The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations



Abstract
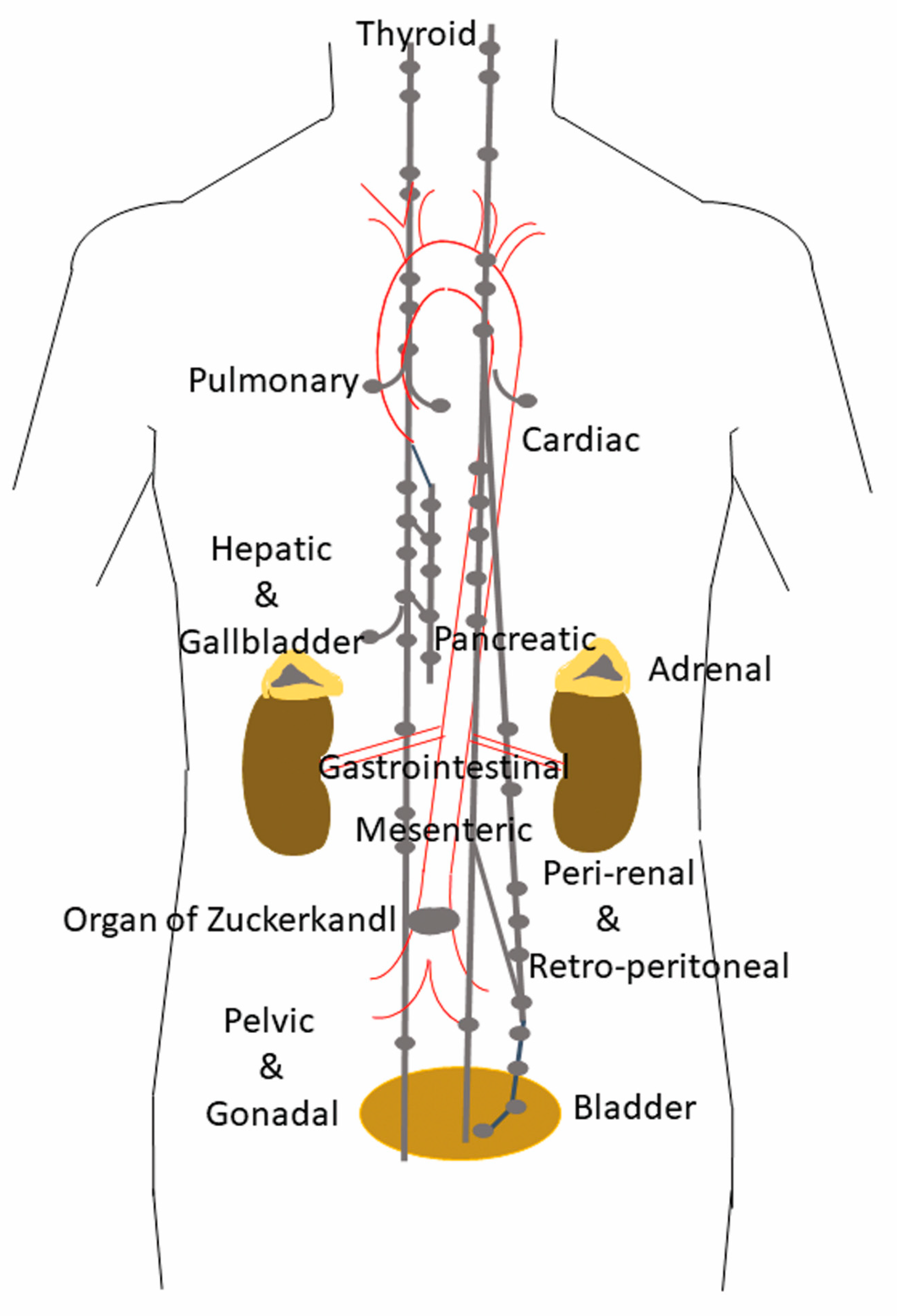
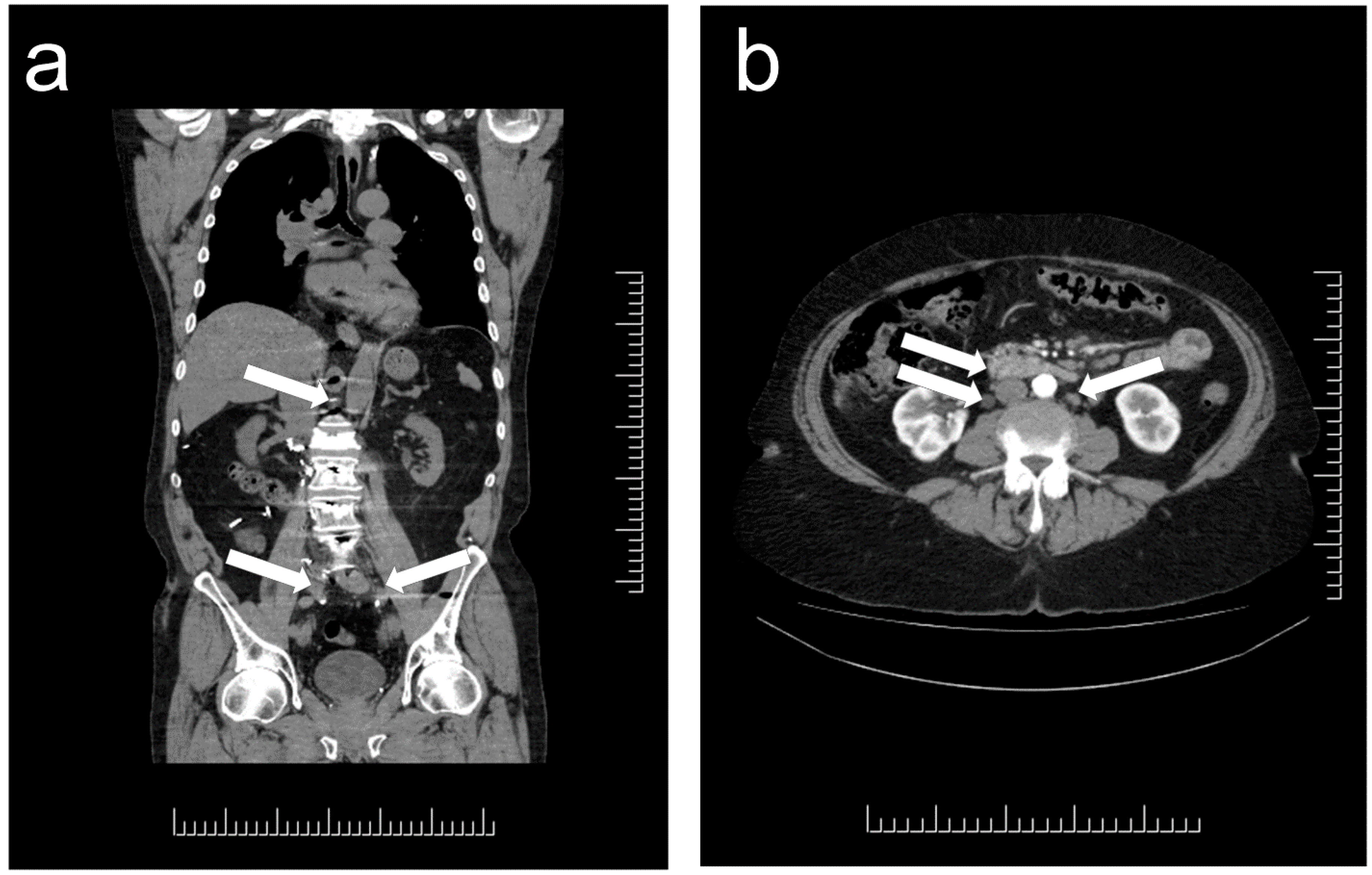
:1. Distribution and Localization of Paragangliomas
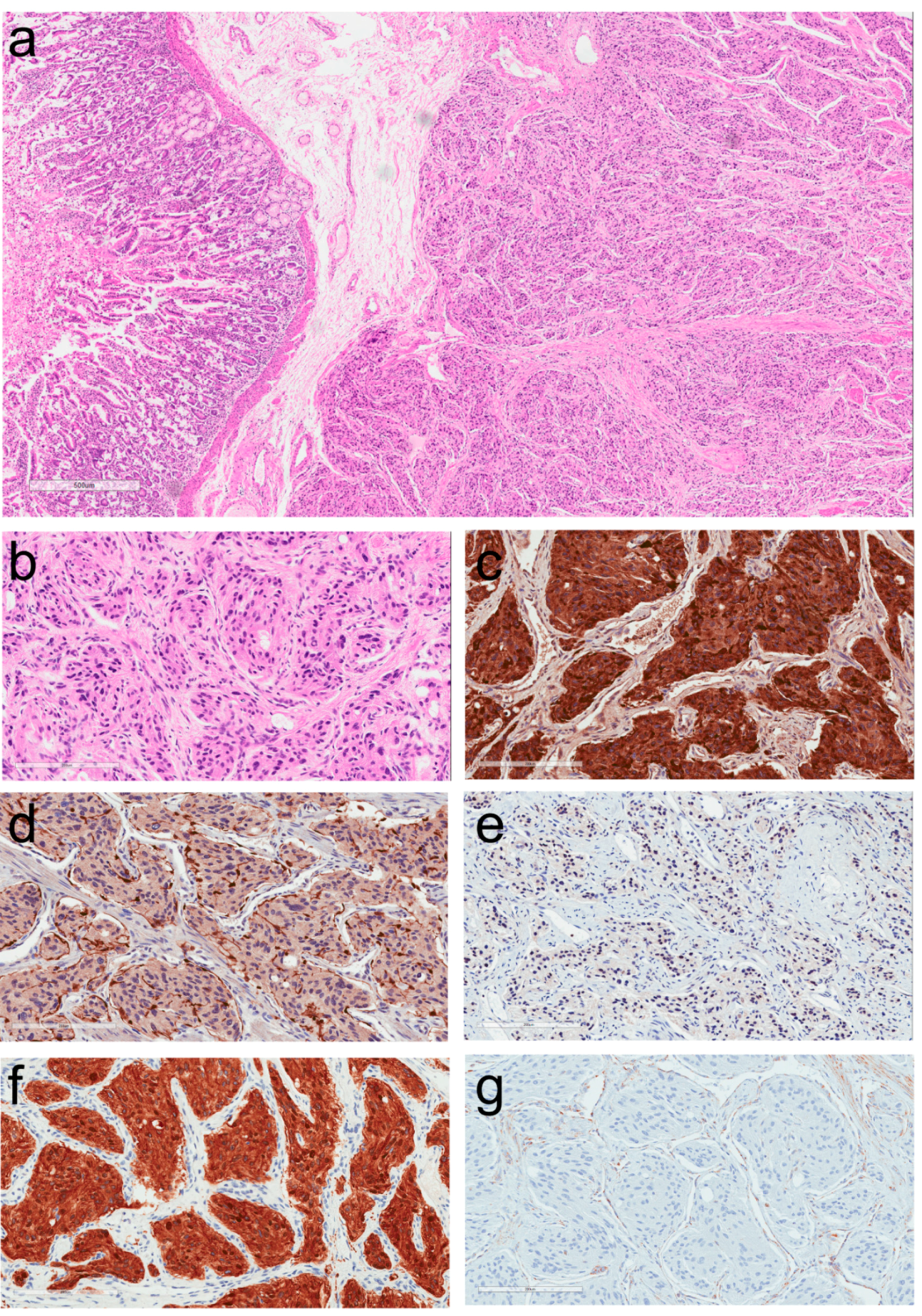
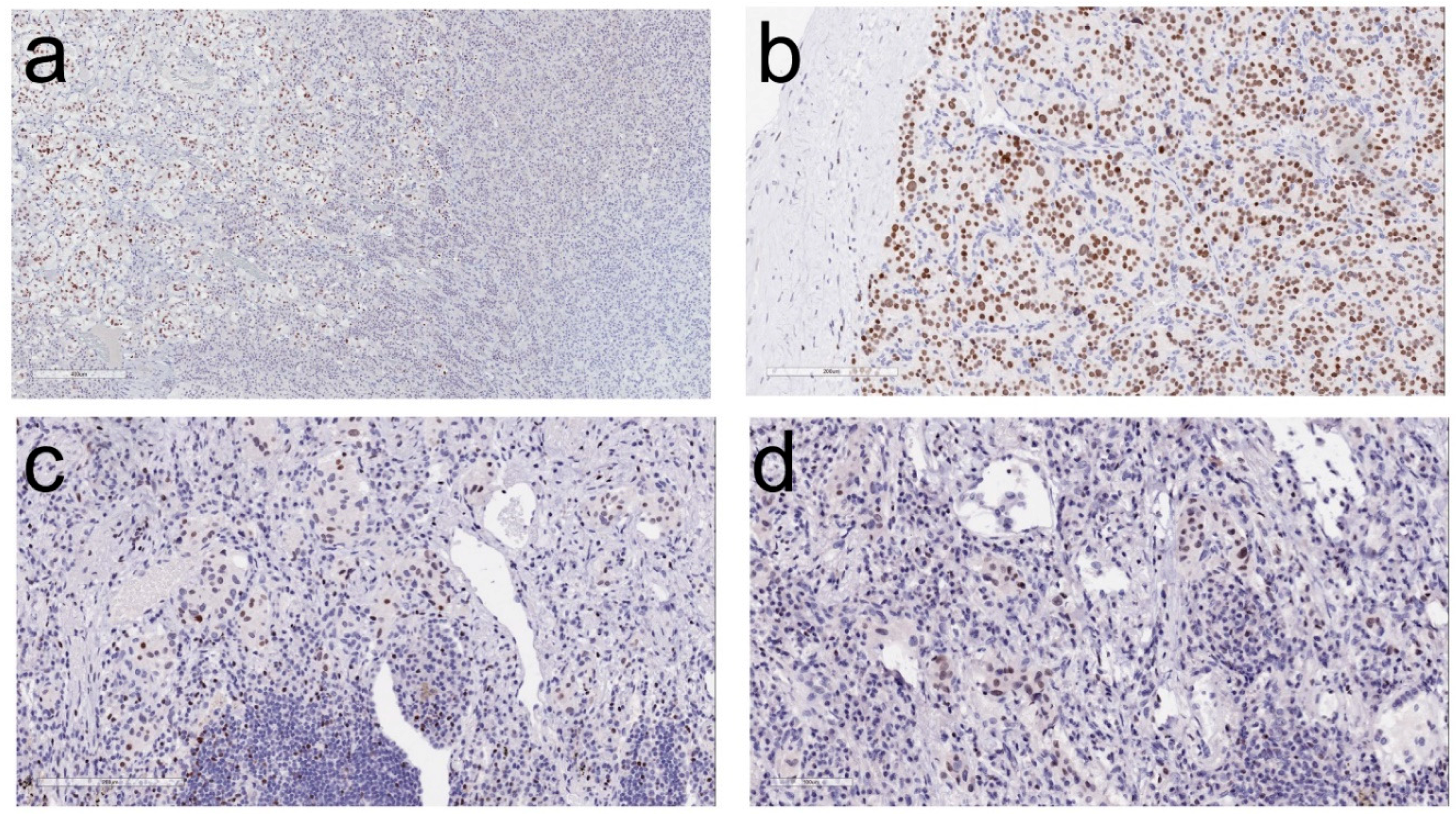
2. Morphologic Diagnosis of Paragangliomas
3. Clinical Implications of the Diagnosis of Paraganglioma
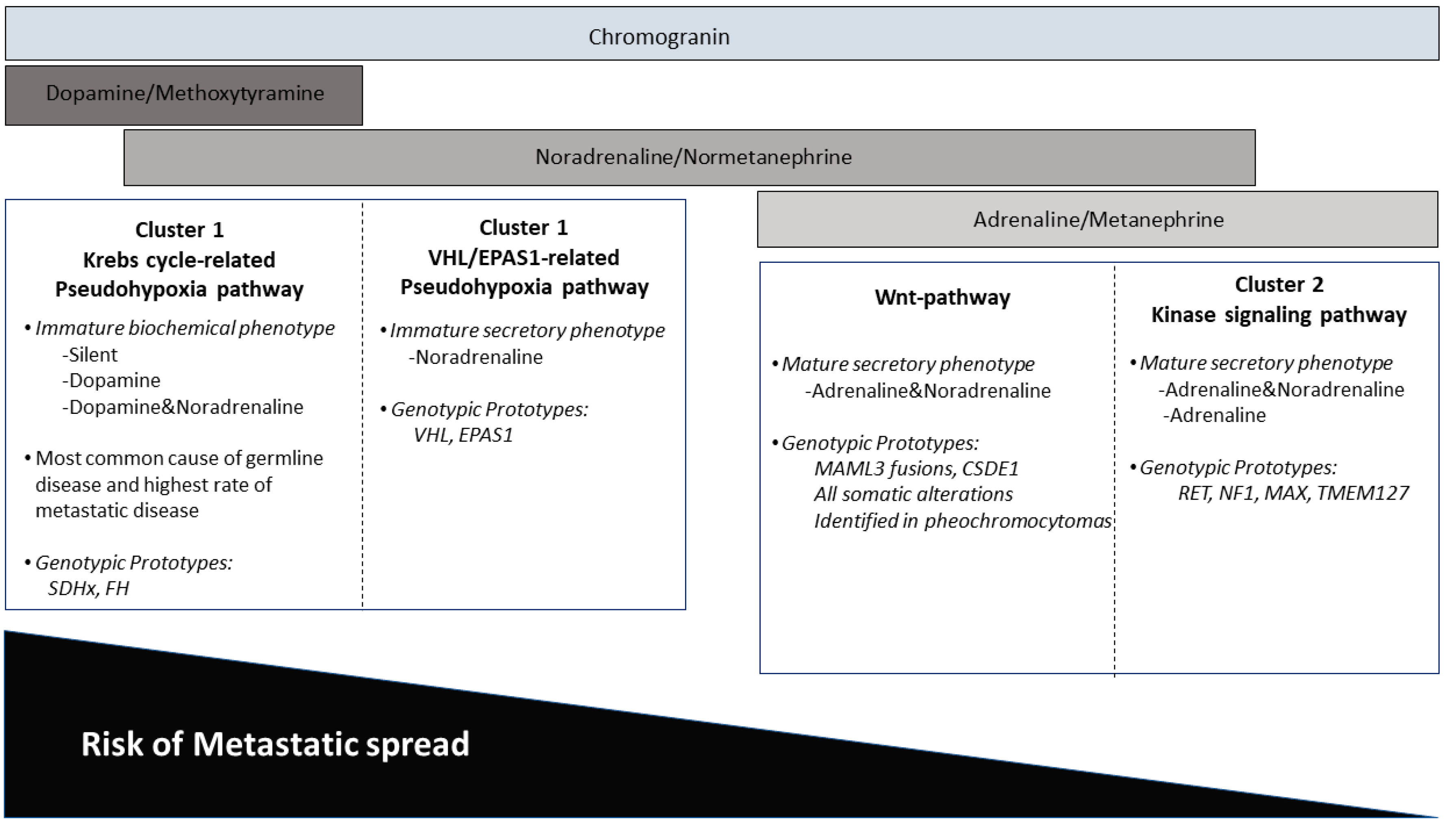
4. Impact of Genotype-Phenotype Correlations in Paraganglioma
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Rindi, G.; Klimstra, D.S.; Abedi-Ardekani, B.; Asa, S.L.; Bosman, F.T.; Brambilla, E.; Busam, K.J.; de Krijger, R.R.; Dietel, M.; El-Naggar, A.K.; et al. A common classification framework for neuroendocrine neoplasms: An international Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lack, E.E. Tumors of the Adrenal Gland and Extraadrenal Paraganglia. Armed Forces Institute of Pathology. In Atlas of Tumor Pathology; Series 4, Faccicle 8; American Registry of Pathology Press: Washington, DC, USA, 2007. [Google Scholar]
- Tischler, A.S. The adrenal medulla and extra-adrenal paraganglia. In Functional Endocrine Pathology; Kovacs, K., Asa, S.L., Eds.; Blackwell Science: Hoboken, NJ, USA, 1998; pp. 550–595. [Google Scholar]
- Oudijk, L.; de Krijger, R.R.; Pacak, K.; Tischler, A.S. Adrenal medulla and extra-adrenal paraganglia. In Endocrine Pathology; Mete, O., Asa, S.L., Eds.; Cambridge University Press: Cambridge, UK, 2016; pp. 628–676. [Google Scholar]
- Hayashi, T.; Mete, O. Head and neck paragangliomas: What does the pathologist need to know? Diagn. Histopathol. 2014, 20, 316–325. [Google Scholar] [CrossRef]
- Tischler, A.S.; Asa, S.L. Paraganglia. In Histology for Pathologists; Mills, S.E., Ed.; Wolters Kluwer: Philadelphia, PA, USA, 2018; in press. [Google Scholar]
- Lack, E.E. Tumors of the Adrenal Gland and Extra-Adrenal Paraganglia. In AFIP Atlas of Tumor Pathology; Series 3, Fascicle 19; Armed Forces Institute of Pathology: Washington, DC, USA, 1997. [Google Scholar]
- Mete, O.; Pakbaz, S.; Cassol, C.; Asa, S.L. The Spectrum and Clinical Features of Paragangliomas. Lab. Investig. 2018, 98, 234. [Google Scholar]
- Ober, W.B. Emil Zuckerkandl and his delightful little organ. Pathol. Annu. 1983, 18 Pt 1, 103–119. [Google Scholar]
- Chow, L.T.C.; Chan, M.H.M.; Wong, S.K.C. Functional Ulnar Nerve Paraganglioma: First Documented Occurrence in the Extremity With Hitherto Undescribed Associated Extensive Glomus Cell Hyperplasia and Tumorlet Formation. Int. J. Surg. Pathol. 2018, 26, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Mehra, S.; Chung-Park, M. Gallbladder paraganglioma: A case report with review of the literature. Arch. Pathol. Lab Med. 2005, 129, 523–526. [Google Scholar] [PubMed]
- Ece, I.; Alptekin, H.; Celik, Z.E.; Sahin, M. Gallbladder paraganglioma. Ulus. Cerrahi. Derg. 2015, 31, 244–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, P.S.; Koong, J.K.; Westerhout, C.J.; Yoong, B.K. Hepatobiliary and pancreatic: A huge liver paraganglioma. J. Gastroenterol. Hepatol. 2013, 28, 1075. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Ding, Z.Y.; Zhang, B.; Chen, L.; Li, G.X.; Wu, J.J.; Zhang, B.X.; Chen, X.P.; Zhu, P. Primary functioning hepatic paraganglioma mimicking hepatocellular carcinoma: A case report and literature review. Medicine 2018, 97, e0293. [Google Scholar] [CrossRef] [PubMed]
- Steel, T.R.; Dailey, A.T.; Born, D.; Berger, M.S.; Mayberg, M.R. Paragangliomas of the sellar region: Report of two cases. Neurosurgery 1993, 32, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Scheithauer, B.W.; Parameswaran, A.; Burdick, B. Intrasellar paraganglioma: Report of a case in a sibship of von Hippel-Lindau disease. Neurosurgery 1996, 38, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Mokry, M.; Kleinert, R.; Clarici, G.; Obermayer-Pietsch, B. Primary paraganglioma simulating pituitary macroadenoma: A case report and review of the literature. Neuroradiology 1998, 40, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Sambaziotis, D.; Kontogeorgos, G.; Kovacs, K.; Horvath, E.; Levedis, A. Intrasellar paraganglioma presenting as nonfunctioning pituitary adenoma. Arch. Pathol. Lab. Med. 1999, 123, 429–432. [Google Scholar] [PubMed]
- Naggara, O.; Varlet, P.; Page, P.; Oppenheim, C.; Meder, J.F. Suprasellar paraganglioma: A case report and review of the literature. Neuroradiology 2005, 47, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Boari, N.; Losa, M.; Mortini, P.; Snider, S.; Terreni, M.R.; Giovanelli, M. Intrasellar paraganglioma: A case report and review of the literature. Acta Neurochir. 2006, 148, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Özüm, Ü.; Egilmez, R.; Yildirim, A. Paraganglioma in pituitary fossa. Neuropathology 2008, 28, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Sharma, M.C.; Sharma, B.S. Malignant paraganglioma of the sellar region mimicking a pituitary macroadenoma. J. Clin. Neurosci. 2008, 15, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Haresh, K.P.; Prabhakar, R.; Anand, R.K.D.; Sharma, D.N.; Julka, P.K.; Rath, G.K. A rare case of paraganglioma of the sella with bone metastases. Pituitary 2009, 12, 276–279. [Google Scholar] [CrossRef] [PubMed]
- LaGuette, J.; Matias-Guiu, X.; Rosai, J. Thyroid paraganglioma: A clinicopathologic and immunohistochemical study of three cases. Am. J. Surg. Pathol. 1997, 21, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Nagahama, M.; Sugino, K.; Ito, K.; Kameyama, K.; Ito, K. Paraganglioma of the thyroid: Report of a male case with ultrasonographic imagings, cytologic, histologic, and immunohistochemical features. Thyroid 2007, 17, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Castelblanco, E.; Gallel, P.; Ros, S.; Gatius, S.; Valls, J.; De-Cubas, A.A.; Maliszewska, A.; Yebra-Pimentel, M.T.; Menarguez, J.; Gamallo, C.; et al. Thyroid paraganglioma. Report of 3 cases and description of an immunohistochemical profile useful in the differential diagnosis with medullary thyroid carcinoma, based on complementary DNA array results. Hum. Pathol. 2012, 43, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Policarpio-Nicolas, M.L. Thyroid Paraganglioma. Arch. Pathol. Lab. Med. 2015, 139, 1062–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taweevisit, M.; Bunyayothin, W.; Thorner, P.S. Thyroid Paraganglioma: “Naked” Nuclei as a Clue to Diagnosis on Imprint Cytology. Endocr. Pathol. 2015, 26, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Navaratne, L.; Mathew, R.G.; Kousparos, G.; McCombe, A. The Management of Locally Invasive Primary Thyroid Paraganglioma: A Case Report and Review of the Literature. Head Neck Pathol. 2017, 11, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.T.; Braun, J.T.; Pennant, M.; Thompson, L.D. Primary paraganglioma of the parathyroid: A case report and clinicopathologic review. Head Neck Pathol. 2010, 4, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nogimura, H.; Kuriki, K.; Kobayashi, R. Superior mediastinal paraganglioma associated with von Hippel-Lindau syndrome: Report of a case. World J. Surg. Oncol. 2014, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Mehta, C.K.; Gillespie, C.T.; Lin, X.; Yeldandi, A.; DeCamp, M.; Bharat, A. Rare Middle Mediastinal Paraganglioma Mimicking Metastatic Neuroendocrine Tumor. Ann. Thorac. Surg. 2015, 100, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Kamitani, T.; Kawanami, S.; Nagao, M.; Yonezawa, M.; Yamasaki, Y.; Baba, S.; Yabuuchi, H.; Hida, T.; Kohashi, K.; et al. Mediastinal paraganglioma. Jpn J. Radiol. 2015, 33, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Millar, A.C.; Mete, O.; Cusimano, R.J.; Fremes, S.E.; Keshavjee, S.; Morgan Sylvia, C.D.; Asa, L.A.; Ezzat, S.; Gilbert, J. Functional Cardiac Paraganglioma Associated with a Rare SDHC Mutation. Endocr. Pathol. 2014, 25, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Gucer, H.; Mete, O. Endobronchial gangliocytic paraganglioma: Not all keratin-positive endobronchial neuroendocrine neoplasms are pulmonary carcinoids. Endocr. Pathol. 2014, 25, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liang, Q.L.; Jiang, L.; Liu, Q.L.; Qu, W.T.; Li, D.H.; Zhang, H.J.; Yuan, G.L. Primary Pulmonary Paraganglioma: A Case Report and Review of Literature. Medicine 2015, 94, e1271. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, G.; Annunziata, A.; de Rosa, N. Primary paraganglioma of the lung: A case report. J. Med. Case Rep. 2015, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Samuel, N.; Ejaz, R.; Silver, J.; Ezzat, S.; Cusimano, R.J.; Kim, R.H. Primary mediastinal paraganglioma associated with a familial variant in the succinate dehydrogenase B subunit gene. J. Surg. Oncol. 2018, 117, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, N.; Santos, P.; Nikfarjam, M. Paraganglioma mimicking a pancreatic neoplasm. J. Pancreas 2011, 12, 259–261. [Google Scholar] [CrossRef]
- Meng, L.; Wang, J.; Fang, S.H. Primary pancreatic paraganglioma: A report of two cases and literature review. World J. Gastroenterol. 2015, 21, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Jacob, N.C.; Howard, M.; Kelly, M.; Hale, P.C. Mesenteric paraganglioma’s: An important differential diagnosis in intra-abdominal tumours. BMJ Case Rep. 2012. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Kamiya, K.; Takahashi, Y.; Miyazaki, S.; Iino, I.; Kikuchi, H.; Hiramatsu, Y.; Ohta, M.; Baba, S.; Konno, H. Mesenteric paraganglioma: Report of a case. World J. Gastrointest. Surg. 2013, 5, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.S.; de Krijger, R.R.; Gill, A.; Kawashima, A.; Kimura, N.; Komminoth, P.; Papathomas, T.G.; Thomposon, L.D.R.; Tissier, F.; Williams, M.D.; et al. Pheochromocytoma. In WHO Classification of Tumours of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloeppel, G., Rosai, J., Eds.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 10, pp. 183–189. [Google Scholar]
- Kimura, N.; Capella, C.; DeLellis, R.A.; Epstein, J.I.; Gill, A.; Kawashima, A.; Koch, C.A.; Komminoth, P.; Lam, A.K.Y.; Merino, M.J.; et al. Extra-adrenal paragangliomas. In WHO Classification of Tumours of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloeppel, G., Rosai, J., Eds.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 10, pp. 190–195. [Google Scholar]
- Assadipour, Y.; Sadowski, S.M.; Alimchandani, M.; Quezado, M.; Steinberg, S.M.; Nilubol, N.; Patel, D.; Prodanov, T.; Pacak, K.; Kebebew, E. SDHB mutation status and tumor size but not tumor grade are important predictors of clinical outcome in pheochromocytoma and abdominal paraganglioma. Surgery 2017, 161, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala-Ramirez, M.; Feng, L.; Johnson, M.M.; Ejaz, S.; Habra, M.A.; Rich, T.; Busaidy, N.; Cote, G.J.; Perrier, N.; Phan, A.; et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: Primary tumor size and primary tumor location as prognostic indicators. J. Clin. Endocrinol. Metab. 2011, 96, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Othman, N.H.; Ezzat, S.; Kovacs, K.; Horvath, E.; Poulin, E.; Smyth, H.S.; Asa, S.L. Growth hormone-releasing hormone (GHRH) and GHRH receptor (GHRH-R) isoform expression in ectopic acromegaly. Clin. Endocrinol. 2001, 55, 135–140. [Google Scholar] [CrossRef]
- Gosney, J.R.; Denley, H.; Resl, M. Sustentacular cells in pulmonary neuroendocrine tumours. Histopathology 1999, 34, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Asa, S.L.; Dey, C.; Kennecke, H.; Laidley, D.; Law, C.; Asmis, T.; Chan, D.; Ezzat, S.; Goodwin, R.; et al. Diagnosis and management of gastrointestinal neuroendocrine tumors: An evidence-based Canadian consensus. Cancer Treat. Rev. 2016, 47, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Takako, N.; Hamada, M.; Maeda, A.; Fujioka, Y.; Kuroha, T.; Huber, R.E.; Hasegawa, S.L.; Rao, A.; Yamamoto, M.; et al. Gata3 participates in a complex transcriptional feedback network to regulate sympathoadrenal differentiation. Development 2006, 133, 3871–3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Shi, J.; Wilkerson, M.L.; Lin, F. Immunohistochemical evaluation of GATA3 expression in tumors and normal tissues: A useful immunomarker for breast and urothelial carcinomas. Am. J. Clin. Pathol. 2012, 138, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, N.G. Value of GATA3 immunostaining in tumor diagnosis: A review. Adv. Anat. Pathol. 2013, 20, 352–360. [Google Scholar] [CrossRef] [PubMed]
- So, J.S.; Epstein, J.I. GATA3 expression in paragangliomas: A pitfall potentially leading to misdiagnosis of urothelial carcinoma. Mod. Pathol. 2013, 26, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- Weissferdt, A.; Kalhor, N.; Liu, H.; Rodriguez, J.; Fujimoto, J.; Tang, X.M.; Wistuba, I.I.; Moran, C.A. Thymic neuroendocrine tumors (paraganglioma and carcinoid tumors): A comparative immunohistochemical study of 46 cases. Hum. Pathol. 2014, 45, 2463–2470. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; McCue, P.A.; Sarlomo-Rikala, M.; Rys, J.; Czapiewski, P.; Wazny, K.; Langfort, R.; Waloszczyk, P.; Biernat, W.; Lasota, J.; et al. GATA3: A multispecific but potentially useful marker in surgical pathology: A systematic analysis of 2500 epithelial and nonepithelial tumors. Am. J. Surg. Pathol. 2014, 38, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Kefeli, M.; Çalişkan, S.; Asa, S.L. GATA-3 Immunoreactivity Expands The Transcription Factor Profile of Pituitary Neuroendocrine Tumors. Mod. Pathol. 2018. submitted. [Google Scholar]
- Hervonen, A.; Pickel, V.M.; Joh, T.H.; Reis, D.J.; Linnoila, I.; Kanerva, L.; Miller, R.J. Immunocytochemical demonstration of the catecholamine-synthesizing enzymes and neuropeptides in the catecholamine-storing cells of human fetal sympathetic nervous system. Adv. Biochem. Psychopharmacol. 1980, 25, 373–378. [Google Scholar] [PubMed]
- Eisenhofer, G.; Tischler, A.S.; de Krijger, R.R. Diagnostic tests and biomarkers for pheochromocytoma and extra-adrenal paraganglioma: From routine laboratory methods to disease stratification. Endocr. Pathol. 2012, 23, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.S. Pheochromocytoma and extra-adrenal paraganglioma: Updates. Arch. Pathol. Lab. Med. 2008, 132, 1272–1284. [Google Scholar] [PubMed]
- Mete, O.; Tischler, A.S.; de Krijger, R.; McNicol, A.M.; Eisenhofer, G.; Pacak, K.; Ezzat, S.; Asa, S.L. Protocol for the examination of specimens from patients with pheochromocytomas and extra-adrenal paragangliomas. Arch. Pathol. Lab. Med. 2014, 138, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Lenders, J.W.; Timmers, H.; Mannelli, M.; Grebe, S.K.; Hofbauer, L.C.; Bornstein, S.R.; Tiebel, O.; Adams, K.; Bratslavsky, G.; et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin. Chem. 2011, 57, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.; Peitzsch, M.; Prejbisz, A.; Hanus, K.; Fassnacht, M.; Beuschlein, F.; Brugger, C.; Fliedner, S.; Langton, K.; Pamporaki, C.; et al. Plasma methoxytyramine: Clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur. J. Endocrinol. 2017, 177, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Osinga, T.E.; van der Horst-Schrivers, A.N.A.; van Faassen, M.; Kerstens, M.N.; Dullaart, R.P.; Peters, M.A.; van der Laan, B.F.A.M.; de Bock, G.H.; Linkset, T.P.; Kema, I.P. Dopamine concentration in blood platelets is elevated in patients with head and neck paragangliomas. Clin. Chem. Lab. Med. 2016, 54, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, R.A.; Burnichon, N.; Cascon, A.; Benn, D.E.; Bayley, J.P.; Welander, J.; Tops, C.M.; Firth, H.; Dwight, T.; Ercolino, T.; et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat. Rev. Endocrinol. 2017, 13, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Turchini, J.; Cheung, V.K.Y.; Tischler, A.S.; de Krijger, R.R.; Gill, A.J. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology 2018, 72, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Wadt, K.; Choi, J.; Chung, J.Y.; Kiilgaard, J.; Heegaard, S.; Drzewiecki, K.T.; Trent, J.M.; Hewitt, S.M.; Hayward, N.K.; Gerdes, A.M.; et al. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res. 2012, 25, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Welander, J.; Andreasson, A.; Juhlin, C.C.; Wiseman, R.W.; Bäckdahl, M.; Höög, A.; Larsson, C.; Gimm, O.; Söderkvist, P. Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2014, 99, E1352–E1360. [Google Scholar] [CrossRef] [PubMed]
- Juhlin, C.C.; Stenman, A.; Haglund, F.; Clark, V.E.; Brown, T.C.; Baranoski, J.; Bilguvar, K.; Goh, G.; Welander, J.; Svahn, F.; Rubinstein, J.C.; et al. Whole-exome sequencing defines the mutational landscape of pheochromocytoma and identifies KMT2D as a recurrently mutated gene. Gene Chromosome Cancer 2015, 54, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Remacha, L.; Curras-Freixes, M.; Torres-Ruiz, R.; Schiavi, F.; Torres-Pérez, R.; Calsina, B.; Letón, R.; Comino-Méndez, I.; Roldán-Romero, J.M.; Montero-Conde, C.; et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Calsina, B.; Curras-Freixes, M.; Buffet, A.; Pons, T.; Contreras, L.; Letón, R.; Comino-Méndez, I.; Remacha, L.; Calatayud, M.; Obispo, B.; et al. Role of MDH2 pathogenic variant in pheochromocytoma and paraganglioma patients. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Udager, A.M.; Magers, M.J.; Goerke, D.M.; Vinco, M.L.; Siddiqui, J.; Cao, X.H.; Lucas, D.R.; Myers, J.L.; Chinnaiyan, A.M.; McHugh, J.B.; et al. The utility of SDHB and FH immunohistochemistry in patients evaluated for hereditary paraganglioma-pheochromocytoma syndromes. Hum. Pathol. 2018, 71, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Korpershoek, E.; Favier, J.; Gaal, J.; Burnichon, N.; van Gessel, B.; Oudijk, L.; Badoual, C.; Gadessaud, N.; Venisse, A.; Bayley, J.P.; et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 2011, 96, E1472–E1476. [Google Scholar] [CrossRef] [PubMed]
- Papathomas, T.G.; Oudijk, L.; Persu, A.; Gill, A.J.; Van Nederveen, F.; Tischler, A.S.; Tissier, F.; Volante, M.; Matias-Guiu, X.; Smid, M.; et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: A multicenter interobserver variation analysis using virtual microscopy: A Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod. Pathol. 2015, 28, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Menara, M.; Oudijk, L.; Badoual, C.; Bertherat, J.; Lepoutre-Lussey, C.; Amar, L.; Iturrioz, X.; Sibony, M.; Zinzindohoué, F.; de Krijger, R.; et al. SDHD immunohistochemistry: A new tool to validate SDHx mutations in pheochromocytoma/paraganglioma. J. Clin. Endocrinol. Metab. 2015, 100, E287–E291. [Google Scholar] [CrossRef] [PubMed]
- Roszko, K.L.; Blouch, E.; Blake, M.; Powers, J.F.; Tischler, A.S.; Hodin, R.; Sadow, P.; Lawson, E.A. Case Report of a Prolactinoma in a Patient With a Novel MAX Mutation and Bilateral Pheochromocytomas. J. Endocr. Soc. 2017, 1, 1401–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinato, D.J.; Ramachandran, R.; Toussi, S.T.K.; Vergine, M.; Ngo, N.; Sharma, R.; Lloyd, T.; Meeran, K.; Palazzo, F.; Martin, N.; et al. Immunohistochemical markers of the hypoxic response can identify malignancy in phaeochromocytomas and paragangliomas and optimize the detection of tumours with VHL germline mutations. Br. J. Cancer 2013, 108, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Asa, S.L. Precursor lesions of endocrine system neoplasms. Pathology 2013, 45, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Grogan, R.H.; Pacak, K.; Pasche, L.; Huynh, T.T.; Greco, R.S. Bilateral adrenal medullary hyperplasia associated with an SDHB mutation. J. Clin. Oncol. 2011, 29, e200–e202. [Google Scholar] [CrossRef] [PubMed]
- Toledo, S.P.A.; Lourenco, D.M., Jr.; Sekiya, T.; Lucon, A.M.; Baena, M.E.; Castro, C.C.; Bortolotto, L.A.; Zerbini, M.C.N.; Siqueira, S.A.C.; Toledo, R.A.; et al. Penetrance and clinical features of pheochromocytoma in a six-generation family carrying a germline TMEM127 mutation. J. Clin. Endocrinol. Metab. 2015, 100, E308–E318. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, K.G.; Ezzat, S.; Morel, C.F.; Swallow, C.; Otremba, M.; Dickson, B.C.; Asa, S.L.; Mete, O. Familial pheochromocytoma and renal cell carcinoma syndrome: TMEM127 as a novel candidate gene for the association. Virchows Arch. 2015, 466, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Romanet, P.; Guerin, C.; Pedini, P.; Essamet, W.; Castinetti, F.; Sebag, F.; Roche, P.; Cascon, A.; Tischler, A.S.; Pacak, K.; et al. Pathological and Genetic Characterization of Bilateral Adrenomedullary Hyperplasia in a Patient with Germline MAX Mutation. Endocr. Pathol. 2017, 28, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Crona, J.; Taieb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef] [PubMed]
- Janssen, I.; Blanchet, E.M.; Adams, K.; Chen, C.C.; Millo, C.; Herscovitch, P.; Taïeb, D.; Kebebew, E.; Lehnert, H.; Fojo, A.T.; et al. Superiority of [68Ga]-DOTATATE PET/CT to Other Functional Imaging Modalities in the Localization of SDHB-Associated Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2015, 21, 3888–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, A.; Ling, A.; Millo, C.; Gupta, G.; Viana, B.; Lin, F.I.; Herscovitch, P.; Adams, K.P.; Taieb, D.; Metwalli, A.R.; et al. Superiority of (68)Ga-DOTATATE over (18)F-FDG and anatomic imaging in the detection of succinate dehydrogenase mutation (SDHx)-related pheochromocytoma and paraganglioma in the pediatric population. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.D.R. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am. J. Surg. Pathol. 2002, 26, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Takayanagi, R.; Takizawa, N.; Itagaki, E.; Katabami, T.; Kakoi, N.; Rakugi, H.; Ikeda, Y.; Tanabe, A.; Nigawara, T.; et al. Phaechromocytoma Study Group in Japan. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr. Relat. Cancer 2014, 21, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, O.; Young, W.F., Jr.; Gruber, L.; Smestad, J.; Yan, Q.; Ponce, O.J.; Prokop, L.; Murad, M.H.; Bancos, I. Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: A systematic review and meta-analysis. Clin. Endocrinol. 2017, 87, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Niemeijer, N.D.; Papathomas, T.G.; Korpershoek, E.; de Krijger, R.R.; Oudijk, L.; Morreau, H.; Bayley, J.P.; Hes, F.J.; Jansen, J.C.; Dinjens, W.N.M.; et al. Succinate Dehydrogenase (SDH)-Deficient Pancreatic Neuroendocrine Tumor Expands the SDH-Related Tumor Spectrum. J. Clin. Endocrinol. Metab. 2015, 100, E1386–E1393. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; de Jesus, A.C.; Kerr, D.; Mete, O.; Nose, V.; Papotti, M.; Volante, M. Thyroid. In Endocrine Pathology; Mete, O., Asa, S.L., Eds.; Cambridge University Press: Cambridge, UK, 2016; pp. 398–572. [Google Scholar]
- Duan, K.; Mete, O. Hereditary endocrine tumor syndromes: The clinical and predictive role of molecular hoistopathology. AJSP Rev. Rep. 2017, 22, 246–268. [Google Scholar] [CrossRef]
- Gill, A.J. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018, 72, 106–116. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Transcription Factors Frequently Expressed |
|---|---|
| Pituitary | Pit-1, Tpit, SF-1, ER-alpha, GATA-3, GATA-2 |
| Thyroid | PAX-8 *, TTF-1 |
| Parathyroid | GATA-3, GCM-2 |
| Lung | TTF-1 |
| Stomach | CDX-2 |
| Duodenum | ISL-1, PDX-1, CDX-2 |
| Pancreas | PDX-1, ISL-1, CDX-2 |
| Jejunum/Ileum | CDX-2 |
| Appendix | CDX-2 |
| Colon/Rectum | CDX-2 |
| Paragangliomas | GATA-3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asa, S.L.; Ezzat, S.; Mete, O. The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. J. Clin. Med. 2018, 7, 280. https://doi.org/10.3390/jcm7090280
Asa SL, Ezzat S, Mete O. The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. Journal of Clinical Medicine. 2018; 7(9):280. https://doi.org/10.3390/jcm7090280
Chicago/Turabian StyleAsa, Sylvia L., Shereen Ezzat, and Ozgur Mete. 2018. "The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations" Journal of Clinical Medicine 7, no. 9: 280. https://doi.org/10.3390/jcm7090280
APA StyleAsa, S. L., Ezzat, S., & Mete, O. (2018). The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. Journal of Clinical Medicine, 7(9), 280. https://doi.org/10.3390/jcm7090280