
The Relationship between Mitochondrial Respiratory Chain Activities in Muscle and Metabolites in Plasma and Urine: A Retrospective Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mitochondrial Respiratory Chain Enzymatic Analysis
2.2. Metabolic Tests
2.3. Fibroblast Growth Factor 21
2.4. Statistical Analysis
3. Results
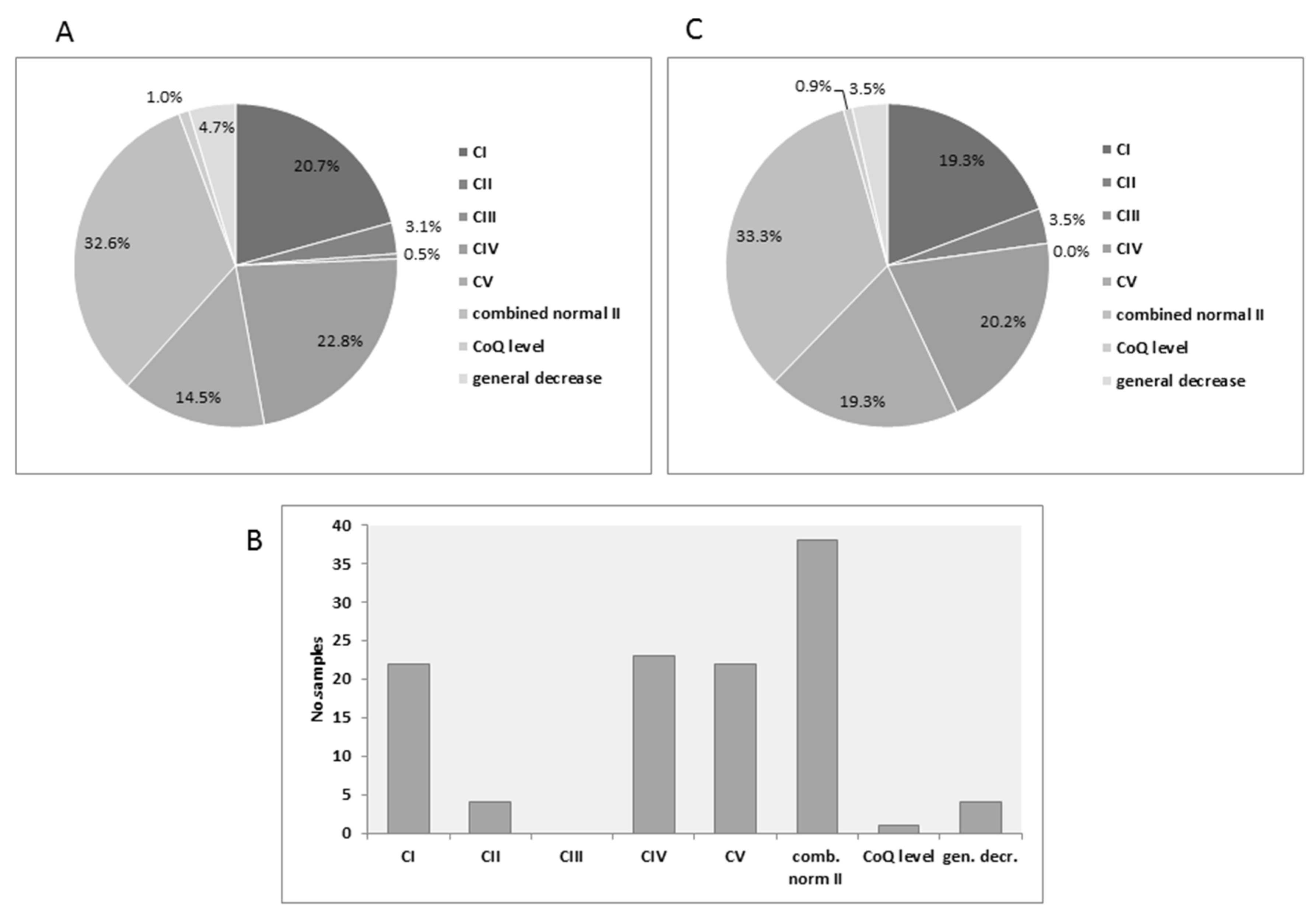
3.1. Distribution of MRC Deficiencies
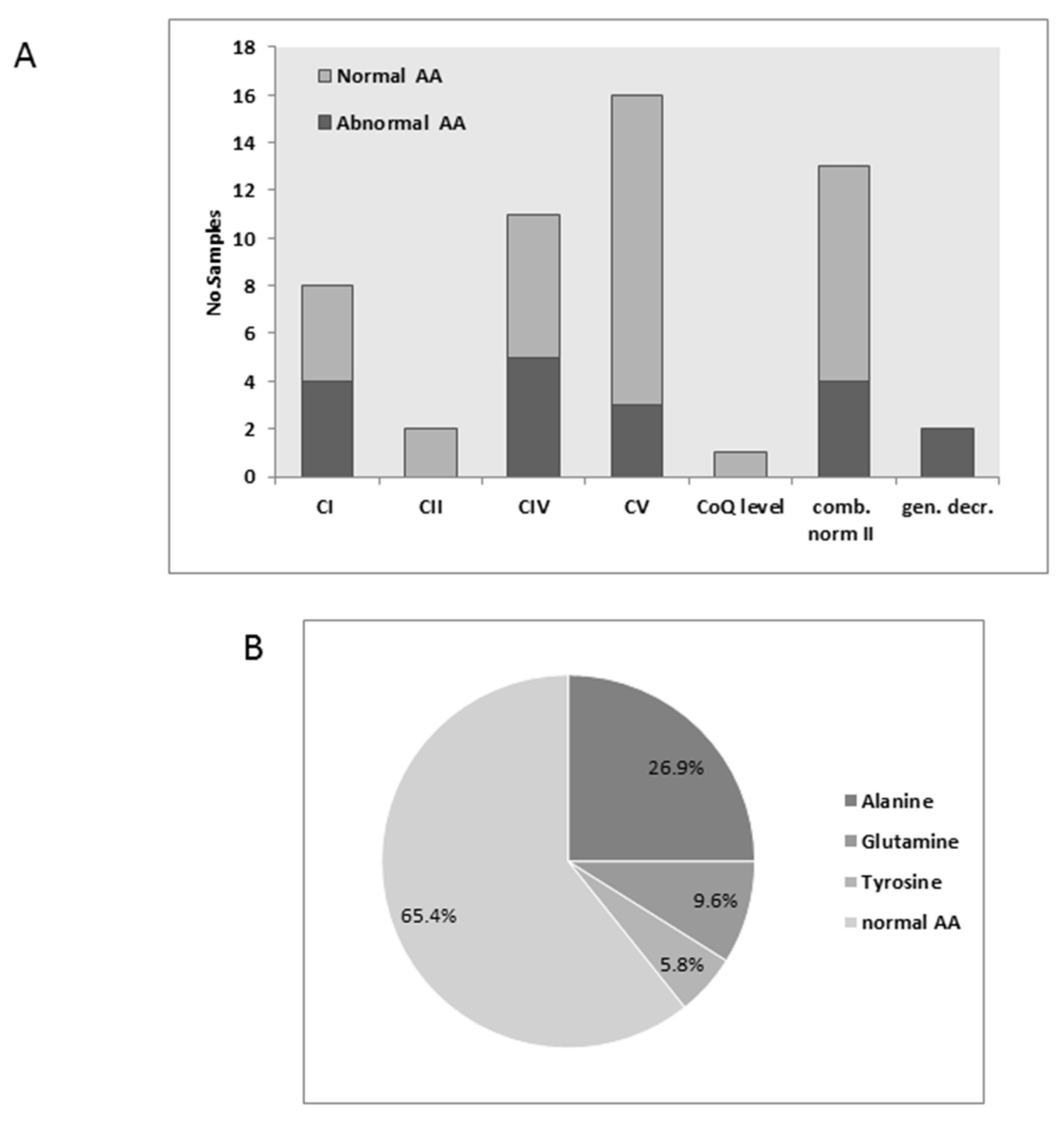
3.2. Plasma Amino Acids
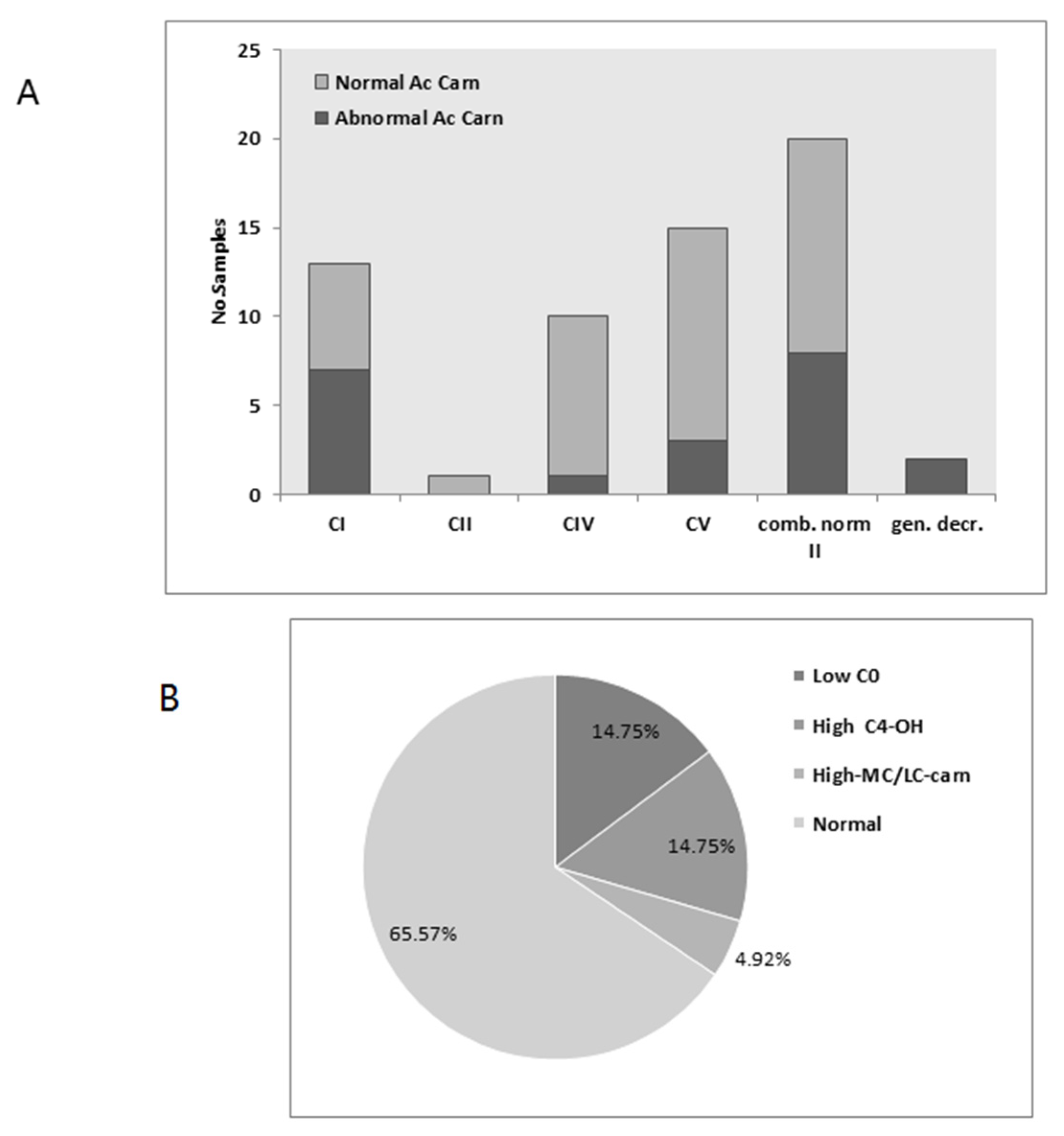
3.3. Acylcarnitines
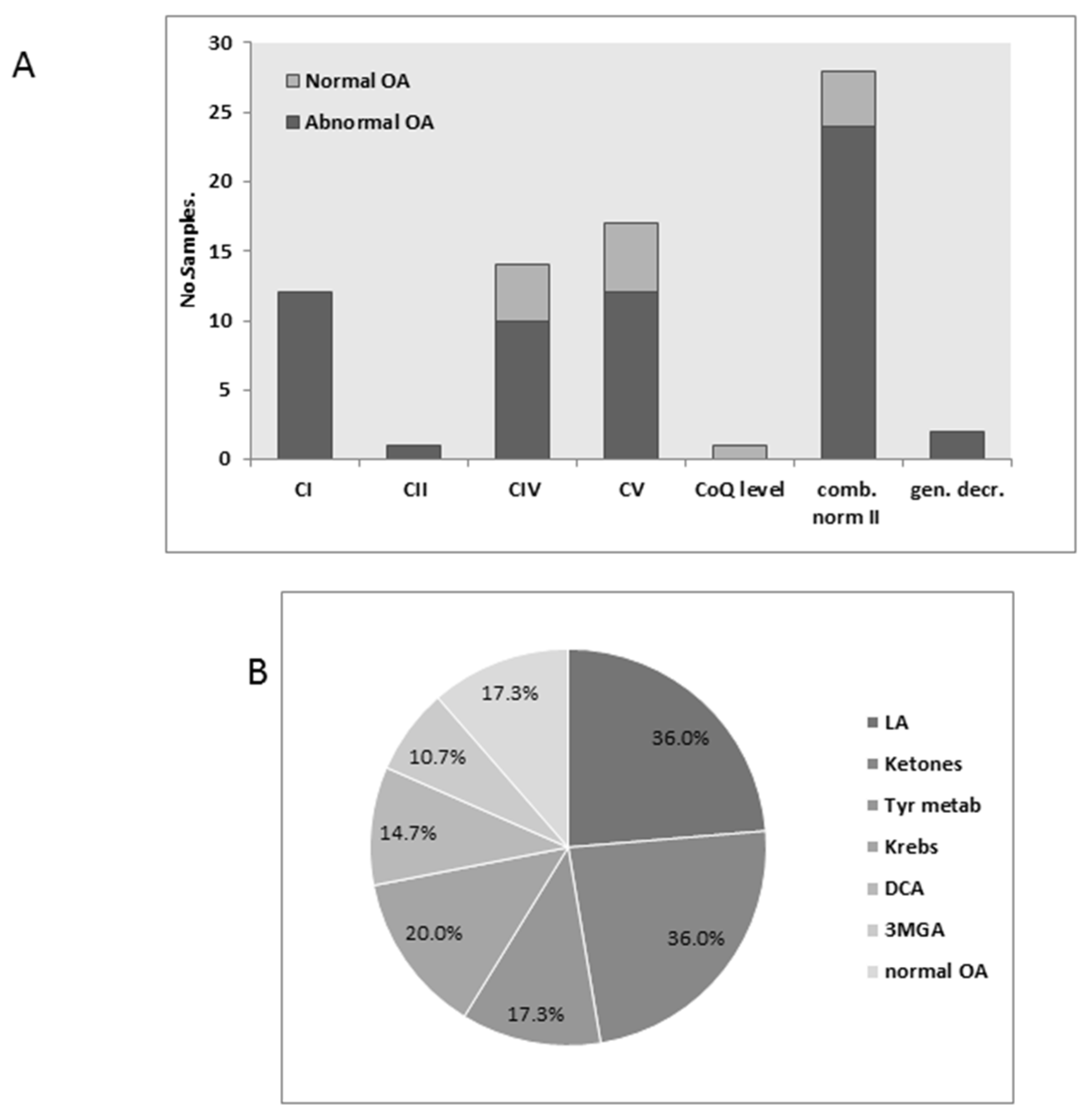
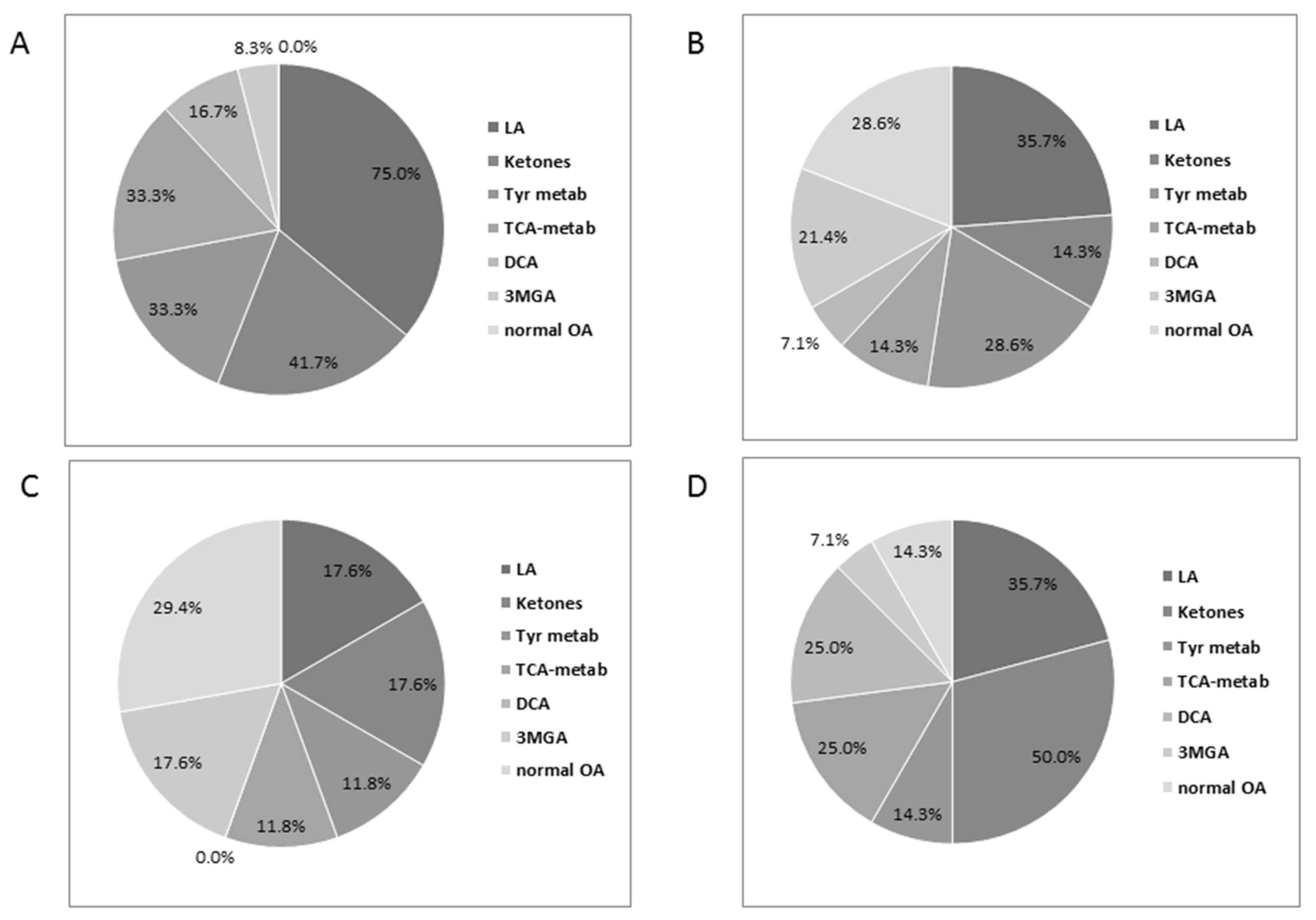
3.4. Urinary Organic Acids
3.5. Plasma FGF21
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 20, 16080. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F. Mitochondrial disorders overview. In GeneReviews; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Stephens, K., et al., Eds.; University of Washington: Seattle, WA, USA, 2014. [Google Scholar]
- Niyazov, D.; Kahler, S.; Frye, R. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Saada, A.; Bar-Meir, M.; Belaiche, C.; Miller, C.; Elpeleg, O. Evaluation of enzymatic assays and compounds affecting ATP production in mitochondrial respiratory chain complex I deficiency. Anal. Biochem. 2004, 335, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Korman, S.H.; Andresen, B.S.; Zeharia, A.; Gutman, A.; Boneh, A.; Pitt, J.J. 2-ethylhydracrylic aciduria in short/branched-chain acyl-CoA dehydrogenase deficiency: Application to diagnosis and implications for the R-pathway of isoleucine oxidation. Clin. Chem. 2005, 51, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Elpeleg, O.N.; Amir, N.; Christensen, E. Variability of clinical presentation in fumarate hydratase deficiency. J. Pediatr. 1992, 121, 752–754. [Google Scholar] [CrossRef]
- Elpeleg, O.; Mandel, H.; Saada, A. Depletion of the other genome-mitochondrial DNA depletion syndromes in humans. J. Mol. Med. 2002, 80, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Hirano, M.; DiMauro, S. Mitochondrial encephalomyopathies: Clinical and molecular analysis. J. Bioenerg. Biomembr. 1994, 26, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Soiferman, D.; Shaag, A.; Shalev, S.; Elpeleg, O.; Saada, A. Novel Homozygous Missense Mutation in SPG20 Gene Results in Troyer Syndrome Associated with Mitochondrial Cytochrome c Oxidase Deficiency. JIMD Rep. 2016. [Google Scholar] [CrossRef]
- Naini, A.; Kaufmann, P.; Shanske, S.; Engelstad, K.; de Vivo, D.C.; Schon, E.A. Hypocitrullinemia in patients with MELAS: An insight into the “MELAS paradox”. J. Neurol. Sci. 2005, 229–230, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; Horvath, R. Mitochondria: Impaired mitochondrial translation in human disease. Int. J. Biochem. Cell. Biol. 2014, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Magner, M.; Dvorakova, V.; Tesarova, M.; Mazurova, S.; Hansikova, H.; Zahorec, M.; Brennerova, K.; Bzduch, V.; Spiegel, R.; Horovitz, Y.; et al. TMEM70 deficiency: Long-term outcome of 48 patients. J. Inherit. Metab. Dis. 2015, 38, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Zeharia, A.; Friedman, J.R.; Tobar, A.; Saada, A.; Konen, O.; Fellig, Y.; Shaag, A.; Nunnari, J.; Elpeleg, O. Mitochondrial hepato-encephalopathy due to deficiency of QIL1/MIC13 (C19 or f70), a MICOS complex subunit. Eur. J. Hum. Genet. 2016, 24, 1778–1782. [Google Scholar] [CrossRef] [PubMed]
- Mandel, H.; Saita, S.; Edvardson, S.; Jalas, C.; Shaag, A.; Goldsher, D.; Vlodavsky, E.; Langer, T.; Elpeleg, O. Deficiency of HTRA2/Omi is associated with infantile neurodegeneration and 3-methylglutaconic aciduria. J. Med. Genet. 2016, 53, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Phypers, B.; Pierce, T. Lactate physiology in health and disease. Contin. Educ. Anaesth. Crit. Care Pain 2006, 6, 128–132. [Google Scholar] [CrossRef]
- Kumps, A.; Duez, P.; Mardens, Y. Metabolic, nutritional, iatrogenic, and artifactual sources of urinary organic acids: A comprehensive table. Clin. Chem. 2002, 48, 708–717. [Google Scholar] [PubMed]
- Suomalainen, A. Fibroblast growth factor 21: A novel biomarker for human muscle-manifesting mitochondrial disorders. Expert Opin. Med. Diagn. 2013, 4, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, J.M.; Forsström, S.; Bottani, E.; Viscomi, C.; Baris, O.R.; Isoniemi, H.; Höckerstedt, K.; Österlund, P.; Hurme, M.; Jylhävä, J.; et al. FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology 2016, 87, 2290–2299. [Google Scholar] [CrossRef] [PubMed]
- Yatsuga, S.; Fujita, Y.; Ishii, A.; Fukumoto, Y.; Arahata, H.; Kakuma, T.; Kojima, T.; Ito, M.; Tanaka, M.; Saiki, R.; et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol. 2015, 78, 814–823. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alban, C.; Fatale, E.; Joulani, A.; Ilin, P.; Saada, A. The Relationship between Mitochondrial Respiratory Chain Activities in Muscle and Metabolites in Plasma and Urine: A Retrospective Study. J. Clin. Med. 2017, 6, 31. https://doi.org/10.3390/jcm6030031
Alban C, Fatale E, Joulani A, Ilin P, Saada A. The Relationship between Mitochondrial Respiratory Chain Activities in Muscle and Metabolites in Plasma and Urine: A Retrospective Study. Journal of Clinical Medicine. 2017; 6(3):31. https://doi.org/10.3390/jcm6030031
Chicago/Turabian StyleAlban, Corinne, Elena Fatale, Abed Joulani, Polina Ilin, and Ann Saada. 2017. "The Relationship between Mitochondrial Respiratory Chain Activities in Muscle and Metabolites in Plasma and Urine: A Retrospective Study" Journal of Clinical Medicine 6, no. 3: 31. https://doi.org/10.3390/jcm6030031
APA StyleAlban, C., Fatale, E., Joulani, A., Ilin, P., & Saada, A. (2017). The Relationship between Mitochondrial Respiratory Chain Activities in Muscle and Metabolites in Plasma and Urine: A Retrospective Study. Journal of Clinical Medicine, 6(3), 31. https://doi.org/10.3390/jcm6030031