015, 160, 261–276. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.Y.; Mav, D.; Shah, R.; Grimm, S.A.; Phadke, D.; Hatzi, K.; Melnick, A.; Geigerman, C.; Sobol, S.E.; Jaye, D.L.; et al. DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome Res. 2013, 23, 2030–2041. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Cortez, V.C.; del Pino-Molina, L.; Rodríguez-Ubreva, J.; Ciudad, L.; Gómez-Cabrero, D.; Company, C.; Urquiza, J.M.; Tegnér, J.; Rodríguez-Gallego, C.; López-Granados, E.; et al. Monozygotic twins discordant for common variable immunodeficiency reveal impaired DNA demethylation during naïve-to-memory B-cell transition. Nat. Commun. 2015, 6, 7335. [Google Scholar] [CrossRef] [PubMed]
- Sterlin, D.; Velasco, G.; Moshous, D.; Touzot, F.; Mahlaoui, N.; Fischer, A.; Suarez, F.; Francastel, C.; Picard, C. Genetic, Cellular and Clinical Features of ICF Syndrome: A French National Survey. J. Clin. Immunol. 2016, 36, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Lana, E.; Mégarbané, A.; Tourrière, H.; Sarda, P.; Lefranc, G.; Claustres, M.; De Sario, A. DNA replication is altered in Immunodeficiency Centromeric instability Facial anomalies (ICF) cells carrying DNMT3B mutations. Eur. J. Hum. Genet. 2012, 20, 1044–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamae, C.; Imai, K.; Kato, T.; Okano, T.; Honma, K.; Nakagawa, N.; Yeh, T.W.; Noguchi, E.; Ohara, A.; Shigemura, T.; et al. Clinical and Immunological Characterization of ICF Syndrome in Japan. J. Clin. Immunol. 2018, 38, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Chouery, E.; Abou-Ghoch, J.; Corbani, S.; El Ali, N.; Korban, R.; Salem, N.; Castro, C.; Klayme, S.; Rjeily, M.A.A.; Khoury-Matar, R.; et al. A novel deletion in ZBTB24 in a Lebanese family with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Clin. Genet. 2012, 82, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, P.E.; Ito, Y.; Grillo, G.; Wang, J.; Velasco, G.; Nitta, H.; Unoki, M.; Yoshihara, M.; Suyama, M.; Sun, Y.; et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat. Commun. 2015, 6, 7870. [Google Scholar] [CrossRef] [Green Version]
- Toubiana, S.; Velasco, G.; Chityat, A.; Kaindl, A.M.; Hershtig, N.; Tzur-Gilat, A.; Francastel, C.; Selig, S. Subtelomeric methylation distinguishes between subtypes of Immunodeficiency, Centromeric instability and Facial anomalies syndrome. Hum. Mol. Genet. 2018, 27, 3568–3581. [Google Scholar] [CrossRef]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Jin, B.; Tao, Q.; Peng, J.; Soo, H.M.; Wu, W.; Ying, J.; Fields, C.R.; Delmas, A.L.; Liu, X.; Qiu, J.; et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum. Mol. Genet. 2008, 17, 690–709. [Google Scholar] [CrossRef] [Green Version]
- de Greef, J.C.; Wang, J.; Balog, J.; den Dunnen, J.T.; Frants, R.R.; Straasheijm, K.R.; Aytekin, C.; van der Burg, M.; Duprez, L.; Ferster, A.; et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am. J. Hum. Genet. 2011, 88, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Jenness, C.; Giunta, S.; Müller, M.M.; Kimura, H.; Muir, T.W.; Funabiki, H. HELLS and CDCA7 comprise a bipartite nucleosome remodeling complex defective in ICF syndrome. Proc. Natl. Acad. Sci. USA 2018, 115, E876–E885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagi, S.; Gulino, A.V.; Lapi, E.; Rigante, D. Epigenetic control of the immune system: A lesson from Kabuki syndrome. Immunol. Res. 2016, 64, 345–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margot, H.; Boursier, G.; Duflos, C.; Sanchez, E.; Amiel, J.; Andrau, J.-C.; Arpin, S.; Brischoux-Boucher, E.; Boute, O.; Burglen, L.; et al. Immunopathological manifestations in Kabuki syndrome: A registry study of 177 individuals. Genet. Med. 2020, 22, 181–188. [Google Scholar] [CrossRef]
- Ming, J.E.; Russell, K.L.; McDonald-McGinn, D.M.; Zackai, E.H. Autoimmune disorders in Kabuki syndrome. Am. J. Med. Genet. A 2005, 132, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.D.; Ciprero, K.L.; Sullivan, K.E.; Kaplan, P.B.; McDonald-McGinn, D.M.; Zackai, E.H.; Ming, J.E. Immune abnormalities are a frequent manifestation of Kabuki syndrome. Am. J. Med. Genet. A 2005, 135, 278–281. [Google Scholar] [CrossRef]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [Green Version]
- Lederer, D.; Grisart, B.; Digilio, M.C.; Benoit, V.; Crespin, M.; Ghariani, S.C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am. J. Hum. Genet. 2012, 90, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaikh, T.H. Kabuki syndrome genes KMT2D and KDM6A: Functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 2015, 24, 4443–4453. [Google Scholar] [CrossRef] [Green Version]
- Lindsley, A.W.; Saal, H.M.; Burrow, T.A.; Hopkin, R.J.; Shchelochkov, O.; Khandelwal, P.; Xie, C.; Bleesing, J.; Filipovich, L.; Risma, K.; et al. Defects of B-cell terminal differentiation in patients with type-1 Kabuki syndrome. J. Allergy Clin. Immunol. 2016, 137, 179–187.e10. [Google Scholar] [CrossRef] [Green Version]
- Aref-Eshghi, E.; Rodenhiser, D.I.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Hood, R.L.; Bulman, D.E.; Kernohan, K.D.; et al. Genomic DNA Methylation Signatures Enable Concurrent Diagnosis and Clinical Genetic Variant Classification in Neurodevelopmental Syndromes. Am. J. Hum. Genet. 2018, 102, 156–174. [Google Scholar] [CrossRef] [Green Version]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; de la Morena, M.T.; van Oers, N.S.C. The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front. Genet. 2019, 10, 1365. [Google Scholar] [CrossRef] [PubMed]
- Rooney, K.; Levy, M.A.; Haghshenas, S.; Kerkhof, J.; Rogaia, D.; Tedesco, M.G.; Imperatore, V.; Mencarelli, A.; Squeo, G.M.; Di Venere, E.; et al. Identification of a DNA Methylation Episignature in the 22q11.2 Deletion Syndrome. Int. J. Mol. Sci. 2021, 22, 8611. [Google Scholar] [CrossRef] [PubMed]
- Baldini, A.; Fulcoli, F.G.; Illingworth, E. Tbx1: Transcriptional and Developmental Functions. Curr. Top Dev. Biol. 2017, 122, 223–243. [Google Scholar] [CrossRef]
- Fulcoli, F.G.; Franzese, M.; Liu, X.; Zhang, Z.; Angelini, C.; Baldini, A. Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nat. Commun. 2016, 7, 11688. [Google Scholar] [CrossRef] [Green Version]
- Boerkoel, C.F.; Takashima, H.; John, J.; Yan, J.; Stankiewicz, P.; Rosenbarker, L.; André, J.L.; Bogdanovic, R.; Burguet, A.; Cockfield, S.; et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat. Genet. 2002, 30, 215–220. [Google Scholar] [CrossRef]
- Bertulli, C.; Marzollo, A.; Doria, M.; Di Cesare, S.; La Scola, C.; Mencarelli, F.; Pasini, A.; Affinita, M.C.; Vidal, E.; Magini, P.; et al. Expanding Phenotype of Schimke Immuno-Osseous Dysplasia: Congenital Anomalies of the Kidneys and of the Urinary Tract and Alteration of NK Cells. Int. J. Mol. Sci. 2020, 21, 8604. [Google Scholar] [CrossRef]
- Saraiva, J.M.; Dinis, A.; Resende, C.; Faria, E.; Gomes, C.; Correia, A.J.; Gil, J.; da Fonseca, N. Schimke immuno-osseous dysplasia: Case report and review of 25 patients. J. Med. Genet. 1999, 36, 786–789. [Google Scholar] [CrossRef]
- Indumathi, C.K.; Bustamante, J. Clinical and immunological profile of children with Mendelian Susceptibility to Mycobacterial Diseases (MSMD) from an Indian tertiary care hospital. Indian J. Tuberc. 2021, 68, 292–297. [Google Scholar] [CrossRef]
- Bustamante, J.; Boisson-Dupuis, S.; Abel, L.; Casanova, J.L. Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin. Immunol. 2014, 26, 454–470. [Google Scholar] [CrossRef] [Green Version]
- Bustamante, J. Mendelian susceptibility to mycobacterial disease: Recent discoveries. Hum. Genet. 2020, 139, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Pacis, A.; Mailhot-Léonard, F.; Tailleux, L.; Randolph, H.E.; Yotova, V.; Dumaine, A.; Grenier, J.C.; Barreiro, L.B. Gene activation precedes DNA demethylation in response to infection in human dendritic cells. Proc. Natl. Acad. Sci. USA 2019, 116, 6938–6943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacis, A.; Tailleux, L.; Morin, A.M.; Lambourne, J.; MacIsaac, J.L.; Yotova, V.; Dumaine, A.; Danckaert, A.; Luca, F.; Grenier, J.C.; et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res. 2015, 25, 1801–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaasinen, E.; Kuismin, O.; Rajamäki, K.; Ristolainen, H.; Aavikko, M.; Kondelin, J.; Saarinen, S.; Berta, D.G.; Katainen, R.; Hirvonen, E.A.M.; et al. Impact of constitutional TET2 haploinsufficiency on molecular and clinical phenotype in humans. Nat. Commun. 2019, 10, 1252. [Google Scholar] [CrossRef]
- Spegarova, J.S.; Lawless, D.; Mohamad, S.M.B.; Engelhardt, K.R.; Doody, G.; Shrimpton, J.; Rensing-Ehl, A.; Ehl, S.; Rieux-Laucat, F.; Cargo, C.; et al. Germline TET2 loss of function causes childhood immunodeficiency and lymphoma. Blood 2020, 136, 1055–1066. [Google Scholar] [CrossRef]
- Muto, H.; Sakata-Yanagimoto, M.; Nagae, G.; Shiozawa, Y.; Miyake, Y.; Yoshida, K.; Enami, T.; Kamada, Y.; Kato, T.; Uchida, K.; et al. Reduced TET2 function leads to T-cell lymphoma with follicular helper T-cell-like features in mice. Blood Cancer J. 2014, 4, e264. [Google Scholar] [CrossRef]
- Rosikiewicz, W.; Chen, X.; Dominguez, P.M.; Ghamlouch, H.; Aoufouchi, S.; Bernard, O.A.; Melnick, A.; Li, S. TET2 deficiency reprograms the germinal center B cell epigenome and silences genes linked to lymphomagenesis. Sci. Adv. 2020, 6, eaay5872. [Google Scholar] [CrossRef]
- Lio, C.W.; Zhang, J.; González-Avalos, E.; Hogan, P.G.; Chang, X.; Rao, A. Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. Elife 2016, 5, e18290. [Google Scholar] [CrossRef] [Green Version]


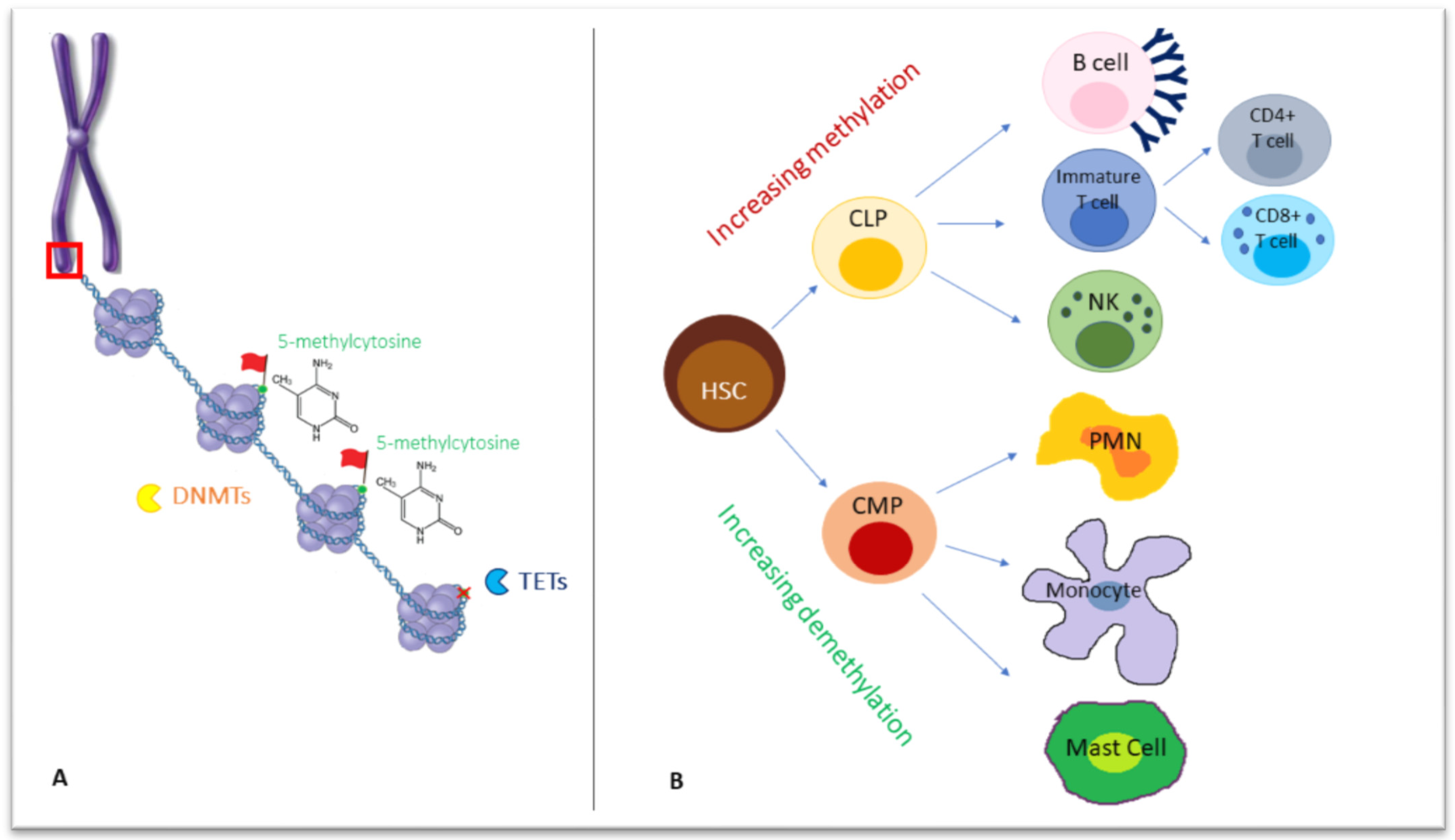{kind=link}
| Humoral Immunity | Disorder | Altered Epigenetic Mechanism | Genes | Major Immunological Alteration | |
|---|---|---|---|---|---|
| CVID | DNA methylation | PAX5, PIK3CD, BCL2L1, RPS6KB2, TCF3, KCNN4 | Agammaglobulinemia, impaired response to vaccines, autoimmunity, CLD, enteropathy | ||
| ICF1 | DNA methylation | DNMT3B | Agammaglobulinemia or hypogammaglobulinemia, recurrent infections | ||
| ICF2 | DNA methylation | ZBTB24 | Agammaglobulinemia or hypogammaglobulinemia, recurrent infections | ||
| ICF3 | DNA methylation | CDCA7 | Agammaglobulinemia or hypogammaglobulinemia, recurrent infections | ||
| ICF4 | DNA methylation | HELLS | Agammaglobulinemia or hypogammaglobulinemia, recurrent infections | ||
| KS1 | Histone modification | KMT2D | Hypogammaglobulinemia, autoimmune cytopenia | ||
| KS2 | Histone modification | KDM6A | Hypogammaglobulinemia, autoimmune cytopenia | ||
| Adaptive immunity | |||||
| 22q11.2 DS | DNA methylation Non-coding RNAs | TBX1 | Lymphopenia, recurrent infections, autoimmunity | ||
| Schimke immuno-osseous dysplasia | Chromatin remodeling | SMARCAL1 | Lymphopenia, recurrent infections | ||
| Immune dysregulation | |||||
| TET2 loss-of-function | DNA methylation | TET2 | Hepatosplenomegaly, lymphadenopathy, autoimmunity | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romano, R.; Cillo, F.; Moracas, C.; Pignata, L.; Nannola, C.; Toriello, E.; De Rosa, A.; Cirillo, E.; Coppola, E.; Giardino, G.; et al. Epigenetic Alterations in Inborn Errors of Immunity. J. Clin. Med. 2022, 11, 1261. https://doi.org/10.3390/jcm11051261
Romano R, Cillo F, Moracas C, Pignata L, Nannola C, Toriello E, De Rosa A, Cirillo E, Coppola E, Giardino G, et al. Epigenetic Alterations in Inborn Errors of Immunity. Journal of Clinical Medicine. 2022; 11(5):1261. https://doi.org/10.3390/jcm11051261
Chicago/Turabian StyleRomano, Roberta, Francesca Cillo, Cristina Moracas, Laura Pignata, Chiara Nannola, Elisabetta Toriello, Antonio De Rosa, Emilia Cirillo, Emma Coppola, Giuliana Giardino, and et al. 2022. "Epigenetic Alterations in Inborn Errors of Immunity" Journal of Clinical Medicine 11, no. 5: 1261. https://doi.org/10.3390/jcm11051261
APA StyleRomano, R., Cillo, F., Moracas, C., Pignata, L., Nannola, C., Toriello, E., De Rosa, A., Cirillo, E., Coppola, E., Giardino, G., Brunetti-Pierri, N., Riccio, A., & Pignata, C. (2022). Epigenetic Alterations in Inborn Errors of Immunity. Journal of Clinical Medicine, 11(5), 1261. https://doi.org/10.3390/jcm11051261