
Autoimmune Heparin-Induced Thrombocytopenia


Abstract
:1. Introduction
2. Methods
3. Results
3.1. Five aHIT Disorders
3.2. Delayed-Onset HIT
3.2.1. Terminology
3.2.2. Delayed-Onset HIT as an aHIT Disorder
3.2.3. Definition of Delayed-Onset HIT
3.2.4. Delayed-Onset HIT Reports with aHIT Antibodies
3.2.5. Delayed-Onset HIT Reports without Laboratory Documentation of aHIT Antibodies
3.3. Persisting (Refractory) HIT
3.4. Heparin Flush HIT
3.4.1. Heparin Flushes during Stem Cell Transplantation
3.4.2. Are Heparin Flushes Helpful in Maintaining Catheter Patency?
3.5. Fondaparinux-Associated HIT
3.5.1. Fondaparinux and Anti-PF4 Antibodies
3.5.2. Fondaparinux as a Treatment of HIT
3.5.3. Fondaparinux-Associated aHIT
3.6. Unusually Severe HIT
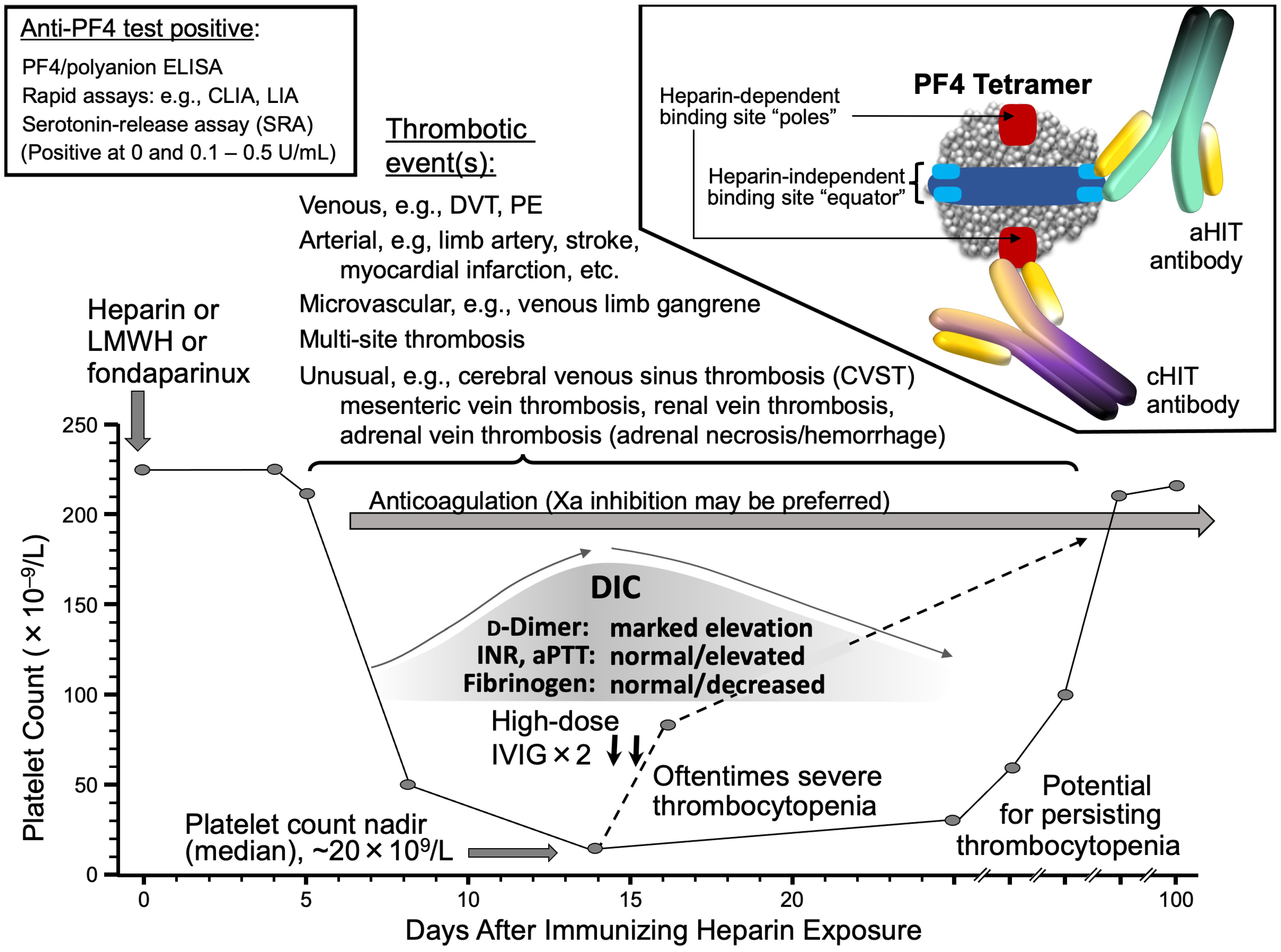
3.7. Laboratory Diagnosis
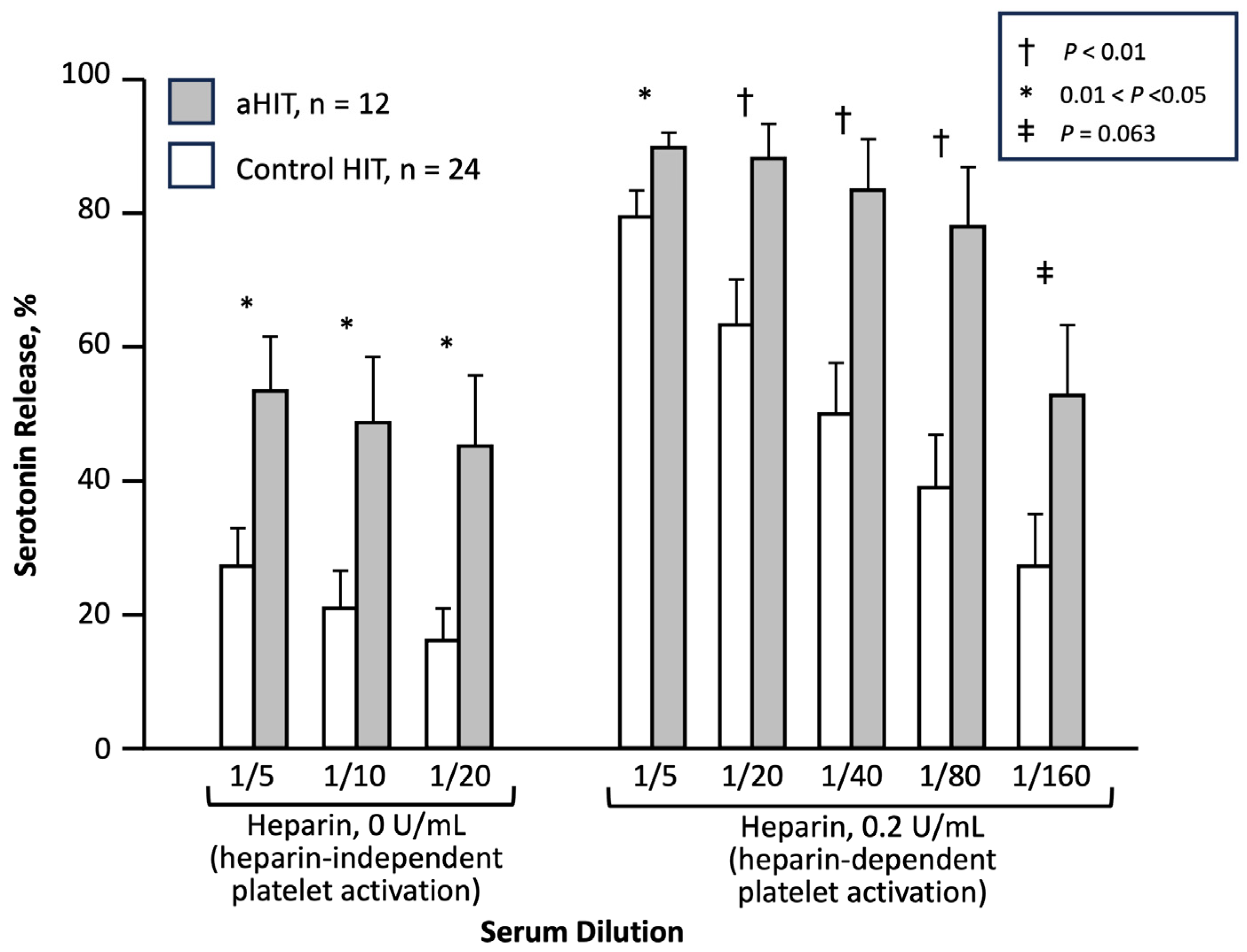
3.7.1. Heparin-Independent Platelet-Activating Properties
3.7.2. Immunoassays for aHIT Antibodies
3.7.3. Technical Challenges
3.7.4. False-Positive Detection of Cross-Reactivity
3.8. Pathogenesis of aHIT
3.8.1. aHIT Antibodies
3.8.2. Patient (Platelet) Risk Factors for aHIT
3.9. Treatment of aHIT
3.9.1. General Considerations
3.9.2. Choice of Anticoagulation
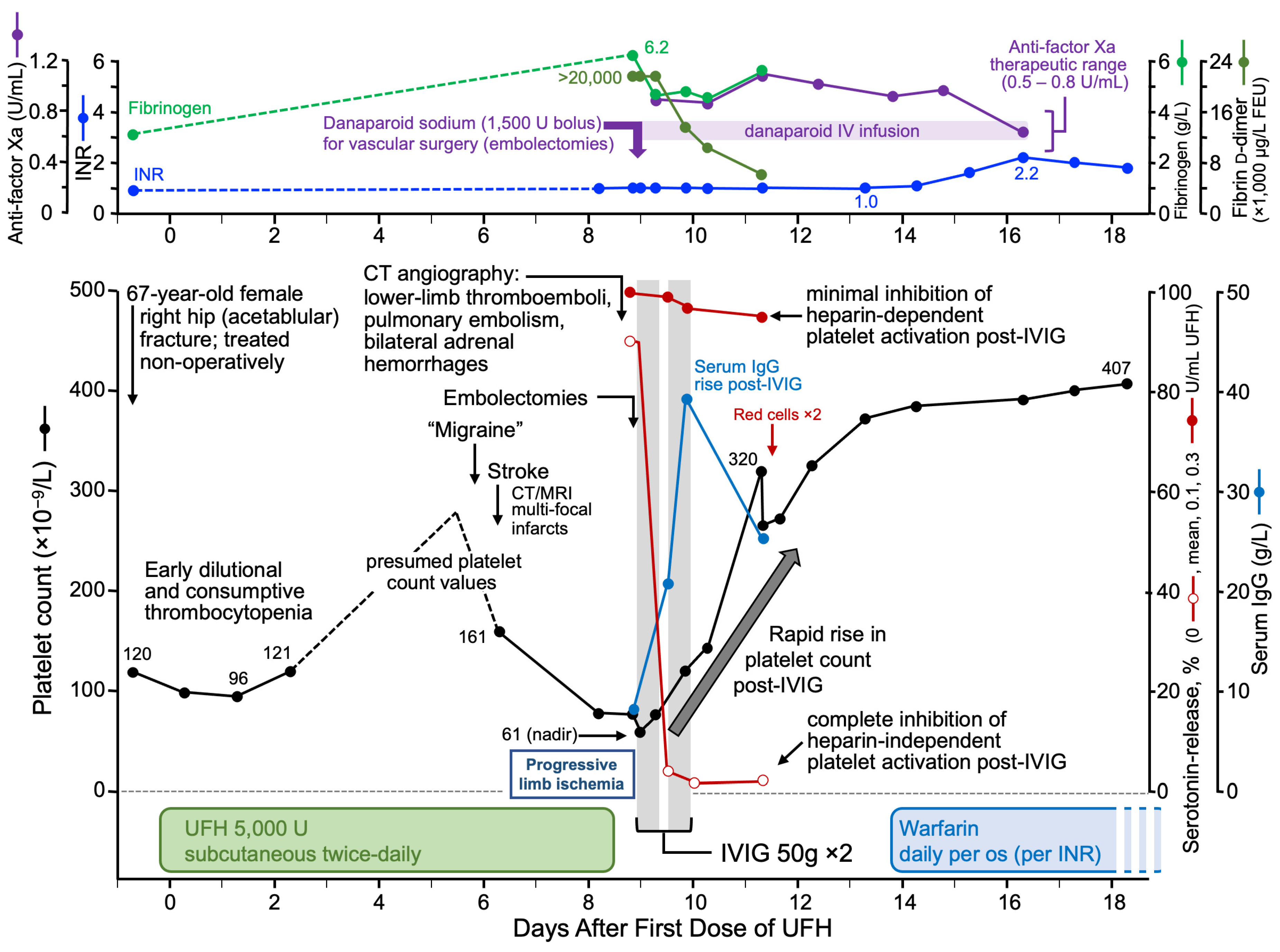
3.9.3. High-Dose IVIG
3.9.4. Therapeutic Plasma Exchange (TPE)
3.9.5. Heparin Rechallenge and Monitoring for aHIT
4. Discussion
- Autoimmune HIT (aHIT) presents with an atypical clinical picture (e.g., onset or worsening of thrombocytopenia despite stopping heparin; slow platelet count recovery after stopping heparin; multi-site thromboses; unusual sites of thrombosis [CVST, mesenteric vein, adrenal vein/adrenal necrosis/hemorrhage]; microthrombosis [e.g., venous limb gangrene, symmetrical peripheral gangrene]; overt DIC, and so forth).
- aHIT features highly pathological antibodies with heparin-independent platelet-activating properties (type 3 anti-PF4 antibodies).
- aHIT antibodies appear to recognize the heparin-binding site of PF4 (i.e., aHIT antibodies resemble VITT antibodies in this respect).
- HIT laboratories should be encouraged to perform tests that can demonstrate heparin-independent platelet-activating properties, e.g., performing the SRA (or another platelet activation assay) in the absence of heparin (0 U/mL heparin, or “buffer control”); the addition of PF4 may be required in some instances to optimize detection of the heparin-independent antibodies.
- Laboratories with a clinical and/or research interest in HIT should collaborate to determine whether standardization of the SRA or other platelet activation assays can be achieved, so as to help make the diagnosis of HIT more consistent.
- Anticoagulation: factor Xa inhibitors may have advantages over direct thrombin inhibitors (e.g., avoiding risk of APTT confounding).
- In addition to the frequent measurement of platelet counts, regular d-dimer and fibrinogen levels should also be assessed when managing a patient with aHIT, as a way to gauge whether HIT hypercoagulability is being adequately controlled (fibrinogen levels should be stable or rising, and d-dimer levels should be steadily decreasing, if a patient with aHIT is well-anticoagulated with an alternative non-heparin anticoagulant).
- Adjunct therapies: high-dose IVIG is an important option to de-escalate hypercoagulability in aHIT disorders, with a rapid platelet count increase (if observed) an indirect marker of IVIG efficacy.
- Therapeutic plasma exchange (TPE) is a potential treatment option for IVIG-refractory patients.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warkentin, T.E. Platelet-activating anti-PF4 disorders: An overview. Semin. Hematol. 2022, 59, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Warkentin, T.E. Thrombotic anti-PF4 immune disorders: HIT, VITT and beyond. Hematol. Am. Soc. Hematol. Educ. Program 2023, in press.
- Warkentin, T.E. HIT paradigms and paradoxes. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 105–117. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Selleng, K.; Warkentin, T.E. Autoimmune heparin-induced thrombocytopenia. J. Thromb. Haemost. 2017, 15, 2099–2114. [Google Scholar] [CrossRef]
- Bauman, M.M.J.; Naylor, R.M.; Wijdicks, E.F. HIT in the head: A systematic review of cerebral venous sinus thrombosis in classical and autoimmune heparin-induced thrombocytopenia. J. Thromb. Thrombolysis 2021, 52, 952–961. [Google Scholar] [CrossRef]
- May, J.; Westbrook, B.; Cuker, A. Heparin-induced thrombocytopenia: An illustrated review. Res. Pract. Thromb. Hemost. 2023, 7, 100283. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Greinacher, A. Spontaneous HIT syndrome: Knee replacement, infection, and parallels with vaccine-induced immune thrombotic thrombocytopenia. Thromb. Res. 2021, 204, 40–51. [Google Scholar] [CrossRef]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Scully, M.; Singh, D.; Lown, R.; Poles, A.; Solomon, T.; Levi, M.; Goldblatt, D.; Kotoucek, P.; Thomas, W.; Lester, W. Pathologic antibodies to platelet factor 4 after ChAdOx1 nCoV-19 vaccination. N. Engl. J. Med. 2021, 384, 2202–2211. [Google Scholar] [CrossRef]
- Pavord, S.; Scully, M.; Hunt, B.J.; Lester, W.; Bagot, C.; Craven, B.; Rampotas, A.; Ambler, G.; Makris, M. Clinical features of vaccine-induced immune thrombocytopenia and thrombosis. N. Engl. J. Med. 2021, 385, 1680–1689. [Google Scholar] [CrossRef]
- Greinacher, A.; Langer, F.; Schönborn, L.; Thiele, T.; Haddad, M.; Renné, T.; Rollin, J.; Gruel, Y.; Warkentin, T.E. Platelet-activating anti-PF4 antibodies mimicking VITT antibodies in an unvaccinated patient with monoclonal gammopathy. Haematologica 2021, 107, 1219–1221. [Google Scholar] [CrossRef] [PubMed]
- Kanack, A.J.; Shaefer, J.K.; Sridharan, M.; Splinter, N.P.; Kohlhagen, M.C.; Singh, B.; De Lorenzo, S.B.; Mauch, E.E.; Hussein, M.A.; Shaikh, M.; et al. Monoclonal gammopathy of thrombotic/thrombocytopenic significance. Blood 2023, 141, 1772–1776. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Baskin-Miller, J.; Raybould, A.L.; Sheppard, J.I.; Daka, M.; Nazy, I.; Moll, S. Adenovirus-associated thrombocytopenia, thrombosis, and VITT-like antibodies. N. Engl. J. Med. 2023, 389, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Campello, E.; Biolo, M.; Simioni, P. More on adenovirus-associated thrombocytopenia, thrombosis, and VITT-like antibodies. N. Engl. J. Med. 2023, 389, 1729–1731. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Arnold, D.M.; Sheppard, J.I.; Moore, J.C.; Kelton, J.G.; Nazy, I. Investigation of anti-PF4 versus anti-PF4/heparin reactivity using fluid-phase enzyme immunoassay for 4 anti-PF4 disorders: Classic heparin-induced thrombocytopenia (HIT), autoimmune HIT, vaccine-induced immune thrombotic thrombocytopenia, and spontaneous HIT. J. Thromb. Haemost. 2023, 21, 2268–2276. [Google Scholar] [CrossRef] [PubMed]
- Gruel, Y.; Vayne, C.; Rollin, J.; Weber, P.; Faille, D.; Bauters, A.; Macchi, L.; Alhenc-Gelas, M.; Lebreton, A.; De Maistre, E.; et al. Comparative analysis of a French prospective series of 144 patients with heparin-induced thrombocytopenia (FRIGTIH) and the literature. Thromb. Haemost. 2020, 120, 1096–1107. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Kelton, J.G. A 14-year study of heparin-induced thrombocytopenia. Am. J. Med. 1996, 101, 502–507. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Roberts, R.S.; Hirsh, J.; Kelton, J.G. An improved definition of immune heparin-induced thrombocytopenia in postoperative orthopedic patients. Arch. Intern. Med. 2003, 163, 2518–2524. [Google Scholar] [CrossRef]
- Warkentin, T.E. Heparin-induced thrombocytopenia: Pathogenesis and management. Br. J. Haematol. 2003, 121, 535–555. [Google Scholar] [CrossRef]
- Tahata, T.; Miki, S.; Kusuhara, K.; Ueda, Y.; Okita, Y.; Matsuo, S. Delayed onset of heparin induced thrombocytopenia—A case report. Nihon Kyobu Geka Gakkai Zasshi 1992, 40, 456–458. (In Japanese) [Google Scholar]
- Balendran, S.; Harrison, A.; Morel-Kopp, M.C.; Ward, C.; Forsyth, C. Delayed-onset heparin-induced thrombocytopenia complicated by arterial and venous thromboses. Intern. Med. J. 2018, 48, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Furuto, Y.; Kawamura, M.; Yamashita, J.; Yoshikawa, T.; Namikawa, A.; Isshiki, R.; Takahashi, H.; Shibuya, Y. Anti-neutrophil cytoplasmic antibody-associated vasculitis accompanied by type II heparin-induced thrombocytopenia resulting in asymptomatic cerebral infarction: A case report. BMC Nephrol. 2021, 22, 220. [Google Scholar] [CrossRef] [PubMed]
- Seiler, J.A.; Durrani, A.K.; Ahmeti, M. A case of argatroban refractory heparin induced thrombocytopenia and thrombosis. Am. Surg. 2023, 89, 3574–3575. [Google Scholar] [CrossRef] [PubMed]
- Omran, A.S.; Karimi, A.; Ahmadi, H.; Yazdanifard, P. Delayed-onset heparin-induced thrombocytopenia presenting with multiple arteriovenous thromboses: Case report. J. Med. Case Rep. 2007, 1, 131. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, G.R.; Dixon, R.H.; Silver, D. Heparin-induced thrombocytopenia with thrombotic and hemorrhagic manifestations. Surg. Gynecol. Obstet. 1973, 136, 409–416. [Google Scholar]
- Babcock, R.B.; Dumper, C.W.; Scharfman, W.B. Heparin-induced immune thrombocytopenia. N. Engl. J. Med. 1976, 295, 237–241. [Google Scholar] [CrossRef]
- Green, D.; Harris, K.; Reynolds, N.; Roberts, M.; Patterson, R. Heparin immune thrombocytopenia: Evidence for a heparin-platelet complex as the antigenic determinant. J. Lab. Clin. Med. 1978, 91, 167–175. [Google Scholar]
- Nelson, J.C.; Lerner, R.G.; Goldstein, R.; Cagin, N.A. Heparin-induced thrombocytopenia. Arch. Intern. Med. 1978, 138, 548–552. [Google Scholar] [CrossRef]
- Trowbridge, A.A.; Caraveo, J.; Green, J.B.; Amaral, B.; Stone, M.J. Heparin-related immune thrombocytopenia. Studies of antibody-heparin specificity. Am. J. Med. 1978, 65, 277–283. [Google Scholar] [CrossRef]
- Wahl, T.O.; Lipschitz, D.A.; Stechschulte, D.J. Thrombocytopenia associated with antiheparin antibody. JAMA 1978, 240, 2560–2562. [Google Scholar] [CrossRef]
- Cimo, P.L.; Moake, J.L.; Weinger, R.S.; Ben-Menachem, Y.B.; Khalil, K.G. Heparin-induced thrombocytopenia: Association with a platelet aggregating factor and arterial thromboses. Am. J. Hematol. 1979, 6, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Towne, J.B.; Bernhard, V.M.; Hussey, C.; Garancis, J.C. White clot syndrome. Peripheral vascular complications of heparin use. Arch. Surg. 1979, 114, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Hussey, C.V.; Bernhard, V.M.; McLean, M.R.; Fobian, J.E. Heparin induced platelet aggregation; in vitro confirmation of thrombotic complications associated with heparin therapy. Ann. Clin. Lab. Sci. 1979, 9, 487–493. [Google Scholar] [PubMed]
- Cines, D.B.; Kaywin, P.; Bina, M.; Tomaski, A.; Schreiber, A.D. Heparin-associated thrombocytopenia. N. Engl. J. Med. 1980, 303, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Chong, B.H.; Pitney, W.R.; Castaldi, P.A. Heparin-induced thrombocytopenia: Association of thrombotic complications with heparin-dependent IgG antibody that induces thromboxane synthesis in platelet aggregation. Lancet 1982, 2, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Van der Weyden, M.B.; Hunt, H.; McGrath, K.; Fawcett, T.; Fitzmaurice, A.; Sawers, R.J.; Rosengarten, D.S. Delayed-onset heparin-induced thrombocytopenia. A potentially malignant disorder. Med. J. Aust. 1983, 2, 132–135. [Google Scholar] [CrossRef]
- Chong, B.H.; Berndt, M.C. Heparin-induced thrombocytopenia. Blut 1989, 58, 53–57. [Google Scholar] [CrossRef]
- Warkentin, T.E. History of heparin-induced thrombocytopenia. In Heparin-Induced Thrombocytopenia, 5th ed.; Warkentin, T.E., Greinacher, A., Eds.; CRC Press: Boca Raton, FL, USA, 2013; pp. 1–23. ISBN 9781841848600. [Google Scholar]
- Guay, D.R.; Richard, A. Heparin-induced thrombocytopenia–association with a platelet aggregating factor and cross-sensitivity to bovine and porcine heparin. Drug Intell. Clin. Pharm. 1984, 18, 398–401. [Google Scholar] [CrossRef]
- Gouault-Heilmann, M.; Huet, Y.; Adnot, S.; Contant, G.; Bonnet, F.; Intrator, L.; Payen, D.; Levent, M. Low molecular weight heparin fractions as an alternative therapy in heparin-induced thrombocytopenia. Haemostasis 1987, 17, 134–140. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Kelton, J.G. Delayed-onset heparin-induced thrombocytopenia and thrombosis. Ann. Intern. Med. 2001, 135, 502–506. [Google Scholar] [CrossRef]
- Hirsh, J.; Bauer, K.A.; Donati, M.B.; Gould, M.; Samama, M.M.; Weitz, J.I. Parenteral anticoagulants: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th edition). Chest 2008, 133, 141S–159S. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.; Attisha, W.K.; Drexler, A.; Francis, J.L. Delayed-onset heparin-induced thrombocytopenia. Ann. Intern. Med. 2002, 136, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E. Clinical picture of heparin-induced thrombocytopenia. In Heparin-Induced Thrombocytopenia, 2nd ed.; Warkentin, T.E., Greinacher, A., Eds.; Marcel Dekker Inc.: New York, NY, USA, 2001; pp. 43–86. ISBN 0824706587. [Google Scholar]
- Jackson, M.R.; Neilson, W.J.; Lary, M.; Baay, P.; Web, K.; Clagett, G.P. Delayed-onset heparin-induced thrombocytopenia and thrombosis after intraoperative heparin anticoagulation. Four case reports. Vasc. Endovasc. Surg. 2006, 40, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Greinacher, A.; Koster, A.; Lincoff, A.M. Treatment and prevention of heparin-induced thrombocytopenia. American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest 2008, 133, 340S–380S. [Google Scholar] [CrossRef] [PubMed]
- LaMuraglia, G.M.; Houbballah, R.; Lapasata, M. The identification and management of heparin-induced thrombocytopenia in the vascular patient. J. Vasc. Surg. 2012, 55, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Kelton, J.G. Temporal aspects of heparin-induced thrombocytopenia. N. Engl. J. Med. 2001, 344, 720–728. [Google Scholar] [CrossRef]
- Lubenow, N.; Kempf, R.; Eichner, A.; Eichler, P.; Carlsson, L.E.; Greinacher, A. Heparin-induced thrombocytopenia: Temporal pattern of thrombocytopenia in relation to initial use or reexposure to heparin. Chest 2002, 122, 37–42. [Google Scholar] [CrossRef]
- Warkentin, T.E. Heparin-induced thrombocytopenia. Curr. Hematol. Rep. 2002, 1, 63–72. [Google Scholar]
- Warkentin, T.E.; Greinacher, A. Heparin-induced thrombocytopenia and cardiac surgery. Ann. Thorac. Surg. 2003, 76, 2121–2131. [Google Scholar] [CrossRef]
- Warkentin, T.E. Agents for the treatment of heparin-induced thrombocytopenia. Hematol. Oncol. Clin. N. Am. 2010, 24, 755–775. [Google Scholar] [CrossRef]
- Warkentin, T.E. Ischemic limb gangrene with pulses. N. Engl. J. Med. 2015, 373, 642–655. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Linkins, L.A. Heparin-induced thrombocytopenia: Real-world issues. Semin. Thromb. Hemost. 2011, 37, 653–663. [Google Scholar] [CrossRef]
- Mian, H.; Warkentin, T.E.; Sheppard, J.I.; MacDonald, A.; Linkins, L.A.; Benger, A.; Foley, R. Autoimmune HIT due to apheresis catheter heparin flushes for stem cell harvesting before autotransplantation for myeloma. Blood 2017, 130, 1679–1682. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Jones, C.G.; Pechauer, S.M.; Curtis, B.R.; Bougie, D.W.; Irani, M.S.; Bryant, B.J.; Alperin, J.B.; Deloughery, T.G.; Mulvey, K.P.; et al. IVIg for treatment of severe refractory heparin-induced thrombocytopenia. Chest 2017, 152, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Sheppard, J.I.; Whitlock, R.P. Temporal presentations of heparin-induced thrombocytopenia following cardiac surgery: A single-centre, retrospective cohort study. J. Thromb. Haemost. 2022, 20, 2601–2616. [Google Scholar] [CrossRef] [PubMed]
- Rollin, J.; Charuel, N.; Gruel, Y.; Billy, S.; Guéry, E.A.; May, M.A.; Pouplard, C.; Vayne, C. Variable serotonin release assay pattern and specificity of PF4-specific antibodies in HIT, and clinical relevance. J. Thromb. Haemost. 2022, 20, 2646–2655. [Google Scholar] [CrossRef]
- Kopolovic, I.; Warkentin, T.E. Progressive thrombocytopenia after cardiac surgery in a 67-year-old man. CMAJ 2014, 186, 929–933. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Pai, M.; Linkins, L.A. Direct oral anticoagulants for treatment of HIT: Update of Hamilton experience and literature review. Blood 2017, 130, 1104–1113. [Google Scholar] [CrossRef]
- Warkentin, T.E. High-dose intravenous immunoglobulin for the treatment and prevention of heparin-induced thrombocytopenia: A review. Expert Rev. Hematol. 2019, 12, 685–698. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Whitlock, R.P.; Teoh, K.H.T. Warfarin-associated multiple digital necrosis complicating heparin-induced thrombocytopenia and Raynaud’s phenomenon after aortic valve replacement for adenocarcinoma-associated thrombotic endocarditis. Am. J. Hematol. 2004, 75, 56–62. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Sheppard, J.I. Clinical sample investigation (CSI) hematology: Pinpointing the precise onset of heparin-induced thrombocytopenia (HIT). J. Thromb. Haemost. 2007, 5, 636–637. [Google Scholar] [CrossRef] [PubMed]
- Alsaleh, K.A.; Al-Nasser, S.M.; Bates, S.M.; Patel, A.; Warkentin, T.E.; Arnold, D.M. Delayed-onset HIT caused by low-molecular-weight heparin manifesting during fondaparinux prophylaxis. Am. J. Hematol. 2008, 83, 876–878. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Moore, J.C.; Vogel, S.; Sheppard, J.I.; Warkentin, N.I.; Eikelboom, J.W. The serological profile of early-onset and persisting post-cardiac surgery thrombocytopenia complicated by “true” heparin-induced thrombocytopenia. Thromb. Haemost. 2012, 107, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Sheppard, J.I. Serological investigation of patients with a previous history of heparin-induced thrombocytopenia who are reexposed to heparin. Blood 2014, 123, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Safyan, E.L.; Linkins, L.A. Heparin-induced thrombocytopenia presenting as bilateral adrenal hemorrhages. N. Engl. J. Med. 2015, 372, 492–494. [Google Scholar] [CrossRef]
- Warkentin, T.E. Heparin-induced thrombocytopenia in critically ill patients. Semin. Thromb. Hemost. 2015, 41, 49–60. [Google Scholar] [CrossRef]
- Tvito, A.; Bakchoul, T.; Rowe, J.M.; Greinacher, A.; Ganzel, C. Severe and persistent heparin-induced thrombocytopenia despite fondaparinux treatment. Am. J. Hematol. 2015, 90, 675–678. [Google Scholar] [CrossRef]
- Tardy-Poncet, B.; Piot, M.; Montmartin, A.; Burdier, A.; Chalayer, E. Delayed-onset heparin-induced thrombocytopenia without thrombosis in a patient receiving postoperative thromboprophylaxis with rivaroxaban. Thromb. Haemost. 2015, 114, 652–654. [Google Scholar] [CrossRef]
- Warkentin, T.E. Heparin-induced thrombocytopenia. Curr. Opin. Crit. Care 2015, 21, 576–585. [Google Scholar] [CrossRef]
- Bakchoul, T.; Jouni, R.; Warkentin, T.E. Protamine (heparin)-induced thrombocytopenia: A review of the serological and clinical features associated with anti-protamine/heparin antibodies. J. Thromb. Haemost. 2016, 14, 1685–1695. [Google Scholar] [CrossRef]
- Ezekwudo, D.E.; Chacko, R.; Gbadamosi, B.; Batool, S.; Gaikazian, S.; Warkentin, T.E.; Sheppard, J.I.; Jaiyesimi, I. Apixaban for treatment of confirmed heparin-induced thrombocytopenia: A case report and review of literature. Exp. Hematol. Oncol. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Nero, J.; Araneta, P.; Warkentin, T.; Moodley, O. Severe autoimmune LMWH-induced thrombocytopenia presenting with aortic thromboses, adrenal hemorrhage and pulmonary embolism: Response to high-dose intravenous immunoglobulin. Can. J. Gen. Intern. Med. 2018, 13, 29–34. [Google Scholar] [CrossRef]
- Kuitunen, A.; Sinisalo, M.; Vahtera, A.; Hiltunen, L.; Javela, K.; Laine, O. Autoimmune heparin-induced thrombocytopenia of delayed-onset: A clinical challenge. Transfusion 2018, 58, 2757–2760. [Google Scholar] [CrossRef] [PubMed]
- Arcinas, L.A.; Manji, R.A.; Hrymak, C.; Dao, V.; Sheppard, J.I.; Warkentin, T.E. Autoimmune heparin-induced thrombocytopenia and venous limb gangrene following aortic dissection repair: In vitro and in vivo effects of intravenous immunoglobulin. Transfusion 2019, 59, 1924–1933. [Google Scholar] [CrossRef]
- Bakchoul, T.; Borst, O.; Riessen, R.; Lucic, J.; Gawaz, M.; Althaus, K.; Aidery, P. Autoimmune heparin-induced thrombocytopenia after transcatheter aortic valve implantation: Successful treatment with adjunct high-dose intravenous immunoglobulin. TH Open 2019, 3, e200–e202. [Google Scholar] [CrossRef]
- Roberge, G.; Tritschler, T.; MacGillivray, C.; Dufresne, L.; Nagpal, S.K.; Scarvelis, D. Persisting autoimmune heparin-induced thrombocytopenia after elective abdominal aortic aneurysm repair: A case report. J. Thromb. Thrombolysis 2020, 50, 674–677. [Google Scholar] [CrossRef]
- Sheridan, D.; Carter, C.; Kelton, J.G. A diagnostic test for heparin-induced thrombocytopenia. Blood 1986, 67, 27–30. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Arnold, D.M.; Nazi, I.; Kelton, J.G. The platelet serotonin-release assay. Am. J. Hematol. 2015, 90, 564–572. [Google Scholar] [CrossRef]
- Falk, G.; Winans, C.G.; Bowens, K.; Bougie, D.W.; Curtis, B.R.; Aster, R.H. An unexpected development after surgery—Post-transfusion purpura! Am. J. Hematol. 2016, 91, 848–851. [Google Scholar] [CrossRef]
- Smythe, M.A.; Stephens, J.L.; Mattson, J.C. Delayed-onset heparin-induced thrombocytopenia. Ann. Emerg. Med. 2005, 45, 417–419. [Google Scholar] [CrossRef]
- Warkentin, T.E. Should vitamin K be administered when HIT is diagnosed after administration of coumarin? J. Thromb. Haemost. 2006, 4, 894–896. [Google Scholar] [CrossRef] [PubMed]
- Doucette, K.; DeStefano, C.B.; Jain, N.A.; Cruz, A.L.; Malkovska, V.; Fitzpatrick, K. Treatment of refractory delayed onset heparin-induced thrombocytopenia after thoracic endovascular aortic repair with intravenous immunoglobulin (IVIG). Res. Pract. Thromb. Haemost. 2017, 1, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Park, B.D.; Kumar, M.; Nagalla, S.; De Simone, N.; Aster, R.H.; Padmanabhan, A.; Sarode, R.; Rambally, S. Intravenous immunoglobulin as an adjunct therapy in persisting heparin-induced thrombocytopenia. Transfus. Apher. Sci. 2018, 57, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Frame, J.N.; Mulvey, K.P.; Phares, J.C.; Anderson, M.J. Correction of severe heparin-associated thrombocytopenia with intravenous immunoglobulin. Ann. Intern. Med. 1989, 111, 946–947. [Google Scholar] [CrossRef]
- Castaman, G.; Ruggeri, M.; Girardello, R.; Rodeghiero, F. An unusually prolonged case of heparin-induced thrombocytopenia and disseminated intravascular coagulation. Haematologica 1992, 77, 174–176. [Google Scholar]
- Warkentin, T.E.; Bernstein, R.A. Delayed-onset heparin-induced thrombocytopenia and cerebral thrombosis after a single injection of unfractionated heparin. N. Engl. J. Med. 2003, 348, 1067–1069. [Google Scholar] [CrossRef]
- Shaw, M.R.; Spencer, J.P. Heparin-induced thrombocytopenia beginning after discontinuation of heparin. J. Am. Board Fam. Pract. 2003, 16, 148–150. [Google Scholar] [CrossRef]
- Ayala, E.; Rosado, M.F.; Morgensztern, D.; Kharfan-Dabaja, M.A.; Byrnes, J.J. Heparin-induced thrombocytopenia presenting with thrombosis of multiple saphenous veins grafts and myocardial infarction. Am. J. Hematol. 2004, 76, 383–385. [Google Scholar] [CrossRef]
- Cormack, G.M.; Kaufman, L.J. Severe heparin-induced thrombocytopenia: When the obvious is not obvious, a case report. J. Med. Case Rep. 2007, 1, 13. [Google Scholar] [CrossRef]
- Greinacher, A.; Warkentin, T.E. The direct thrombin inhibitor hirudin. Thromb. Haemost. 2008, 99, 819–829. [Google Scholar] [CrossRef]
- Kumar, M.; Abrina, V.M.; Chittimireddy, S. Pulmonary embolism caused by delayed heparin-induced thrombocytopenia in a patient who received prophylactic LMWH. Am. J. Case Rep. 2012, 13, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Kinoshita, H.; Sugano, M.; Kurobe, H.; Kanbara, T.; Fujimoto, E.; Kitaichi, T.; Fujita, H.; Sogabe, H.; Kitigawa, T. Delayed-onset severe heparin-induced thrombocytopenia after total arch replacement under cardiopulmonary bypass. J. Med. Investig. 2013, 60, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Schell, A.M.; Petras, M.; Szczepiorkowski, Z.M.; Ornstein, D.L. Refractory heparin induced thrombocytopenia with thrombosis (HITT) treated with therapeutic plasma exchange and rituximab as adjuvant therapy. Transfus. Apher. Sci. 2013, 49, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Casan, J.M.L.; Grigoriadis, G.; Chan, N.; Chunilal, S. Rivaroxaban in treatment refractory heparin-induced thrombocytopenia. BMJ Case Rep. 2016, 2016, bcr2016216110. [Google Scholar] [CrossRef]
- Horlait, G.; Minet, V.; Mullier, F.; Michaux, I. Persistent heparin-induced thrombocytopenia: Danaparoid cross-reactivity or delayed-onset heparin-induced thrombocytopenia? A case report. Blood Coagul. Fibrinolysis 2017, 28, 193–197. [Google Scholar] [CrossRef]
- Lei, B.Z.; Statzel, J.J.; Sendowski, M. Rapid and durable response to intravenous immunoglobulin in delayed heparin-induced thrombocytopenia. Transfusion 2017, 57, 919–923. [Google Scholar] [CrossRef]
- Azimov, M.B.; Slater, E.D. Persistent heparin-induced thrombocytopenia treated with IVIg. Chest 2017, 152, 679–680. [Google Scholar] [CrossRef]
- Ibrahim, I.F.; Rice, L. Intravenous immunoglobulin for heparin-induced thrombocytopenia. Chest 2017, 152, 906–907. [Google Scholar] [CrossRef]
- Gleichgerrcht, E.; Lim, M.Y.; Turan, T.N. Cerebral venous sinus thrombosis due to low-molecular-weight heparin-induced thrombocytopenia. Neurologist 2017, 22, 241–244. [Google Scholar] [CrossRef]
- Gonzales, M.; Pipalia, A.; Weil, A. Refractory heparin-induced thrombocytopenia with cerebral venous sinus thrombosis treated with IVIg, steroids, and a combination of anticoagulants: A case report. J. Investig. Med. High Impact Case Rep. 2019, 7, 2324709619832324. [Google Scholar] [CrossRef]
- Ramachandran, P.; Farag, F.; Morcus, R.; Gottlieb, V. Autoimmune heparin-induced thrombocytopenia: Treatment obstacles and challenging length of stay. Am. J. Case Rep. 2019, 20, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Newton, F.; Glaser, K.; Reeves, J.; Sheperd, L.; Ray, B. Refractory heparin-induced thrombocytopenia in a patient with subarachnoid hemorrhage—A clinical conundrum. Neurohospitalist 2021, 11, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.M.J.; Naylor, R.M.; Santilli, A.R.; Wijdicks, E.F. Delayed-onset heparin-induced thrombocytopenia with cerebral venous sinus thrombosis following total knee arthroplasty: Case report. Neurohospitalist 2022, 12, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Blanc-Vannet, S.; Vautrin, E.; Piliero, N. Massive device thrombosis, multiple embolisms, and delayed-onset heparin-induced thrombocytopenia: A rare but severe complication of patent foramen ovale closure. Eur. Heart J. 2022, 43, 2425. [Google Scholar] [CrossRef]
- Chittal, A.R.; Kumar, A.; Rao, S.J.; Lakra, P.; Nacu, N. Arterial and venous thrombosis from delayed-onset heparin-induced thrombocytopenia. J. Community Hosp. Intern. Med. Perspect. 2022, 12, 102–106. [Google Scholar] [CrossRef]
- Abu Kar, S.; Kaur, A.; Khan, A.M.; Bloomfield, D. Early utilization of intravenous immunoglobulin in heparin-induced thrombocytopenia for limb salvaging purposes. Cureus 2022, 14, e23202. [Google Scholar] [CrossRef]
- Naji, F.S.; Shafie, M.; Issaiy, M.; Jalalabadi, N.Z.; Parsa, S. Delayed-onset heparin-induced thrombocytopenia complicated with saddle embolus. Clin. Case Rep. 2022, 10, e6085. [Google Scholar] [CrossRef]
- Deen, I.U.; Jha, S.A.; Mustafa, S. A case report and literature review on argatroban refractory heparin-induced thrombocytopenia. J. Community Hosp. Intern. Med. Perspect. 2022, 12, 102–105. [Google Scholar] [CrossRef]
- Makita, N.; Ohara, T.; Tsuji, Y.; Ueda, T.; Nakamura, T.; Mizuno, T.; Makino, M. Early high-dose intravenous immunoglobulin for refractory heparin-induced thrombocytopenia with stroke: Two case reports. J. Stroke Cerebrovasc. Dis. 2023, 32, 107032. [Google Scholar] [CrossRef]
- Ling, E.; Warkentin, T.E. Intraoperative heparin flushes and subsequent acute heparin-induced thrombocytopenia. Anesthesiology 1998, 89, 1567–1569. [Google Scholar] [CrossRef]
- Mayo, D.J.; Cullinane, A.M.; Merryman, P.K.; Horne, M.K. III. Serologic evidence of heparin sensitization in cancer patients receiving heparin flushes of venous access devices. Support. Care Cancer 1999, 7, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Gettings, E.M.; Brush, K.A.; Van Cott, E.M.; Hurford, W.E. Outcome of postoperative critically ill patients with heparin-induced thrombocytopenia: An observational retrospective case-control study. Crit. Care 2006, 10, R161. [Google Scholar] [CrossRef] [PubMed]
- Heeger, P.S.; Backstrom, J.T. Heparin flushes and thrombocytopenia. Ann. Intern. Med. 1986, 105, 143. [Google Scholar] [CrossRef]
- Rizzoni, W.E.; Miller, K.; Rick, M.; Lotze, M.T. Heparin-induced thrombocytopenia and thromboembolism in the postoperative period. Surgery 1988, 103, 470–476. [Google Scholar]
- Kadidal, V.V.; Mayo, D.J.; Horne, M.K. Heparin-induced thrombocytopenia (HIT) due to heparin flushes: A report of three cases. J. Intern. Med. 1999, 246, 325–329. [Google Scholar] [CrossRef]
- Parney, I.F.; Steinke, D.E. Heparin-induced thrombocytopenia and thrombosis following subarachnoid hemorrhage. Case report. J. Neurosurg. 2000, 93, 136–139. [Google Scholar] [CrossRef]
- Vezali, E.; Elefsiniotis, I.; Pirounaki, M.; Boltsis, N.; Paizis, V.; Moulakakis, A. Heparin-induced thrombocytopenia due to heparin flushes: Report of two cases. Int. J. Clin. Pract. 2007, 61, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Muslimani, A.A.; Ricaurte, B.; Daw, H.A. Immune heparin-induced thrombocytopenia resulting from preceding exposure to heparin catheter flushes. Am. J. Hematol. 2007, 82, 652–655. [Google Scholar] [CrossRef]
- Refaai, M.A.; Warkentin, T.E.; Axelson, M.; Matevosyan, K.; Sarode, R. Delayed-onset heparin-induced thrombocytopenia, venous thromboembolism, and cerebral venous thrombosis: A consequence of heparin “flushes”. Thromb. Haemost. 2007, 98, 1139–1140. [Google Scholar] [CrossRef]
- Chan, K.M.; Cheung, C.Y.; Chau, K.F. Heparin-induced thrombocytopenia due to heparin lock in a hemodialysis patient: A case report. Hemodial. Int. 2014, 18, 555–558. [Google Scholar] [CrossRef]
- De Bree, L.C.; Alings, A.M.; van Wijngaarden, P. Heparin-induced thrombocytopenia after ICD-lead flushing. Acta Cardiol. 2014, 69, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Tanaka, A.; Yoshioka, N.; Yokote, J. Heparin ‘flush’ induced thrombocytopenia triggered by total hip replacement: A case report. Eur. Heart J. Case Rep. 2020, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Agharazii, M.; Plamondon, I.; Lebel, M.; Douville, P.; Desmeules, S. Estimation of heparin leak into the systemic circulation after central venous catheter heparin lock. Nephrol. Dial. Transplant. 2005, 20, 1238–1240. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.P.; Cook, D.J.; Sigouin, C.S.; Warkentin, T.E. Central venous catheters and upper-extremity deep-vein thrombosis complicating immune heparin-induced thrombocytopenia. Blood 2003, 101, 3049–3051. [Google Scholar] [CrossRef]
- Tezcan, A.Z.; Tezcan, H.; Gastineau, D.A.; Armitage, J.O.; Haire, W.D. Heparin-induced thrombocytopenia after bone marrow transplantation: Report of two cases. Bone Marrow Transplant. 1994, 14, 487–490. [Google Scholar]
- Sarosiek, S.; Quillen, K.; Sloan, J.M.; Brauneis, D.; Sanchorawala, V. Heparin-induced thrombocytopenia and thrombosis during high dose melphalan and autologous stem cell transplantation. Blood 2018, 132, 755–757. [Google Scholar] [CrossRef]
- McKenzie, D.S.; Anuforo, J.; Morgan, J.; Neculiseanu, E. Successful use of intravenous immunoglobulin G to treat refractory heparin-induced thrombocytopenia with thrombosis complicating peripheral blood stem cell harvest. J. Investig. Med. High Impact Case Rep. 2018, 6, 2324709618755414. [Google Scholar] [CrossRef]
- Bavli, N.; Christensen, B.; Sarode, R.; Hofmann, S.; Ibrahim, I. Therapeutic plasma exchange in severe refractory autoimmune heparin-induced thrombocytopenia. Br. J. Haematol. 2022, 196, e44–e47. [Google Scholar] [CrossRef]
- Thein, K.Z.; Elsaim, S.A.; Ma, M.Q.; Rojas Hernandez, C.M.; Elsayem, A. Heparin-induced thrombocytopenia at the emergency department due to intermittent heparin flush in a patient undergoing stem cell transplant. Cureus 2022, 14, e31798. [Google Scholar] [CrossRef]
- Stephens, L.C.; Haire, W.D.; Tarantolo, S.; Reed, E.; Schmit-Pokorny, K.; Kessinger, A.; Klein, R. Normal saline versus heparin flush for maintaining central venous catheter patency during apheresis collection of peripheral blood stem cells (PBSC). Transfus. Sci. 1997, 18, 187–193. [Google Scholar] [CrossRef]
- Mitchell, M.D.; Anderson, B.J.; Williams, K.; Umscheid, C.A. Heparin flushing flushing and other interventions to maintain patency of central venous catheters: A systematic review. J. Adv. Nurs. 2009, 65, 2007–2021. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Wang, H.L.; Xu, B.; Yuan, Y.; Wang, X.; Zhang, Y.Y.; Ji, L.; Pan, Z.M.; Hu, Z.S. Normal saline versus heparin for patency of central venous catheters in adult patients—A systematic review and meta-analysis. Crit. Care 2017, 21, 5. [Google Scholar] [CrossRef] [PubMed]
- Kordzadeh, A.; Austin, T.; Panayiotopoulos, Y. Efficacy of normal saline in the maintenance of the arterial lines in comparison to heparin flush: A comprehensive review of the literature. J. Vasc. Access 2014, 15, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Mudgal, S.K.; Gaur, R.; Sharma, R.; Sharma, M.; Thakur, K. Heparin flush vs. normal saline flush to maintain the patency of central venous catheter among adult patients: A systematic review and meta-analysis. J. Family Med. Prim. Care 2019, 8, 2779–2792. [Google Scholar] [CrossRef]
- Schallom, M.E.; Prentice, D.; Sona, C.; Micek, S.T.; Skrupky, L.P. Heparin or 0.9% sodium chloride to maintain central venous catheter patency: A randomized trial. Crit. Care Med. 2012, 40, 1820–1826. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Ling, E.; Ho, A.; Sheppard, J.I. “Incidental” unfractionated heparin (UFH) vs. normal saline (NS) flushes for intraoperative invasive catheters and the frequency of formation of heparin-induced thrombocytopenia IgG antibodies (HIT-IgG): A randomized, controlled trial [abstr]. Blood 1998, 92 (Suppl. 1), 91b. [Google Scholar]
- Warkentin, T.E.; Eikelboom, J.W. Fondaparinux and other emerging anticoagulants to treat heparin-induced thrombocytopenia. In Heparin-Induced Thrombocytopenia, 5th ed.; Warkentin, T.E., Greinacher, A., Eds.; CRC Press: Boca Raton, FL, USA, 2013; pp. 489–519. ISBN 13 978-1841848600. [Google Scholar]
- Warkentin, T.E. Fondaparinux: Does it cause HIT? can it treat HIT? Expert Rev. Hematol. 2010, 3, 567–581. [Google Scholar] [CrossRef]
- Visentin, G.P. Heparin-induced thrombocytopenia: Molecular pathogenesis. Thromb. Haemost. 1999, 82, 448–456. [Google Scholar] [CrossRef]
- Mikhailov, D.; Young, H.C.; Linhardt, R.J.; Mayo, K.H. Heparin dodecasaccharide binding to platelet factor-4 and growth-related protein-α: Induction of a partially folded state and implications for heparin-induced thrombocytopenia. J. Biol. Chem. 1999, 274, 25317–25329. [Google Scholar] [CrossRef]
- Savi, P.; Chong, B.H.; Greinacher, A.; Gruel, Y.; Kelton, J.G.; Warkentin, T.E.; Eichler, P.; Meuleman, D.; Petitou, M.; Herault, J.P.; et al. Effect of fondaparinux on platelet activation in the presence of heparin-dependent antibodies: A blinded comparative multicenter study with unfractionated heparin. Blood 2005, 105, 139–144. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Davidson, B.L.; Büller, H.R.; Gallus, A.; Gent, M.; Lensing, A.W.A.; Piovella, F.; Prins, M.H.; Segers, A.E.M.; Kelton, J.G. Prevalence and risk of preexisting heparin-induced thrombocytopenia antibodies in patients with acute VTE. Chest 2011, 140, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Cook, R.J.; Marder, V.J.; Sheppard, J.I.; Moore, J.C.; Eriksson, B.I.; Greinacher, A.; Kelton, J.G. Anti-platelet factor 4/heparin antibodies in orthopedic surgery patients receiving antithrombotic prophylaxis with fondaparinux or enoxaparin. Blood 2005, 106, 3791–3796. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Sheppard, J.I.; Sun, J.C.J.; Jung, H.; Eikelboom, J.W. Anti-PF4/heparin antibodies and venous graft occlusion in postcoronary artery bypass surgery patients randomized to postoperative unfractionated heparin or fondaparinux thromboprophylaxis. J. Thromb. Haemost. 2013, 11, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Pouplard, C.; Couvret, C.; Regina, S.; Gruel, Y. Development of antibodies specific to polyanion modified platelet factor 4 during treatment with fondaparinux. J. Thromb. Haemost. 2005, 3, 2813–2815. [Google Scholar] [CrossRef]
- Greinacher, A.; Gopinadhan, M.; Guenther, J.U.; Omer-Adam, M.A.; Strobel, U.; Warkentin, T.E.; Papastavrou, G.; Weitschies, W.; Helm, C.A. Close approximation of two platelet factor 4 tetramers by charge neutralization forms the antigens recognized by HIT antibodies. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2386–2393. [Google Scholar] [CrossRef]
- Chen, L.Y.; Khan, N.; Lindenbauer, A.; Nguyen, T.H. When will fondaparinux induce thrombocytopenia? Bioconjug. Chem. 2022, 33, 1574–1583. [Google Scholar] [CrossRef]
- Greinacher, A.; Alban, S.; Omer-Adam, M.A.; Weitschies, W.; Warkentin, T.E. Heparin-induced thrombocytopenia: A stoichiometry-based model to explain the differing immunogenicities of unfractionated heparin, low-molecular-weight heparin, and fondaparinux in different clinical settings. Thromb. Res. 2008, 122, 211–220. [Google Scholar] [CrossRef]
- Parody, R.; Oliver, A.; Souto, J.C.; Fontcuberta, J. Fondaparinux (ARIXTRA) as an alternative anti-thrombotic prophylaxis when there is hypersensitivity to low molecular weight and unfractionated heparins. Haematologica 2003, 88, ECR32. [Google Scholar]
- D’Angelo, A.; Della Valle, P.; Fattorini, A.; Luciano, C. Disappearance of anti-PF4/heparin antibodies under prolonged fondaparinux administration was observed in a patient with DVT associated with LMWH-induced thrombocytopenia. Thromb. Haemost. 2006, 95, 573–575. [Google Scholar] [CrossRef]
- Efird, L.E.; Kockler, D.R. Fondaparinux for thromboembolic treatment and prophylaxis of heparin-induced thrombocytopenia. Ann. Pharmacother. 2006, 40, 1383–1387. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Pai, M.; Sheppard, J.I.; Schulman, S.; Spyropoulos, A.C.; Eikelboom, J.W. Fondaparinux treatment of acute heparin-induced thrombocytopenia confirmed by the serotonin-release assay: A 30-month, 16-patient case series. J. Thromb. Haemost. 2011, 9, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Al-Rossaies, A.; Alkharfy, K.M.; Al-Ayoubi, F.; Al-Momen, A. Heparin-induced thrombocytopenia: Comparison between response to fondaparinux and lepirudin. Int. J. Clin. Pharm. 2011, 33, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Schindewolf, M.; Steindl, J.; Beyer-Westendorf, J.; Schellong, S.; Dohmen, P.M.; Brachmann, J.; Madlener, K.; Pötzsch, B.; Klamroth, R.; Hankowitz, J.; et al. Frequent off-label use of fondaparinux in patients with suspected heparin-induced thrombocytopenia (HIT)—Findings from the GerHIT multi-centre registry study. Thromb. Res. 2014, 134, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Schindewolf, M.; Steindl, J.; Beyer-Westendorf, J.; Schellong, S.; Dohmen, P.M.; Brachmann, J.; Madlener, K.; Pötzsch, B.; Klamroth, R.; Hankowitz, J.; et al. Use of fondaparinux off-label or approved anticoagulants for management of heparin-induced thrombocytopenia. J. Am. Coll. Cardiol. 2017, 70, 2636–2648. [Google Scholar] [CrossRef]
- Dulicek, P.; Ivanova, E.; Kostal, M.; Fiedlerova, Z.; Sadilek, P.; Hirmerova, J. Heparin-induced thrombocytopenia treated with fondaparinux: Single center experience. Int. Angiol. 2020, 39, 76–81. [Google Scholar] [CrossRef]
- Linkins, L.A.; Hu, G.; Warkentin, T.E. Systematic review of fondaparinux for heparin-induced thrombocytopenia: When there are no randomized controlled trials. Res. Pract. Thromb. Haemost. 2018, 2, 678–683. [Google Scholar] [CrossRef]
- Manji, F.; Warkentin, T.E.; Sheppard, J.I.; Lee, A. Fondaparinux cross-reactivity in heparin-induced thrombocytopenia successfully treated with high-dose intravenous immunoglobulin and rivaroxaban. Platelets 2020, 31, 124–127. [Google Scholar] [CrossRef]
- Warkentin, T.E. Clinical picture of heparin-induced thrombocytopenia (HIT) and its differentiation from non-HIT thrombocytopenia. Thromb. Haemost. 2016, 116, 813–822. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Lim, W. Can heparin-induced thrombocytopenia be associated with fondaparinux use? Reply to a rebuttal. J. Thromb. Haemost. 2008, 6, 1243–1246. [Google Scholar] [CrossRef]
- Pistulli, R.; Oberle, V.; Figulla, H.R.; Yilmaz, A.; Pfeifer, R. Fondaparinux cross-reacts with heparin antibodies in vitro in a patient with fondaparinux-related thrombocytopenia. Blood Coagul. Fibrinolysis 2011, 22, 76–78. [Google Scholar] [CrossRef]
- Sartori, M.; Cosmi, B. Failure of fondaparinux in autoimmune heparin-induced thrombocytopenia. TH Open 2020, 4, e305–e308. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Maurer, B.T.; Aster, R.H. Heparin-induced thrombocytopenia associated with fondaparinux. N. Engl. J. Med. 2007, 356, 2653–2654. [Google Scholar] [CrossRef] [PubMed]
- Rota, E.; Bazzan, M.; Fantino, G. Fondaparinux-related thrombocytopenia in a previous low-molecular-weight heparin (LMWH)-induced heparin-induced thrombocytopenia. Thromb. Haemost. 2008, 99, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Modi, C.; Satani, D.; Cervellione, K.L.; Cervantes, J.; Gintautas, J. Delayed-onset heparin-induced thrombocytopenia type-2 during fondiparinux [sic] (Arixtra) therapy. Proc. West Pharmacol. Soc. 2009, 52, 5–7. [Google Scholar] [PubMed]
- Salem, M.; Elrefai, S.; Shrit, M.A.; Warkentin, T.E. Fondaparinux thromboprophylaxis-associated heparin-induced thrombocytopenia syndrome complicated by arterial thrombotic stroke. Thromb. Haemost. 2010, 104, 1071–1072. [Google Scholar] [CrossRef] [PubMed]
- Burch, M.; Cooper, B. Fondaparinux-associated heparin-induced thrombocytopenia. Bayl. Univ. Med. Cent. Proc. 2012, 25, 13–15. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Chakraborty, A.K.; Sheppard, J.I.; Griffin, D.K. The serological profile of fondaparinux-associated heparin-induced thrombocytopenia syndrome. Thromb. Haemost. 2012, 108, 394–396. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Sheppard, J.I.; Manheim, J.C. HIT complicating fondaparinux prophylaxis: Fondaparinux-dependent platelet activation as a marker for fondaparinux-induced HIT. Thromb. Haemost. 2014, 112, 1319–1322. [Google Scholar] [CrossRef]
- Greinacher, A.; Michels, I.; Kiefel, V.; Meuller-Eckhardt, C. A rapid and sensitive test for diagnosing heparin-associated thrombocytopenia. Thromb. Haemost. 1991, 66, 734–736. [Google Scholar] [CrossRef]
- Eichler, P.; Budde, U.; Haas, S.; Kroll, H.; Loreth, R.M.; Meyer, O.; Pachmann, U.; Pötzsch, B.; Schabel, A.; Albrecht, D.; et al. First workshop for detection of heparin-induced antibodies: Validation of the heparin-induced platelet-activation test (HIPA) in comparison with a PF4/heparin ELISA. Thromb. Haemost. 1999, 81, 625–629. [Google Scholar] [CrossRef]
- Prechel, M.M.; McDonald, M.K.; Jeske, W.P.; Messmore, H.L.; Walenga, J.M. Activation of platelets by heparin-induced thrombocytopenia antibodies in the serotonin release assay is not dependent on the presence of heparin. J. Thromb. Haemost. 2005, 3, 2168–2175. [Google Scholar] [CrossRef] [PubMed]
- Prechel, M.M.; Jeske, W.P.; Walenga, J.M. Physiological changes in membrane-expressed platelet factor 4: Implications in heparin-induced thrombocytopenia. Thromb. Res. 2010, 125, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Caple, J.F.; Kottke-Marchant, K.; Miller, M.L. The effect of a heparin removal filter on platelet aggregation studies in heparin-induced thrombocytopenia. Am. J. Clin. Pathol. 1995, 103, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Socher, I.; Kroll, H.; Jorks, S.; Santoso, S.; Sachs, U.J.H. Heparin-independent activation of platelets by heparin-induced thrombocytopenia antibodies: A common occurrence. J. Thromb. Haemost. 2008, 6, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Tanaka, Y.; Okazaki, Y.; Kaburaki, J.; Ikeda, Y.; Kuwana, M. Heparin-dependent and -independent anti-platelet factor 4 autoantibodies in patients with systemic lupus erythematosus. Rheumatology 2012, 51, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Arepally, G.M.; Kamei, S.; Park, K.S.; Kamei, K.; Li, Z.Q.; Liu, W.; Siegel, D.L.; Kisiel, W.; Cines, D.B.; Poncz, M. Characterization of a murine monoclonal antibody that mimics heparin-induced thrombocytopenia antibodies. Blood 2000, 95, 1533–1540. [Google Scholar] [CrossRef]
- Pouplard, C.; Rollin, J.; Vayne, C.; Charuel, N.; Ahmadi, Z.; Alberio, L.; Azjenberg, N.; Althaus, K.; Bakchoul, T.; Chong, B.; et al. Multicentre evaluation of 5B9, a monoclonal anti-PF4/heparin IgG mimicking human IgG antibodies, as an internal quality control in HIT functional assays: Communication from the ISTH SSC Subcommittee on Platelet Immunology. J. Thromb. Haemost. 2022, 20, 252–259. [Google Scholar] [CrossRef]
- Vayne, C.; Nguyen, T.H.; Rollin, J.; Charuel, N.; Poupon, A.; Pouplard, C.; Normann, N.; Gruel, Y.; Greinacher, A. Characterization of new monoclonal PF4-specific antibodies as useful tools for studies on typical and autoimmune heparin-induced thrombocytopenia. Thromb. Haemost. 2021, 121, 322–331. [Google Scholar] [CrossRef]
- Ziporen, L.; Li, Z.Q.; Park, K.S.; Sabnekar, P.; Liu, W.Y.; Arepally, G.; Shoenfeld, Y.; Kieber-Emmons, T.; Cines, D.B.; Poncz, M. Defining an antigenic epitope on platelet factor 4 associated with heparin-induced thrombocytopenia. Blood 1998, 92, 3250–3259. [Google Scholar] [CrossRef]
- Li, Z.Q.; Liu, W.; Park, K.S.; Sachais, B.S.; Arepally, G.M.; Cines, D.B.; Poncz, M. Defining a second epitope for heparin-induced thrombocytopenia/thrombosis antibodies using KKO, a murine HIT-like monoclonal antibody. Blood 2002, 99, 1230–1236. [Google Scholar] [CrossRef]
- Huynh, A.; Arnold, D.M.; Kelton, J.G.; Smith, J.W.; Horsewood, P.; Clare, R.; Guarné, A.; Nazy, I. Characterization of platelet factor 4 amino acids that bind pathogenic antibodies in heparin-induced thrombocytopenia. J. Thromb. Haemost. 2019, 17, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Huynh, A.; Kelton, J.G.; Arnold, D.M.; Daka, M.; Nazy, I. Antibody epitopes in vaccine-induced immune thrombotic thrombocytopenia. Nature 2021, 596, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Huynh, A.; Arnold, D.M.; Michael, J.V.; Clare, R.; Smith, J.W.; Daka, M.; Ianosi-Irimie, M.; McKenzie, S.E.; Kelton, J.G.; Nazy, I. Characteristics of VITT antibodies in patients vaccinated with Ad26.COV2.S. Blood Adv. 2023, 7, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Huynh, A.; Arnold, D.M.; Ivetic, N.; Clare, R.; Hadzi-Tosev, M.; Liu, Y.; Smith, J.W.; Bissola, A.L.; Daka, M.; Kelton, J.G.; et al. Antibodies against platelet factor 4 and the risk of cerebral venous sinus thrombosis in patients with vaccine-induced immune thrombotic thrombocytopenia. J. Thromb. Haemost. 2023, 21, 2833–2843. [Google Scholar] [CrossRef]
- Schönborn, L.; Esteban, O.; Wesche, J.; Dobosz, P.; Broto, M.; Puig, S.R.; Fuhrmann, J.; Torres, R.; Serra, J.; Llevadot, R.; et al. Anti-PF4 immunothrombosis without proximate heparin or adenovirus vector vaccine exposure. Blood 2023. online ahead of print. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Sheppard, J.I.; Linkins, L.A.; Arnold, D.M.; Nazy, I. High sensitivity and specificity of an automated IgG-specific chemiluminescence immunoassay for diagnosis of HIT. Blood 2018, 132, 1345–1349. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Sheppard, J.I.; Smith, J.W.; Li, N.; Moore, J.C.; Arnold, D.M.; Nazy, I. Combination of two complementary automated rapid assays for diagnosis of heparin-induced thrombocytopenia (HIT). J. Thromb. Haemost. 2020, 18, 1435–1446. [Google Scholar] [CrossRef]
- Kanack, A.J.; Athale, J.; Leger, R.R.; Saadalla, A.; Heikal, N.M.; Chen, D.; Garcia, D.A.; Singh, R.; Pruthi, R.K.; Padmanabhan, A. “Autoimmune HIT” antibodies in diagnostic samples are a potential artifact and not associated with more severe outcomes. Blood Adv. 2023, 7, 4431–4434. [Google Scholar] [CrossRef]
- Northam, K.A.; Chen, S.L.; Stivers, A.P.; Cicci, J.D.; Hedrick, T.L.; Rollins-Raval, M.A.; Kasthuri, R.S. Impact of a multidisciplinary workflow on safety and management of patients with heparin-induced thrombocytopenia. Am. J. Health Syst. Pharm. 2021, 78, 49–59. [Google Scholar] [CrossRef]
- Greinacher, A.; Warkentin, T.E. Platelet factor 4 triggers thrombo-inflammation by bridging innate and adaptive immunity. Int. J. Lab. Hematol. 2023, 45 (Suppl. 2), 11–22. [Google Scholar] [CrossRef]
- Delcia, M.; Greinacher, A. Biophysical tools to assess the interaction of PF4 with polyanions. Thromb. Haemost. 2016, 116, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Krauel, K.; Gottschalk, K.E.; Renné, T.; Helm, C.A.; Greinacher, A.; Block, S. Characterisation of the conformational changes in platelet factor 4 induced by polyanions: Towards in vitro predictivity of antigenicity. Thromb. Haemost. 2014, 112, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Krauel, K.; Jaax, M.; Renné, T.; Helm, C.A.; Hammerschmidt, S.; Delcia, M.; Greinacher, A. Polyphosphates form antigenic complexes with platelet factor 4 (PF4) and enhance PF4-binding to bacteria. Thromb. Haemost. 2015, 114, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Greinacher, A.; Delcea, M. Quantitative description of thermodynamic and kinetic properties of the platelet factor 4/heparin bonds. Nanoscale 2015, 7, 10130–10139. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Medvedev, N.; Delcea, M.; Greinacher, A. Anti-platelet factor 4/polyanion antibodies mediate a new mechanism of autoimmunity. Nat. Commun. 2017, 8, 14945. [Google Scholar] [CrossRef]
- Kreimann, M.; Brandt, S.; Krauel, K.; Block, S.; Helm, C.A.; Weitschies, W.; Greinacher, A.; Delcea, M. Binding of anti-platelet factor 4/heparin antibodies depends on the thermodynamics of conformational changes in platelet factor 4. Blood 2014, 124, 2442–2449. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Greinacher, A. Platelet factor 4/heparin complexes present epitopes differently on solid-phase vs platelet surfaces. Blood 2017, 129, 3498–3501. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Greinacher, A. Distinct binding characteristics of pathogenic anti-platelet factor-4/polyanion antibodies to antigens coated on different substrates: A perspective on clinical application. ACS Nano 2018, 12, 12030–12041. [Google Scholar] [CrossRef]
- O’Brien, J.R.; Etherington, M.D.; Pashley, M. Intra-platelet platelet factor 4 (IP.PF4) and the heparin-mobilisable pool of PF4 in health and atherosclerosis. Thromb. Haemost. 1984, 51, 354–357. [Google Scholar] [CrossRef]
- Lambert, M.; Reznikov, A.; Grubbs, A.; Nguyen, Y.; Xiao, L.; Aplenc, R.; Rauova, L.; Poncz, M. Platelet factor 4 platelet levels are inversely correlated with steady-state platelet counts and with platelet transfusion needs in pediatric leukemia patients. J. Thromb. Haemost. 2012, 10, 1442–1446. [Google Scholar] [CrossRef]
- Cines, D.B.; Rauova, L.; Arepally, G.; Reilly, M.P.; McKenzie, S.E.; Sachais, B.S.; Poncz, M. Heparin-induced thrombocytopenia: An autoimmune disorder regulated through dynamic autoantigen assembly/disassembly. J. Clin. Apher. 2007, 22, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Pötzsch, B.; Amiral, J.; Dummel, V.; Eichner, A.; Mueller-Eckhardt, C. Heparin-associated thrombocytopenia: Isolation of the antibody and characterization of a multimolecular PF4-heparin complex as the major antigen. Thromb. Haemost. 1994, 71, 247–251. [Google Scholar] [PubMed]
- Rauova, L.; Poncz, M.; McKenzie, S.E.; Reilly, M.P.; Arepally, G.; Weisel, J.W.; Nagaswami, C.; Cines, D.B.; Sachais, B.S. Ultra large complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005, 105, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Yarovoi, S.V.; Zaitsev, S.V.; Lebedeva, T.; Rauova, L.; Poncz, M.; Arepally, G.M.; Khandelwal, S.; Stepanova, V.; Rux, A.H.; et al. Polyphosphate/platelet factor 4 complexes can mediate heparin-independent platelet activation in heparin-induced thrombocytopenia. Blood Adv. 2016, 1, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Rauova, L.; Zhai, L.; Kowalska, M.A.; Arepally, G.M.; Cines, D.B.; Poncz, M. Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis: Diagnostic and therapeutic implications. Blood 2006, 107, 2346–2353. [Google Scholar] [CrossRef]
- Nader, H.B. Characterization of a heparan sulfate and a peculiar chondroitin 4-sulfate proteoglycan from platelets: Inhibition of the aggregation process by platelet chondroitin sulfate proteoglycan. J. Biol. Chem. 1991, 266, 10518–10523. [Google Scholar] [CrossRef]
- Warkentin, T.E. Anticoagulant failure in coagulopathic patients: PTT confounding and other pitfalls. Exp. Opin. Drug Saf. 2014, 13, 25–43. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Elavathil, L.J.; Hayward, C.P.M.; Johnston, M.A.; Russett, J.I.; Kelton, J.G. The pathogenesis of venous limb gangrene associated with heparin-induced thrombocytopenia. Ann. Intern. Med. 1997, 127, 804–812. [Google Scholar] [CrossRef]
- Furugohri, T.; Sugiyama, N.; Morishima, Y.; Shibano, T. Antithrombin-dependent thrombin inhibitors, but not direct factor Xa inhibitors, enhance thrombin generation in plasma through inhibition of thrombin-thrombomodulin-protein C system. Thromb. Haemost. 2011, 106, 1076–1083. [Google Scholar] [CrossRef]
- Perzborn, E.; Heitmeier, S.; Buetehorn, U.; Laux, V. Direct thrombin inhibitors, but not the direct factor Xa inhibitor rivaroxaban, increase tissue factor-induced hypercoagulability in vitro and in vivo. J. Thromb. Haemost. 2014, 12, 1054–1065. [Google Scholar] [CrossRef]
- Warkentin, T.E. How I diagnose and manage HIT. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 143–149. [Google Scholar] [CrossRef]
- Ning, S.; Warkentin, T.E. IV immunoglobulin for autoimmune heparin-induced thrombocytopenia. Chest 2017, 152, 453–455. [Google Scholar] [CrossRef]
- Aryal, M.R.; Gosain, R.; Donato, A.; Katel, A.; Chakradhar, R.; Dhital, R.; Kouides, P.A. Effectiveness of intravenous immunoglobulin use in heparin-induced thrombocytopenia. Blood Coagul. Fibrinolysis 2020, 31, 287–292. [Google Scholar] [CrossRef]
- Onuoha, C.; Barton, K.D.; Wong, E.C.C.; Raval, J.S.; Rollins-Raval, M.A.; Ipe, T.S.; Kiss, J.E.; Boral, L.I.; Adamksi, J.; Zantek, N.D.; et al. Therapeutic plasma exchange and intravenous immune globulin in the treatment of heparin-induced thrombocytopenia. Transfusion 2020, 60, 2714–2736. [Google Scholar] [CrossRef]
- Dougherty, J.A.; Yarsley, R.L. Intravenous immune globulin (IVIG) for treatment of autoimmune heparin-induced thrombocytopenia: A systematic review. Ann. Pharmacother. 2021, 55, 198–215. [Google Scholar] [CrossRef]
- Bourguignon, A.; Arnold, D.M.; Warkentin, T.E.; Smith, J.W.; Pannu, T.; Shrum, J.M.; Al Maqrashi, Z.A.A.; Shroff, A.; Lessard, M.C.; Blais, N.; et al. Adjunct immune globulin for vaccine-induced immune thrombotic thrombocytopenia. N. Engl. J. Med. 2021, 385, 720–728. [Google Scholar] [CrossRef]
- Scutelnic, A.; Krzywicka, K.; Mbroh, J.; van de Munckhof, A.; van Kammen, S.M.; de Sousa, D.A.; Lindgren, E.; Jood, K.; Günther, A.; Hiltunen, S.; et al. Management of cerebral venous thrombosis due to adenoviral COVID-19 vaccination. Ann. Neurol. 2022, 92, 562–573. [Google Scholar] [CrossRef]
- Gabarin, N.; Arnold, D.M.; Nazy, I.; Warkentin, T.E. Treatment of vaccine-induced immune thrombotic thrombocytopenia (VITT). Semin. Hematol. 2022, 59, 89–96. [Google Scholar] [CrossRef]
- Mohanty, E.; Nazir, S.; Sheppard, J.I.; Forman, D.A.; Warkentin, T.E. High-dose intravenous immunoglobulin to treat spontaneous heparin-induced thrombocytopenia syndrome. J. Thromb. Haemost. 2019, 17, 841–844. [Google Scholar] [CrossRef]
- Hwang, S.R.; Wang, Y.; Weil, E.L.; Padmanabhan, A.; Warkentin, T.E.; Pruthi, R.K. Cerebral venous sinus thrombosis associated with spontaneous heparin-induced thrombocytopenia syndrome after total knee arthroplasty. Platelets 2021, 32, 936–940. [Google Scholar] [CrossRef]
- Jones, C.G.; Pechauer, S.M.; Curtis, B.R.; Bougie, D.W.; Aster, R.H.; Padmanabhan, A. Normal plasma IgG inhibits antibody-mediated platelet activation: Implications for therapeutic plasma exchange. Blood 2018, 131, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Anderson, J.A. How I treat patients with a history of heparin-induced thrombocytopenia. Blood 2016, 128, 348–359. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of aHIT Disorder | Concept (Definition) |
|---|---|
| Delayed-onset HIT | Platelet count fall that begins or worsens despite stopping heparin |
| Persisting (refractory) HIT | Delayed platelet count recovery despite stopping heparin (>1 week) |
| Heparin “flush” HIT | HIT that occurs with exposure only to small amounts of heparin |
| Fondaparinux-induced HIT | HIT associated with proximate exposure to fondaparinux |
| Unusually severe HIT | Marked thrombocytopenia, multiple-site thromboses, overt DIC, etc. |
| Study | Key Observations, Including Heparin-Independent Serotonin-Release (HISR) | HISR (%) |
|---|---|---|
| Case-series with controls | ||
| [41] | Greater HISR in 12 aHIT patients vs. 24 HIT controls (p < 0.05) | 50% (mean) |
| [54] | Delayed platelet count recovery: aHIT vs. cHIT (5/5 vs. 1/6; p = 0.015) | >50% |
| [55] | Higher frequency of HISR in aHIT pts vs. controls (4/4 vs. 34/100; p = 0.016) | >80% |
| [56] | 3 cases with SRA-positive refractory HIT (IVIG-responsive) | >40% PEA [low PF4] a |
| [57] | 3/3 pts with post-discharge HIT had HISR (80%, 83%, 99%) | >80% |
| [58] | aHIT: higher thrombosis rate, lower platelet nadirs, slower platelet count recovery | ≥30% |
| Relationship between platelet counts and HISR (serial blood samples) | ||
| [59] | Inverse relationship between HISR and platelet count (n = 1 pt) | >80% (peak) |
| [55] | Inverse relationship between HISR and platelet count (n = 2 pts) | >90% (peak) |
| [60] | Inverse relationship between HISR and platelet count (n = 1 pt) | >90% (peak) |
| [61] | Abrupt plt count rise and parallel decrease in HISR post-IVIG (n = 1 pt) | 90% (peak) |
| aHIT pts (case reports) with laboratory evidence of HISR | ||
| [52] | Delayed/persisting with DIC, microvascular ischemia, nadir = 2 | >80% b |
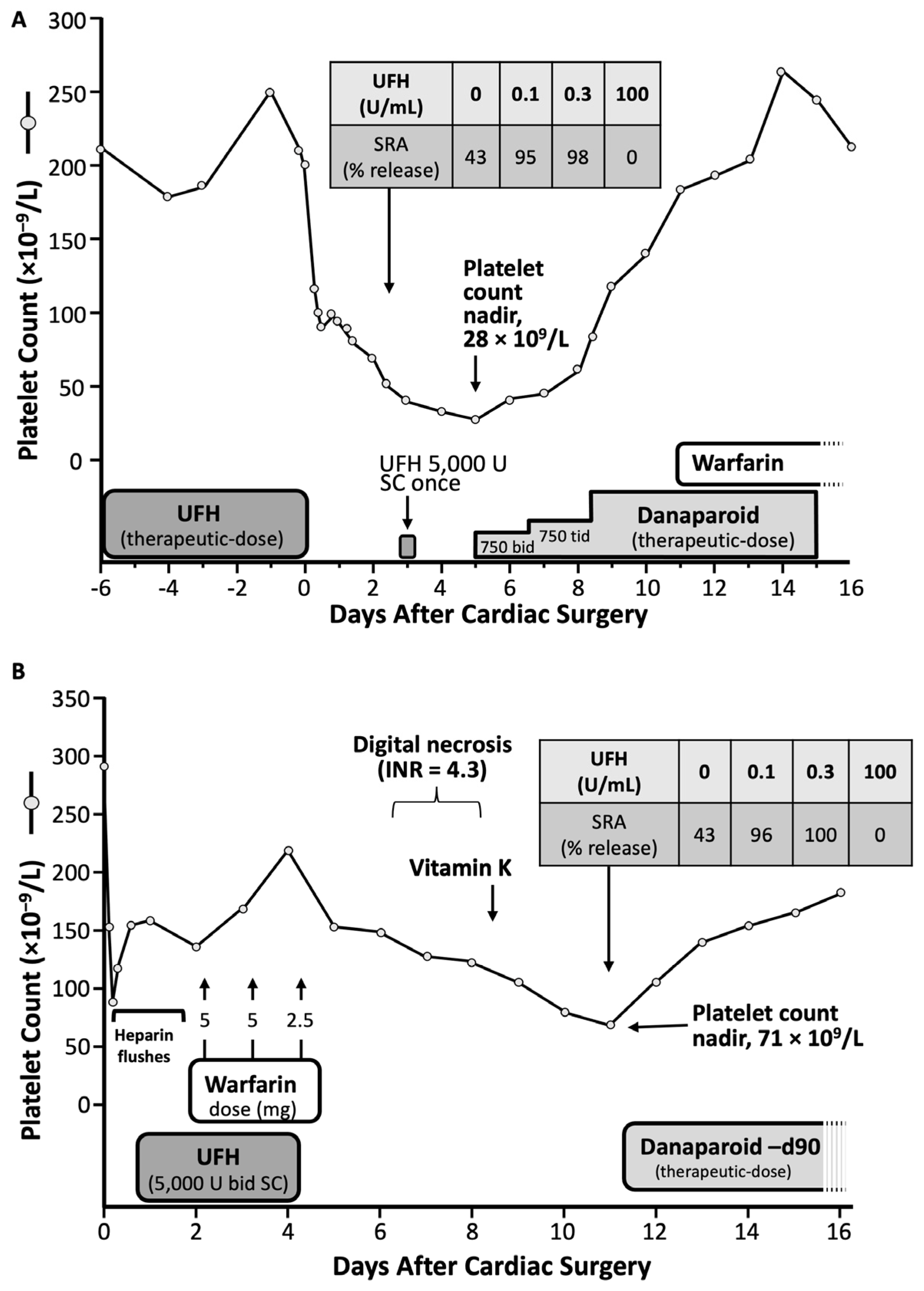
| [62] | Delayed; warfarin-induced microthrombosis (see Figure 3A), nadir = 71 | >40% |
| [63] | Delayed; postoperative platelet count fall, DVT (see Figure 3B), nadir = 28 | >40% |
| [64] | Delayed/persisting; stroke, adrenal hemorrhages, DVT, DIC, nadir = 68 | >90% |
| [65] | Delayed, DVT, while on Fx; serial rise and fall of HISR, nadir = 13 (post-UFH) | >70% |
| [66] | Delayed/persisting, recurrent (i.e., repeat UFH; previous HIT), DVT, nadir = 20 | >90% |
| [67] | Delayed, adrenal hemorrhages, DVT, nadir = 117 | >90% |
| [68] | Delayed/severe, DIC, multilimb microvascular gangrene, death; nadir = 10 | >90% |
| [69] | Delayed, persisting, DVTs, Fx cross-reactivity, nadir = 3 | HIPA + 5 min c |
| [70] | Delayed/persisting; no thrombosis (ascribed to rivaroxaban), nadir = 56 | >70% |
| [71] | Severe, DIC with coagulation factor depletion, death, nadir = 65 | >60% |
| [72] | Delayed/persisting, DIC, microvascular limb ischemia, death, nadir = 16 | >80% |
| [73] | Delayed/persisting, DVTs (argatroban failure), nadir = 16 | 100% |
| [74] | Delayed, multiple arterial/venous (incl. adrenal) thrombi, IVIG, nadir = 10 | 100% |
| [75] | Delayed, CVST, DIC, argatroban failure, death, nadir = 7 | HIPA + c |
| [76] | Delayed/persisting, venous limb gangrene (bivalirudin failure), IVIG, nadir = 9 | 100% |
| [77] | Delayed/persisting, DVT, IVIG, nadir = 12 | HIPA + 5 min c |
| [78] | Persisting, multiple venous/arterial thrombi (argatroban failure), IVIG, nadir = 25 | ATP release c |
| Study | Trigger | Nadir | HIT Thrombosi(e)s | Treatment | Other | Refractory |
|---|---|---|---|---|---|---|
| [45] | U-cpb | 51 a | LAT | Warf, aspirin | Post-D/C; amp | |
| [45] | U-cpb | 29 | DVT | DS | Post-D/C | |
| [45] | U-cpb | 25 | LAT | DS, Lep | Post-D/C; amp | Yes |
| [45] | U-cpb | 40 | DVT, PE | Lep; IVC filter | Post-D/C | |
| [82] | U-rx | 15 a | DVTs × 3 | Lep; IVC filter | Post-D/C | |
| [82] | U-cpb | 54 a | SVG-thrombosis × 1 | Lep | Post-D/C | |
| [83] | U-cpb | 7 | VLG × 2 limbs | Arg | VLG (Warf) | Yes |
| [83] | U-cpb | 18 | VLG × 2 limbs | Lep | VLG (Warf) | Yes |
| [84] | U-vasc | 5 | DVT; graft thrombus | Arg, CS, IVIG | Arg-fail (DVT) | Yes |
| [84] | U-vasc | 16 | DVTs × 2 | Arg, CS, IVIG, Fx | DIC | Yes |
| [85] | U-rx | 19 | DVT | Arg, Fx, CS, IVIG | In Vitro IVIG studies | Yes |
| [85] | U-pr | 18 | DVT | Arg, IVIG | In Vitro IVIG studies | Yes |
| [86] | U-rx | 7 | DVT, PE, ecchymoses | IVC filter, Warf, IVIG | DIC | Yes |
| [87] | U-rx | 24 | DVT, purpura | Warf | DIC | Yes |
| [88] | U-pr | 7 | CVST, DVT, PE | Nil | Post-D/C, DIC | Yes |
| [89] | U-cpb | 39 | DVT × 2, VLG × 2 | Lep | Post-D/C; amps | |
| [90] | U-cpb | 69 | MI, SVG-thrombosis × 4 | Lep | Post-D/C | |
| [91] | U-cpb | 8 | DVT, SPG × 3 limbs | Arg | Amps × 3 limbs | Yes |
| [92] | U-cpb | 40 | PE; SPG × 4 limbs | Lep | DIC, Lep-fail; amps | Yes |
| [93] | L-pr | 58 a | PE | Arg | Post-D/C; death | Yes |
| [94] | U-cpb | 3 | Ecchymoses | Arg | No sequelae | Yes |
| [95] | U-rx | 8 | DVT progr, IVC filter | Arg, Lep, CS, TPE, Ritux | Lep/Arg-fail, amps × 2 | Yes |
| [96] | L-pr | 23 | DVT, PE | Fx, DS, Riv | Fx/DS-fail (↑dD) | Yes |
| [97] | U-cpb | 12 | Testicular, b PE | DS, Biv, TPE | X-R assays conducted | Yes |
| [98] | U-cpb | 8 | LAT, DVT/PE, Lt atrial | Arg, Biv, CS, IVIG | Post-D/C | Yes |
| [99] | U-rx | 26 | Nil HIT thrombosis | IVC filter, Fx, Arg, IVIG | No sequelae | Yes |
| [100] | U-hd | 15 | DVT | Arg, IVIG, apix | Post-D/C | Yes |
| [101] | L-pr | 25 | CVST | Arg, CS, IVIG, Warf | Post-D/C | Yes |
| [102] | L-pr | <10 | CVST, DVT | Arg, Biv, CS, IVIG | Arg-fail (worse DVT) | Yes |
| [103] | U-hd | 16 | DVT | Arg, IVIG, Apix | Arg-fail (DVT) | Yes |
| [104] | U-pr | 4 | DVTs × 4 limbs | Arg, Biv, CS, IVIG | Arg-fail (DVT ischemia) | Yes |
| [105] | L-pr | 32 a | CVST | Biv | Post-D/C | |
| [106] | U-pr | 36 | LAT, PE, PFO c | Arg, IVIG | Post-D/C | |
| [107] | U-MI | ~100 | DVT, LAT | Heparin continued | Amp × 1 | |
| [108] | U-pr | 28 | DVT | Arg, tPA, IVIG | Post-D/C | Yes |
| [109] | U-pr | <12 | DVT × 3, PE | Riv | Post-D/C | |
| [110] | L-pr | 8 | Bilateral LAT | Arg, IVIG | Post-D/C | |
| [111] | U-cpb | 24 | Strokes (arterial) | Arg, IVIG | DIC; Arg fail (↑dD) | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warkentin, T.E. Autoimmune Heparin-Induced Thrombocytopenia. J. Clin. Med. 2023, 12, 6921. https://doi.org/10.3390/jcm12216921
Warkentin TE. Autoimmune Heparin-Induced Thrombocytopenia. Journal of Clinical Medicine. 2023; 12(21):6921. https://doi.org/10.3390/jcm12216921
Chicago/Turabian StyleWarkentin, Theodore E. 2023. "Autoimmune Heparin-Induced Thrombocytopenia" Journal of Clinical Medicine 12, no. 21: 6921. https://doi.org/10.3390/jcm12216921
APA StyleWarkentin, T. E. (2023). Autoimmune Heparin-Induced Thrombocytopenia. Journal of Clinical Medicine, 12(21), 6921. https://doi.org/10.3390/jcm12216921