
Potential Utility of Cerebrospinal Fluid Glycoprotein Nonmetastatic Melanoma Protein B as a Neuroinflammatory Diagnostic Biomarker in Mild Cognitive Impairment and Alzheimer’s Disease


 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Material
2.2. Biochemical Measurements
2.3. Statistical Analysis
3. Results
3.1. Patient Characteristics and CSF Concentrations of GPNMB and YKL-40
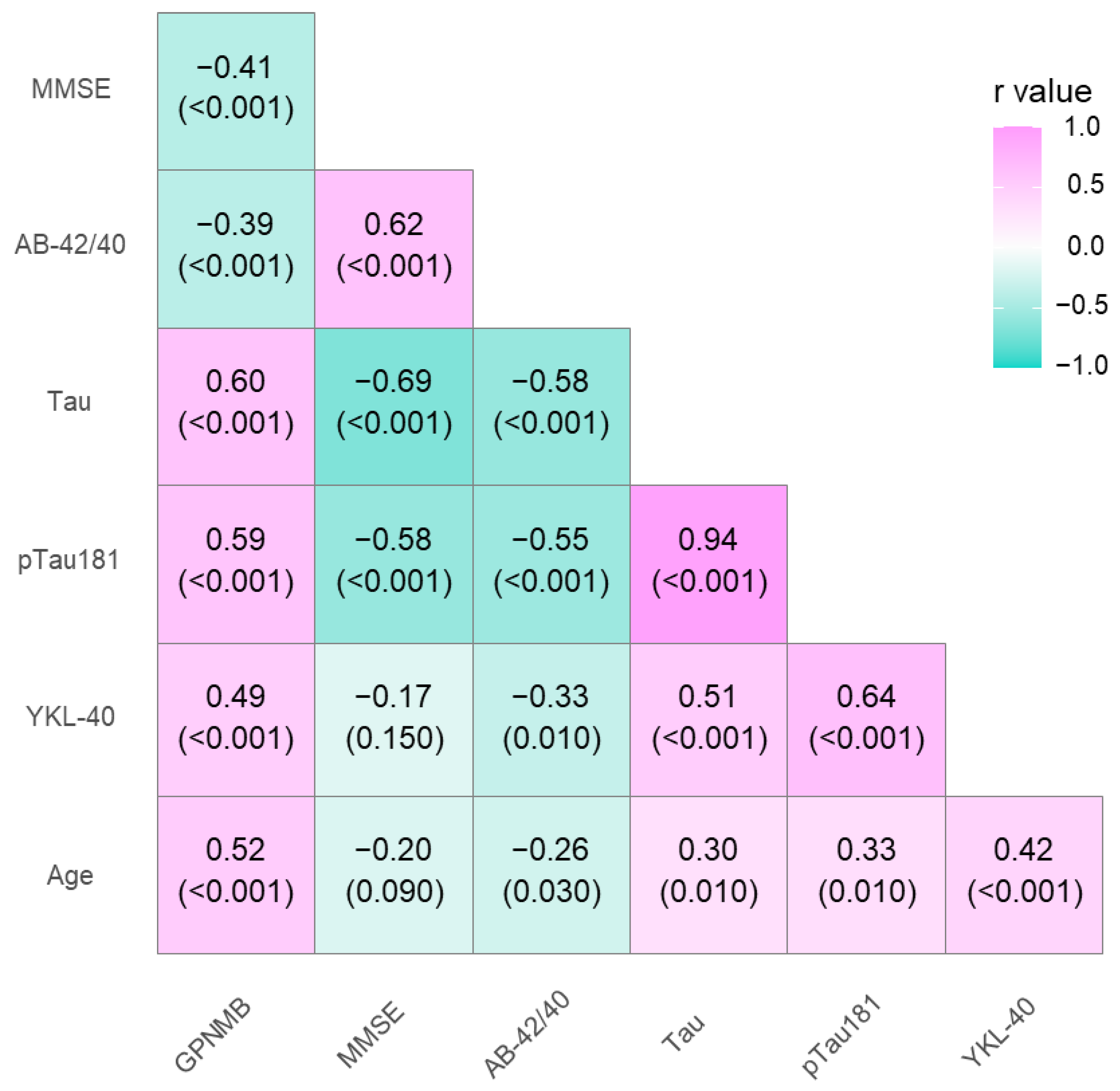
3.2. Association between GPNMB, YKL-40, and CSF Biomarkers
3.3. Diagnostic Usefulness of Candidate Biomarkers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s Disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association Alzheimer’s Facts and Figures Report|Alzheimer’s Association; Alzheimer’s Association: Chicago, IL, USA, 2021; Volume 1.
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. N. Am 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- El Kadmiri, N.; Said, N.; Slassi, I.; El Moutawakil, B.; Nadifi, S. Biomarkers for Alzheimer Disease: Classical and Novel Candidates’ Review. Neuroscience 2018, 370, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [Green Version]
- Kojro, E.; Fahrenholz, F. The Non-Amyloidogenic Pathway: Structure and Function of α-Secretases. In Alzheimer’s Disease; Springer: Boston, MA, USA, 2005; Volume 38, pp. 105–127. [Google Scholar]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The Contribution of Neuroinflammation to Amyloid Toxicity in Alzheimer’s Disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2015, 53, 1181–1194. [Google Scholar] [CrossRef]
- Nazem, A.; Sankowski, R.; Bacher, M.; Al-Abed, Y. Rodent Models of Neuroinflammation for Alzheimer’s Disease. J. Neuroinflamm. 2015, 12, 74. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An Early and Late Peak in Microglial Activation in Alzheimer’s Disease Trajectory. Brain 2017, 140, 792. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 75. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [Green Version]
- Weterman, M.A.J.; Ajubi, N.; van Dinter, I.M.R.; Degen, W.G.J.; van Muijen, G.N.P.; Ruiter, D.J.; Bloemers, H.P.J. Nmb, a Novel Gene, Is Expressed in Low-Metastatic Human Melanoma Cell Lines and Xenografts. Int. J. Cancer 1995, 60, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Ripoll, V.M.; Irvine, K.M.; Ravasi, T.; Sweet, M.J.; Hume, D.A. Gpnmb Is Induced in Macrophages by IFN-γ and Lipopolysaccharide and Acts as a Feedback Regulator of Proinflammatory Responses. J. Immunol. 2007, 178, 6557–6566. [Google Scholar] [CrossRef] [Green Version]
- Saade, M.; Araujo de Souza, G.; Scavone, C.; Kinoshita, P.F. The Role of GPNMB in Inflammation. Front. Immunol. 2021, 12, 674739. [Google Scholar] [CrossRef] [PubMed]
- Layé, S.; Parnet, P.; Goujon, E.; Dantzer, R. Peripheral Administration of Lipopolysaccharide Induces the Expression of Cytokine Transcripts in the Brain and Pituitary of Mice. Brain Res. Mol. Brain Res. 1994, 27, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Hagan, P.; Poole, S.; Bristow, A.F. Endotoxin-Stimulated Production of Rat Hypothalamic Interleukin-1 Beta in Vivo and in Vitro, Measured by Specific Immunoradiometric Assay. J. Mol. Endocrinol. 1993, 11, 31–36. [Google Scholar] [CrossRef]
- Huang, J.J.; Ma, W.J.; Yokoyama, S. Expression and Immunolocalization of Gpnmb, a Glioma-Associated Glycoprotein, in Normal and Inflamed Central Nervous Systems of Adult Rats. Brain Behav. 2012, 2, 85. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.A.N.; Annis, M.G.; Dong, Z.; Pepin, F.; Hallett, M.; Park, M.; Siegel, P.M. ADAM10 Releases a Soluble Form of the GPNMB/Osteoactivin Extracellular Domain with Angiogenic Properties. PLoS ONE 2010, 5, e12093. [Google Scholar] [CrossRef] [Green Version]
- Hüttenrauch, M.; Ogorek, I.; Klafki, H.; Otto, M.; Stadelmann, C.; Weggen, S.; Wiltfang, J.; Wirths, O. Glycoprotein NMB: A Novel Alzheimer’s Disease Associated Marker Expressed in a Subset of Activated Microglia. Acta Neuropathol. Commun. 2018, 6, 108. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Tsuruma, K.; Takata, M.; Shimazawa, M.; Hara, H. Glycoprotein Nonmetastatic Melanoma Protein B Extracellular Fragment Shows Neuroprotective Effects and Activates the PI3K/Akt and MEK/ERK Pathways via the Na+/K+-ATPase. Sci. Rep. 2016, 6, 23241. [Google Scholar] [CrossRef] [Green Version]
- Moloney, E.B.; Moskites, A.; Ferrari, E.J.; Isacson, O.; Hallett, P.J. The Glycoprotein GPNMB Is Selectively Elevated in the Substantia of Parkinson’s Disease Patients and Increases after Lysosomal. Neurobiol. Dis. 2018, 120, 1. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Alboslemy, T.; Safadi, F.; Kim, M.H. Glycoprotein Nonmelanoma Clone B Regulates the Crosstalk between Macrophages and Mesenchymal Stem Cells toward Wound Repair. J. Investig. Dermatol. 2018, 138, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zhuo, H.; Ouyang, H.; Liu, Y.; Yuan, F.; Sun, L.; Liu, F.; Liu, H. Glycoprotein Non-Metastatic Melanoma Protein b (Gpnmb) Is Highly Expressed in Macrophages of Acute Injured Kidney and Promotes M2 Macrophages Polarization. Cell Immunol. 2017, 316, 53–60. [Google Scholar] [CrossRef]
- Manochkumar, J.; Doss, C.G.P.; El-Seedi, H.R.; Efferth, T.; Ramamoorthy, S. The Neuroprotective Potential of Carotenoids in Vitro and in Vivo. Phytomedicine 2021, 91, 153676. [Google Scholar] [CrossRef]
- Calderaro, A.; Patanè, G.T.; Tellone, E.; Barreca, D.; Ficarra, S.; Misiti, F.; Laganà, G. The Neuroprotective Potentiality of Flavonoids on Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 14835. [Google Scholar] [CrossRef]
- Asadbegi, M.; Komaki, H.; Faraji, N.; Taheri, M.; Safari, S.; Raoufi, S.; Kourosh-Arami, M.; Golipoor, Z.; Komaki, A. Effectiveness of Coenzyme Q10 on Learning and Memory and Synaptic Plasticity Impairment in an Aged Aβ-Induced Rat Model of Alzheimer’s Disease: A Behavioral, Biochemical, and Electrophysiological Study. Psychopharmacology 2023, 240, 951–967. [Google Scholar] [CrossRef]
- Nazari, L.; Komaki, S.; Salehi, I.; Raoufi, S.; Golipoor, Z.; Kourosh-Arami, M.; Komaki, A. Investigation of the Protective Effects of Lutein on Memory and Learning Using Behavioral Methods in a Male Rat Model of Alzheimer’s Disease. J. Funct. Foods 2022, 99, 105319. [Google Scholar] [CrossRef]
- Rehli, M.; Niller, H.H.; Ammon, C.; Langmann, S.; Schwarzfischer, L.; Andreesen, R.; Krause, S.W. Transcriptional Regulation of CHI3L1, a Marker Gene for Late Stages of Macrophage Differentiation. J. Biol. Chem. 2003, 278, 44058–44067. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, R.B.; Emery, J.G.; Connor, J.R.; Dodds, R.; Lysko, P.G.; Rosenberg, M. Induction and Expression of Human Cartilage Glycoprotein 39 in Rheumatoid Inflammatory and Peripheral Blood Monocyte-Derived Macrophages. Exp. Cell Res. 1997, 237, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Nicoll, J.A.R.; Holmes, C. Microglia in Neurodegenerative Disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef]
- Colton, C.A.; Mott, R.T.; Sharpe, H.; Xu, Q.; Van Nostrand, W.E.; Vitek, M.P. Expression Profiles for Macrophage Alternative Activation Genes in AD and in Mouse Models of AD. J. Neuroinflamm. 2006, 3, 27. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The Diagnosis of Dementia Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The Diagnosis of Mild Cognitive Impairment Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissmann, R.; Hüttenrauch, M.; Kacprowski, T.; Bouter, Y.; Pradier, L.; Bayer, T.A.; Kuss, A.W.; Wirths, O. Gene Expression Profiling in the APP/PS1KI Mouse Model of Familial Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 50, 397–409. [Google Scholar] [CrossRef]
- Murata, K.; Yoshino, Y.; Tsuruma, K.; Moriguchi, S.; Oyagi, A.; Tanaka, H.; Ishisaka, M.; Shimazawa, M.; Fukunaga, K.; Hara, H. The Extracellular Fragment of GPNMB (Glycoprotein Nonmelanosoma Protein B, Osteoactivin) Improves Memory and Increases Hippocampal GluA1 Levels in Mice. J. Neurochem. 2015, 132, 583–594. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s Disease. J. Cell Biol. 2018, 217, 459. [Google Scholar] [CrossRef]
- Aichholzer, F.; Klafki, H.-W.W.; Ogorek, I.; Vogelgsang, J.; Wiltfang, J.; Scherbaum, N.; Weggen, S.; Wirths, O. Evaluation of Cerebrospinal Fluid Glycoprotein NMB (GPNMB) as a Potential Biomarker for Alzheimer’s Disease. Alzheimer’s Res. Ther. 2021, 13, 94. [Google Scholar] [CrossRef]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105, 975. [Google Scholar] [CrossRef]
- Wang, H.; Dey, K.K.; Chen, P.C.; Li, Y.; Niu, M.; Cho, J.H.; Wang, X.; Bai, B.; Jiao, Y.; Chepyala, S.R.; et al. Integrated Analysis of Ultra-Deep Proteomes in Cortex, Cerebrospinal Fluid and Serum Reveals a Mitochondrial Signature in Alzheimer’s Disease. Mol. Neurodegener. 2020, 15, 43. [Google Scholar] [CrossRef]
- Satoh, J.-I.; Kino, Y.; Yanaizu, M.; Ishida, T.; Saito, Y. Microglia Express GPNMB in the Brains of Alzheimer’s Disease and Nasu-Hakola Disease. Intractable Rare Dis. Res. 2019, 8, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Wuolikainen, A.; Wu, J.; Öhman, A.; Wingsle, G.; Moritz, T.; Andersen, P.M.; Forsgren, L.; Trupp, M. Targeted Multiple Reaction Monitoring Analysis of CSF Identifies UCHL1 and GPNMB as Candidate Biomarkers for ALS. J. Mol. Neurosci. 2019, 69, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeckl, P.; Weydt, P.; Thal, D.R.; Weishaupt, J.H.; Ludolph, A.C.; Otto, M. Proteomics in Cerebrospinal Fluid and Spinal Cord Suggests UCHL1, MAP2 and GPNMB as Biomarkers and Underpins Importance of Transcriptional Pathways in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2020, 139, 119–134. [Google Scholar] [CrossRef]
- Budge, K.M.; Neal, M.L.; Richardson, J.R.; Safadi, F.F. Transgenic Overexpression of GPNMB Protects against MPTP-Induced Neurodegeneration. Mol. Neurobiol. 2020, 57, 2920–2933. [Google Scholar] [CrossRef]
- Mavroudis, I.; Chowdhury, R.; Petridis, F.; Karantali, E.; Chatzikonstantinou, S.; Balmus, I.M.; Luca, I.S.; Ciobica, A.; Kazis, D. YKL-40 as a Potential Biomarker for the Differential Diagnosis of Alzheimer’s Disease. Medicina 2022, 58, 60. [Google Scholar] [CrossRef] [PubMed]
- Muszyński, P.; Groblewska, M.; Kulczyńska-Przybik, A.; Kułakowska, A.; Mroczko, B. YKL-40 as a Potential Biomarker and a Possible Target in Therapeutic Strategies of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janelidze, S.; Hertze, J.; Zetterberg, H.; Landqvist Waldö, M.; Santillo, A.; Blennow, K.; Hansson, O. Cerebrospinal Fluid Neurogranin and YKL-40 as Biomarkers of Alzheimer’s Disease. Ann. Clin. Transl. Neurol. 2016, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Laurent, C.; Buée, L.; Blum, D. Tau and Neuroinflammation: What Impact for Alzheimer’s Disease and Tauopathies? Biomed. J. 2018, 41, 21. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tested Variables | Median (Interquartile Range) | p (Kruskal–Wallis Test) | p (Dwass–Steele–Critchlow–Flinger Test) | ||||
|---|---|---|---|---|---|---|---|
| AD | MCI | Controls | AD vs. CTRL | AD vs. MCI | MCI vs. CTRL | ||
| Aβ1-42 (pg/mL) | 520 (197–994) | 802 (382–1878) | 851 (357–1235) | <0.001 | <0.001 | 0.030 | 0.947 |
| Aβ1-42/1-40 ratio | 0.03 (0.02–0.05) | 0.04 (0.03–0.07) | 0.07 (0.02–0.09) | <0.001 | <0.001 | <0.001 | 0.011 |
| Tau (pg/mL) | 710 (357–1722) | 389 (209–994) | 221 (148–414) | <0.001 | <0.001 | <0.001 | <0.001 |
| pTau181 (pg/mL) | 87.9 (46.4–209) | 57.2 (34.1–97.1) | 36.6 (24.8–55.6) | <0.001 | 0.001 | <0.001 | 0.002 |
| n | GPNMB Median | Interquartile Range | p-Value | |
|---|---|---|---|---|
| AD | Aβ(+) = 17 | 3328 | 2830–3842 | 0.028 * |
| Aβ(−) = 18 | 2559 | 2276–3397 | ||
| MCI | Aβ(+) = 12 | 2658 | 2366–3328 | 0.122 |
| Aβ(−) = 6 | 2174 | 1923–2950 |
| Tested Parameters | ROC Criteria in AD Compared to CTRL | ROC Criteria in MCI Compared to CTRL | ||||||
|---|---|---|---|---|---|---|---|---|
| AUC | SE | 95% C.I. (AUC) | p (AUC = 0.5) | AUC | SE | 95% C.I. (AUC) | p (AUC = 0.5) | |
| GPNMB | 0.869 | 0.054 | 0.763–0.975 | <0.001 | 0.787 | 0.078 | 0.633–0.941 | 0.003 |
| YKL-40 | 0.755 | 0.065 | 0.627–0.882 | 0.001 | 0.812 | 0.075 | 0.665–0.959 | <0.001 |
| Aβ1-42 | 0.836 | 0.063 | 0.713–0.958 | <0.001 | 0.531 | 0.102 | 0.33–0.731 | 0.763 |
| Aβ1-42/1-40 ratio | 0.934 | 0.052 | 0.833–0.964 | <0.001 | 0.784 | 0.078 | 0.631–0.937 | 0.003 |
| Tau | 0.995 | 0.006 | 0.984–1 | <0.001 | 0.901 | 0.052 | 0.8–1 | <0.001 |
| pTau181 | 0.989 | 0.011 | 0.967–1 | <0.001 | 0.883 | 0.057 | 0.77–0.995 | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doroszkiewicz, J.; Kulczyńska-Przybik, A.; Dulewicz, M.; Borawska, R.; Zajkowska, M.; Słowik, A.; Mroczko, B. Potential Utility of Cerebrospinal Fluid Glycoprotein Nonmetastatic Melanoma Protein B as a Neuroinflammatory Diagnostic Biomarker in Mild Cognitive Impairment and Alzheimer’s Disease. J. Clin. Med. 2023, 12, 4689. https://doi.org/10.3390/jcm12144689
Doroszkiewicz J, Kulczyńska-Przybik A, Dulewicz M, Borawska R, Zajkowska M, Słowik A, Mroczko B. Potential Utility of Cerebrospinal Fluid Glycoprotein Nonmetastatic Melanoma Protein B as a Neuroinflammatory Diagnostic Biomarker in Mild Cognitive Impairment and Alzheimer’s Disease. Journal of Clinical Medicine. 2023; 12(14):4689. https://doi.org/10.3390/jcm12144689
Chicago/Turabian StyleDoroszkiewicz, Julia, Agnieszka Kulczyńska-Przybik, Maciej Dulewicz, Renata Borawska, Monika Zajkowska, Agnieszka Słowik, and Barbara Mroczko. 2023. "Potential Utility of Cerebrospinal Fluid Glycoprotein Nonmetastatic Melanoma Protein B as a Neuroinflammatory Diagnostic Biomarker in Mild Cognitive Impairment and Alzheimer’s Disease" Journal of Clinical Medicine 12, no. 14: 4689. https://doi.org/10.3390/jcm12144689
APA StyleDoroszkiewicz, J., Kulczyńska-Przybik, A., Dulewicz, M., Borawska, R., Zajkowska, M., Słowik, A., & Mroczko, B. (2023). Potential Utility of Cerebrospinal Fluid Glycoprotein Nonmetastatic Melanoma Protein B as a Neuroinflammatory Diagnostic Biomarker in Mild Cognitive Impairment and Alzheimer’s Disease. Journal of Clinical Medicine, 12(14), 4689. https://doi.org/10.3390/jcm12144689