
Text-Mining Approach to Identify Hub Genes of Cancer Metastasis and Potential Drug Repurposing to Target Them



Abstract
:1. Introduction
2. Materials and Methods
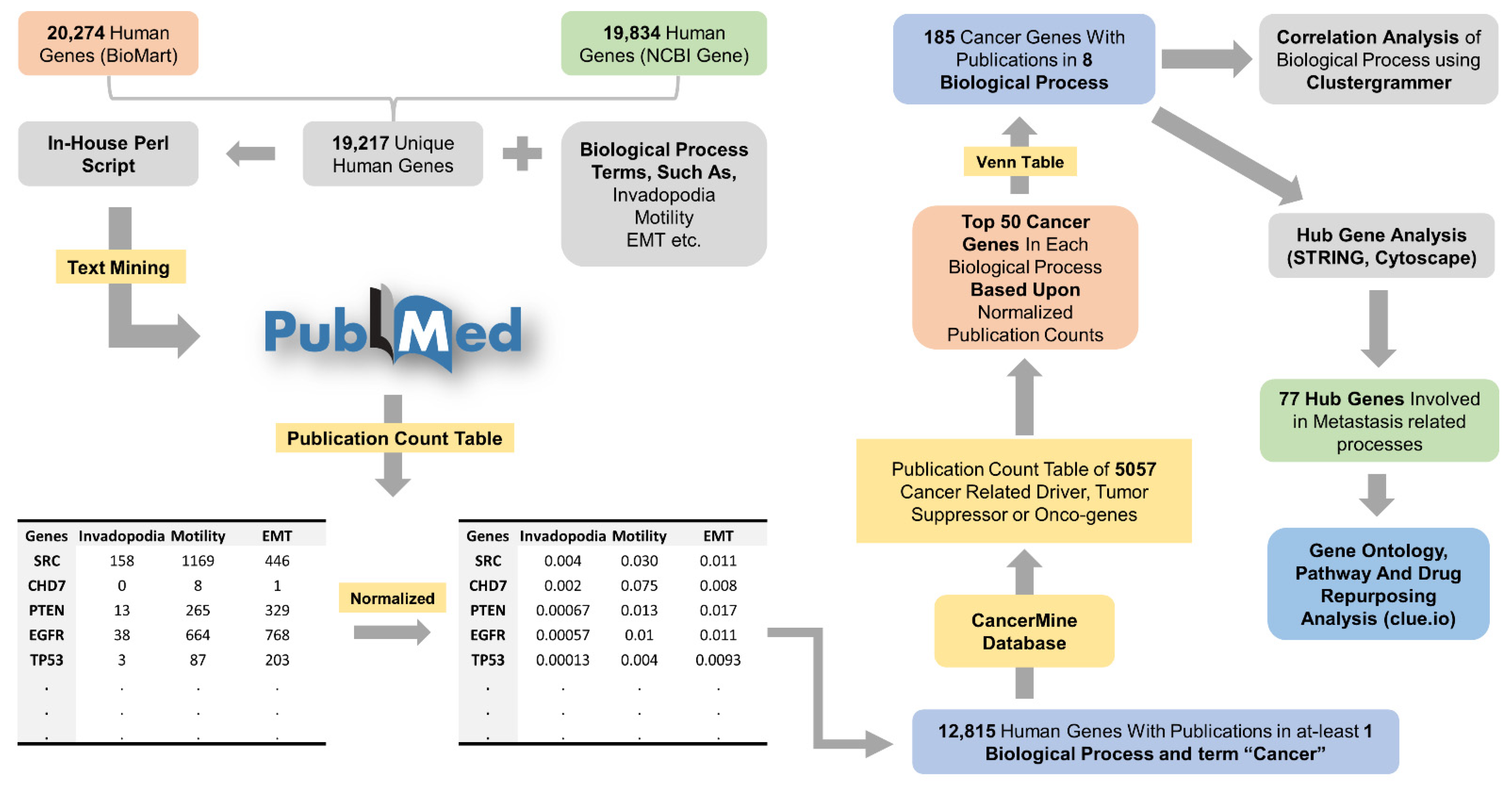
2.1. Preparing a List of Human Genes
2.2. Text-Mining to Determine the Association of Human Genes and Biological Processes
2.3. Filtration of Cancer-Specific Text-Mining Results
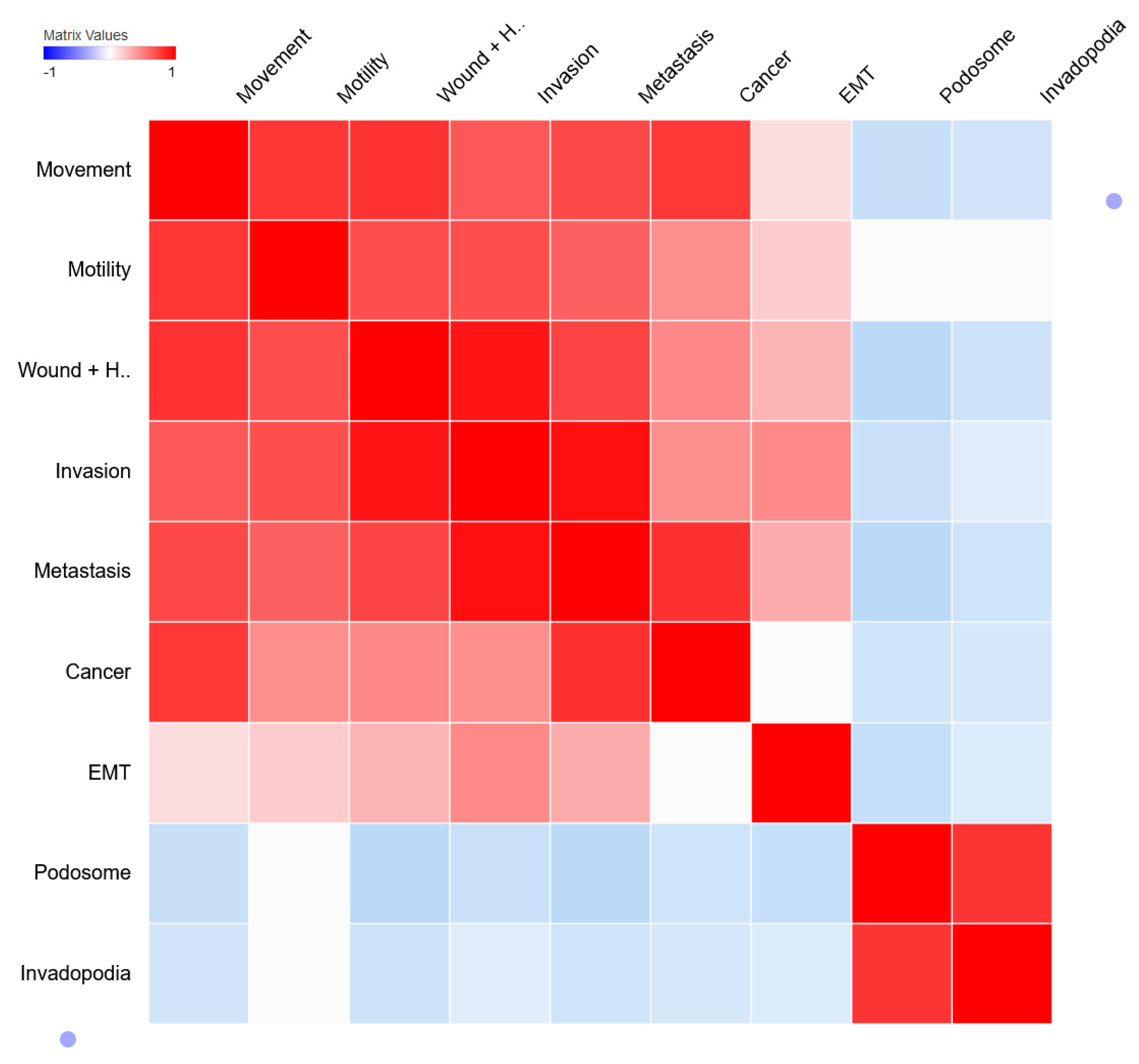
2.4. Correlation Analysis of Cancer-Specific Genes
2.5. PPI Network, Module, and Hub Gene Analysis
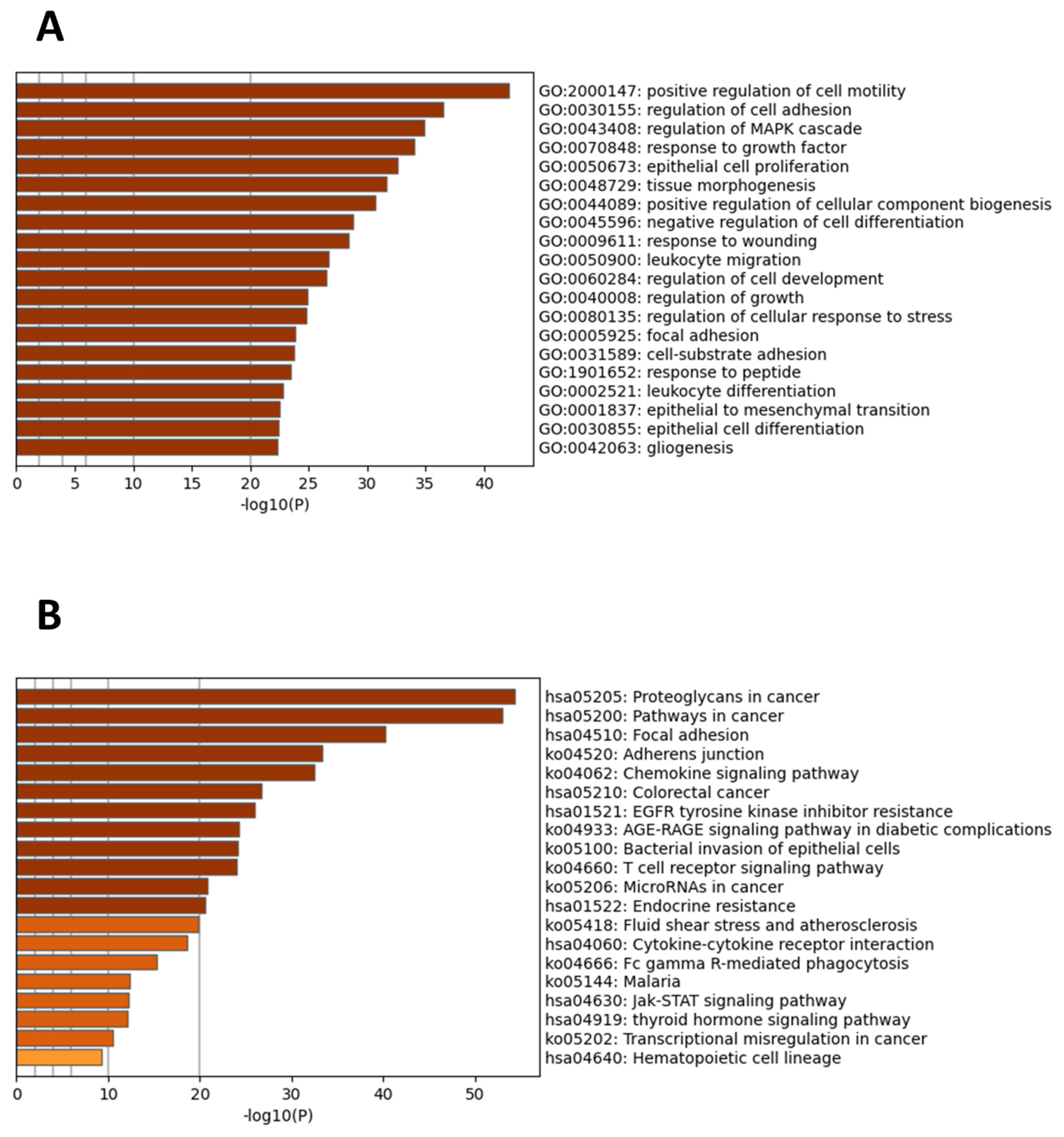
2.6. Gene Ontology and Pathway Analysis
2.7. Drug-Gene Interaction
2.8. Drug Repurposing and Connectivity Map (CMap) Analysis
3. Results
3.1. Association of Human Genes and Biological Processes
3.2. Correlation Analysis of Cancer-Specific Genes and Process
3.3. PPI Network, Module, and Hub Gene Analysis
3.4. Functional and Signaling Pathway Enrichment Analysis
3.5. Drug Target Identification and Drug Repurposing of Potential Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pienta, K.J.; Robertson, B.A.; Coffey, D.S.; Taichman, R.S. The Cancer Diaspora: Metastasis beyond the Seed and Soil Hypothesis. Clin. Cancer Res. 2013, 19, 5849–5855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.P.; Massagué, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Krakhmal, N.V.; Zavyalova, M.V.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V.M. Cancer Invasion: Patterns and Mechanisms. Acta Nat. 2015, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Aiello, N.M.; Kang, Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef] [Green Version]
- Pearson, G.W. Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. J. Clin. Med. 2019, 8, 646. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, Y.; Sheng, G. Epithelial to mesenchymal transition during gastrulation: An embryological view. Dev. Growth Differ. 2008, 50, 755–766. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Bolós, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Tilló, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.-S.; Jiang, J.; Chen, B.-J.; Wang, K.; Tang, Y.-L.; Liang, X.-H. Plasticity of cancer cell invasion: Patterns and mechanisms. Transl. Oncol. 2021, 14, 100899. [Google Scholar] [CrossRef]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Mizutani, T.; Kawabata, K.; Haga, H. Leader cells regulate collective cell migration via Rac activation in the downstream signaling of integrin β1 and PI3K. Sci. Rep. 2015, 5, 7656. [Google Scholar] [CrossRef] [Green Version]
- Nabeshima, K.; Inoue, T.; Shimao, Y.; Okada, Y.; Itoh, Y.; Seiki, M.; Koono, M. Front-cell-specific expression of membrane-type 1 matrix metalloproteinase and gelatinase A during cohort migration of colon carcinoma cells induced by hepatocyte growth factor/scatter factor. Cancer Res. 2000, 60, 3364–3369. [Google Scholar]
- Kumar, S.; Kapoor, A.; Desai, S.; Inamdar, M.M.; Sen, S. Proteolytic and non-proteolytic regulation of collective cell invasion: Tuning by ECM density and organization. Sci. Rep. 2016, 6, 19905. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef]
- Sundaram, G.M.; Quah, S.; Sampath, P. Cancer: The dark side of wound healing. FEBS J. 2018, 285, 4516–4534. [Google Scholar] [CrossRef] [PubMed]
- MacCarthy-Morrogh, L.; Martin, P. The hallmarks of cancer are also the hallmarks of wound healing. Sci. Signal. 2020, 13, 8690. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Basnet, H.; Kaygusuz, Y.; Laughney, A.M.; He, L.; Sharma, R.; O’Rourke, K.P.; Reuter, V.P.; Huang, Y.-H.; Turkekul, M.; et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nat. Cancer 2020, 1, 28. [Google Scholar] [CrossRef] [PubMed]
- Genna, A.; Lapetina, S.; Lukic, N.; Twafra, S.; Meirson, T.; Sharma, V.P.; Condeelis, J.S.; Gil-Henn, H. Pyk2 and FAK differentially regulate invadopodia formation and function in breast cancer cells. J. Cell Biol. 2018, 217, 375–395. [Google Scholar] [CrossRef] [Green Version]
- Meirson, T.; Gil-Henn, H. Targeting invadopodia for blocking breast cancer metastasis. Drug Resist. Updat. 2018, 39, 1–17. [Google Scholar] [CrossRef]
- Takkunen, M.; Hukkanen, M.; Liljeström, M.; Grenman, R.; Virtanen, I. Podosome-like structures of non-invasive carcinoma cells are replaced in epithelial-mesenchymal transition by actin comet-embedded invadopodia. J. Cell. Mol. Med. 2010, 14, 1569–1593. [Google Scholar] [CrossRef] [Green Version]
- Calle, Y.; Burns, S.; Thrasher, A.J.; Jones, G.E. The leukocyte podosome. Eur. J. Cell Biol. 2006, 85, 151–157. [Google Scholar] [CrossRef]
- Day, C.P.; Merlino, G.; Van Dyke, T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Fontebasso, Y.; Dubinett, S.M. Drug Development for Metastasis Prevention. Crit. Rev. Oncog. 2015, 20, 449. [Google Scholar] [CrossRef]
- Ma, W.; Shi, B.; Zhao, F.; Wu, Y.; Jin, F. Systematic analysis of breast atypical hyperplasia-associated hub genes and pathways based on text mining. Eur. J. Cancer Prev. 2019, 28, 507–514. [Google Scholar] [CrossRef]
- Zhou, J.; Fu, B. The research on gene-disease association based on text-mining of PubMed. BMC Bioinform. 2018, 19, 37. [Google Scholar] [CrossRef] [PubMed]
- Elbattah, M.; Arnaud, É.; Gignon, M.; Dequen, G. The Role of Text Analytics in Healthcare: A Review of Recent Developments and Applications. Healthinf 2021, 5, 825–832. [Google Scholar] [CrossRef]
- Pletscher-Frankild, S.; Pallejà, A.; Tsafou, K.; Binder, J.X.; Jensen, L.J. DISEASES: Text mining and data integration of disease–gene associations. Methods 2015, 74, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Musa, A.; Dehmer, M.; Yli-Harja, O.P.; Emmert-Streib, F. Global genetics research in prostate cancer: A text minning and computational network theory approach. Front. Genet. 2019, 10, 70. [Google Scholar] [CrossRef]
- Saha, T.; Solomon, J.; Samson, A.O.; Gil-Henn, H. Invasion and Metastasis as a Central Hallmark of Breast Cancer. J. Clin. Med. 2021, 10, 3498. [Google Scholar] [CrossRef]
- Beckman, M.F.; Brennan, E.J.; Igba, C.K.; Brennan, M.T.; Mougeot, F.B.; Mougeot, J.-L.C.A.; Beckman, M.F.; Brennan, E.J.; Igba, C.K.; Brennan, M.T.; et al. A Computational Text Mining-Guided Meta-Analysis Approach to Identify Potential Xerostomia Drug Targets. J. Clin. Med. 2022, 11, 1442. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Meirson, T.; Gil-Henn, H.; Samson, A.O. Invasion and metastasis: The elusive hallmark of cancer. Oncogene 2019, 39, 2024–2026. [Google Scholar] [CrossRef]
- Lever, J.; Zhao, E.Y.; Grewal, J.; Jones, M.R.; Jones, S.J.M. CancerMine: A literature-mined resource for drivers, oncogenes and tumor suppressors in cancer. Nat. Methods 2019, 16, 505–507. [Google Scholar] [CrossRef]
- Fernandez, N.F.; Gundersen, G.W.; Rahman, A.; Grimes, M.L.; Rikova, K.; Hornbeck, P.; Ma’ayan, A. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci. Data 2017, 4, 170151. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, F.; Zhou, Y.; Zhang, Y.; Wang, Z.; Zhang, R.; Zhu, J.; Ren, Y.; Tan, Y.; et al. Therapeutic target database 2020: Enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2020, 48, D1031–D1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Magalhães, J.P. Every gene can (and possibly will) be associated with cancer. Trends Genet. 2021, 38, 216–217. [Google Scholar] [CrossRef]
- Flynn, D.C.; Cho, Y.; Vincent, D.; Cunnick, J.M. Podosomes and Invadopodia: Related structures with Common Protein Components that May Promote Breast Cancer Cellular Invasion. Breast Cancer 2008, 2, 17. [Google Scholar] [CrossRef]
- Murphy, D.A.; Courtneidge, S.A. The “ins” and “outs” of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, D.; Branch, K.M.; Weaver, A.M. Signaling inputs to invadopodia and podosomes. J. Cell Sci. 2013, 126, 2979–2989. [Google Scholar] [CrossRef] [Green Version]
- Weaver, A.M.; Williams, N. Quick guides Invadopodia. Curr. Biol. 2008, 18, 362–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, T.; Gil-Henn, H. Invadopodia, a Kingdom of Non-Receptor Tyrosine Kinases. Cells 2021, 10, 2037. [Google Scholar] [CrossRef] [PubMed]
- Boyer, B.; Bourgeois, Y.; Poupon, M.F. Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene 2002, 21, 2347–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, S.; Gosens, R.; Wieland, T.; Schmidt, M. Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacol. Ther. 2018, 183, 1–21. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.Y.; Wang, C.Y.; Phan, N.N.; Chiao, C.C.; Li, C.Y.; Sun, Z.; Hung, J.H.; Chen, Y.L.; Yen, M.C.; Weng, T.Y.; et al. PODXL2 maintains cellular stemness and promotes breast cancer development through the Rac1/Akt pathway. Int. J. Med. Sci. 2020, 17, 1639. [Google Scholar] [CrossRef]
- Huang, J.S.; Cho, C.Y.; Hong, C.C.; Yan, M. De; Hsieh, M.C.; Lay, J.D.; Lai, G.M.; Cheng, A.L.; Chuang, S.E. Oxidative stress enhances Axl-mediated cell migration through an Akt1/Rac1-dependent mechanism. Free Radic. Biol. Med. 2013, 65, 1246–1256. [Google Scholar] [CrossRef]
- Chiu, Y.W.; Liou, L.Y.; Chen, P.T.; Huang, C.M.; Luo, F.J.; Hsu, Y.K.; Yuan, T.C. Tyrosine 397 phosphorylation is critical for FAK-promoted Rac1 activation and invasive properties in oral squamous cell carcinoma cells. Lab. Investig. 2016, 96, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Rethinking the war on cancer. Lancet 2014, 383, 558–563. [Google Scholar] [CrossRef]
- Stoletov, K.; Beatty, P.H.; Lewis, J.D. Novel therapeutic targets for cancer metastasis. Expert Rev. Anticancer Ther. 2020, 20, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrens, T.D.; Bang-Christensen, S.R.; Jørgensen, A.M.; Løppke, C.; Spliid, C.B.; Sand, N.T.; Clausen, T.M.; Salanti, A.; Agerbæk, M.Ø. The Role of Proteoglycans in Cancer Metastasis and Circulating Tumor Cell Analysis. Front. Cell Dev. Biol. 2020, 8, 749. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Berdiaki, A.; Spyridaki, I.; Krasanakis, T.; Tsatsakis, A.; Tzanakakis, G.N. Proteoglycans—Biomarkers and Targets in Cancer Therapy. Front. Endocrinol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzanakakis, G.; Neagu, M.; Tsatsakis, A.; Nikitovic, D. Proteoglycans and Immunobiology of Cancer—Therapeutic Implications. Front. Immunol. 2019, 10, 875. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W.; Wong, G.; Earle, C.; Krueger, K.; Spevak, C.C. Hyaluronan-CD44 interaction promotes c-Src-mediated twist signaling, microRNA-10b expression, and RhoA/RhoC up-regulation, leading to Rho-kinase-associated cytoskeleton activation and breast tumor cell invasion. J. Biol. Chem. 2010, 285, 36721–36735. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Hong, S.; Yu, S.; Huang, Y.; Chen, S.; Liu, Y.; Zhang, Q.; Li, Y.; Xiao, H. MiR-195 Inhibits Tumor Growth and Metastasis in Papillary Thyroid Carcinoma Cell Lines by Targeting CCND1 and FGF2. Int. J. Endocrinol. 2017, 2017, 6180425. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.; Johnson, N.; Cooke, L.; Johnson, B.; Chen, Y.; Pandey, M.; Chandler, J.; Mahadevan, D. TP53 Mutations as a Driver of Metastasis Signaling in Advanced Cancer Patients. Cancers 2021, 13, 597. [Google Scholar] [CrossRef]
- Ren, Z.; Liang, S.; Yang, J.; Han, X.; Shan, L.; Wang, B.; Mu, T.; Zhang, Y.; Yang, X.; Xiong, S.; et al. Coexpression of CXCR4 and MMP9 predicts lung metastasis and poor prognosis in resected osteosarcoma. Tumour Biol. 2016, 37, 5089–5096. [Google Scholar] [CrossRef]
- Gu, J.J.; Hoj, J.; Rouse, C.; Pendergast, A.M. Mesenchymal stem cells promote metastasis through activation of an ABL-MMP9 signaling axis in lung cancer cells. PLoS ONE 2020, 15, e0241423. [Google Scholar] [CrossRef]
- Chen, W.; Wu, J.; Shi, W.; Zhang, G.; Chen, X.; Ji, A.; Wang, Z.; Wu, J.; Jiang, C. PRRX1 deficiency induces mesenchymal-epithelial transition through PITX2/miR-200–dependent SLUG/CTNNB1 regulation in hepatocellular carcinoma. Cancer Sci. 2021, 112, 2158. [Google Scholar] [CrossRef]
- Beaver, J.A.; Kluetz, P.G.; Pazdur, R. Metastasis-free Survival—A New End Point in Prostate Cancer Trials. N. Engl. J. Med. 2018, 378, 2458–2460. [Google Scholar] [CrossRef] [PubMed]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-metastatic and Anti-invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmy, L.; Xiang, Y.; Xie, Z.; Tao, C.; Zhi, D. Med-BERT: Pretrained contextualized embeddings on large-scale structured electronic health records for disease prediction. Npj Digit. Med. 2021, 4, 86. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Xiang, H.; Xie, H.; Yu, Y.; Dong, S.; Yang, Z.; Zhao, N. Application of BERT to Enable Gene Classification Based on Clinical Evidence. Biomed Res. Int. 2020, 2020, 5491963. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Gene | Drug | Drug Type | Mechanism of Action | Disease |
|---|---|---|---|---|---|
| 1 | EGFR | Afatinib | Small molecule | Inhibitor | HER2/NEU overexpressing breast cancer |
| Osimertinib | Small molecule | Inhibitor | metastatic non-small cell lung cancer | ||
| 2 | MET | Capmatinib | Small molecule | Inhibitor | non-small cell lung cancer |
| 3 | MTOR | Temsirolimus | Small molecule | Inhibitor | renal cell carcinoma |
| 4 | EZH2 | Tazemetostat | Small molecule | Inhibitor | Follicular lymphoma |
| 5 | KIT | Ripretinib | Small molecule | Inhibitor | gastrointestinal stromal tumor |
| 6 | CXCR4 | Plerixafor | Small molecule | Antagonist | non-Hodgkin’s lymphoma, multiple myeloma |
| 7 | IL2 | Aldesleukin | Protein Based Therapies | Agonist, modulator | renal cell carcinoma |
| 8 | SRC | Bosutinib | Small molecule | Inhibitor | hematologic malignancy |
| Dasatinib | Small molecule | Inhibitor | hematologic malignancy | ||
| 9 | HCK | Bosutinib | Small molecule | inhibitor | hematologic malignancy |
| 10 | ERBB2 | Trastuzumab | monoclonal antibody | Antagonist/Inhibitor | HER2-positive breast, gastroesophageal, and gastric cancers |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Detroja, T.S.; Gil-Henn, H.; Samson, A.O. Text-Mining Approach to Identify Hub Genes of Cancer Metastasis and Potential Drug Repurposing to Target Them. J. Clin. Med. 2022, 11, 2130. https://doi.org/10.3390/jcm11082130
Detroja TS, Gil-Henn H, Samson AO. Text-Mining Approach to Identify Hub Genes of Cancer Metastasis and Potential Drug Repurposing to Target Them. Journal of Clinical Medicine. 2022; 11(8):2130. https://doi.org/10.3390/jcm11082130
Chicago/Turabian StyleDetroja, Trishna Saha, Hava Gil-Henn, and Abraham O. Samson. 2022. "Text-Mining Approach to Identify Hub Genes of Cancer Metastasis and Potential Drug Repurposing to Target Them" Journal of Clinical Medicine 11, no. 8: 2130. https://doi.org/10.3390/jcm11082130
APA StyleDetroja, T. S., Gil-Henn, H., & Samson, A. O. (2022). Text-Mining Approach to Identify Hub Genes of Cancer Metastasis and Potential Drug Repurposing to Target Them. Journal of Clinical Medicine, 11(8), 2130. https://doi.org/10.3390/jcm11082130