Role of Endothelial ADAM17 in Early Vascular Changes Associated with Diabetic Retinopathy



 ,
, 
_Kwok.png) ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Human Samples
2.2. Experimental Animals
2.3. STZ Model of Type I Diabetes
2.4. Assessment of Retinal Vascular Permeability
2.5. ADAM17 Activity
2.6. Analysis of Leukocyte Adhesion
2.7. Retinal Single Cell Suspension Preparation and Flow Cytometry Analysis
2.8. Immunohistochemical Analysis
2.9. Dihydroethidium (DHE) Staining for Detection of Superoxide
2.10. Protein Analysis
2.11. Dot Blot Analysis
2.12. Cell Culture and Treatments
2.13. Quantitative PCR
2.14. Leukocyte Adhesion Assay
2.15. Measurements of TER of EC Monolayer
2.16. Statistical Analysis
3. Results
3.1. ADAM17 Expression and Activity Are Increased in Human and Mouse Diabetic Retinas
3.2. Endothelial ADAM17 Contributes to Hyperglycemia-Induced Vascular Permeability
3.3. Lack of Endothelial ADAM17 Diminishes Hyperglycemia-Induced Retinal Inflammation
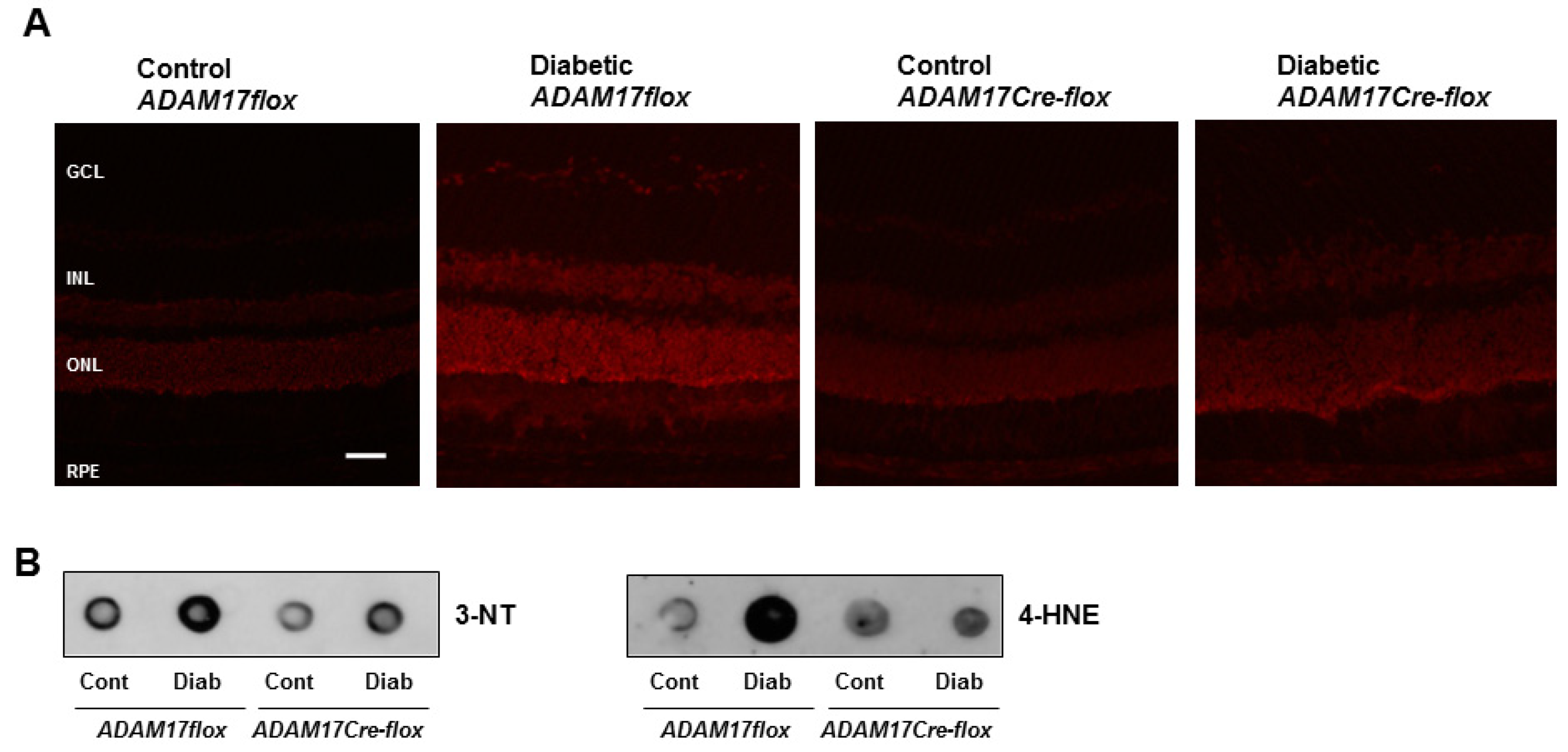
3.4. Endothelial ADAM17 Knockdown Diminishes Hyperglycemia-Induced Oxidative Stress
3.5. Glucidic Stress Upregulates Expression of ADAM17 in HREC
3.6. ADAM17 Contributes to High Glucose-Induced Retinal Endothelial Cell Permeability
3.7. Blocking of ADAM17 Diminishes High Glucose-Induced Leukocyte Adhesion in Vitro
3.8. Blocking of ADAM17 Reduces Oxidative Stress in High Glucose-Stimulated HREC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sabanayagam, C.; Banu, R.; Chee, M.L.; Lee, R.; Wang, Y.X.; Tan, G.; Jonas, J.B.; Lamoureux, E.L.; Cheng, C.-Y.; Klein, B.E.K.; et al. Incidence and progression of diabetic retinopathy: A systematic review. Lancet Diabetes Endocrinol. 2019, 7, 140–149. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaxman, S.R.; A Bourne, R.R.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990–2020: A systematic review and meta-analysis. Lancet Glob. Heal. 2017, 5, e1221–e1234. [Google Scholar] [CrossRef] [Green Version]
- Gardner, T.W.; Larsen, M.; Girach, A.; Zhi, X. on behalf of the Protein Kinase C Diabetic Retinopathy Study (PKC-DRS2) Study Group. Diabetic macular oedema and visual loss: Relationship to location, severity and duration. Acta Ophthalmol. 2009, 87, 709–713. [Google Scholar] [CrossRef]
- Moss, S. The 14-year incidence of visual loss in a diabetic population. Ophthalmology 1998, 105, 998–1003. [Google Scholar] [CrossRef]
- Sander, B.; Thornit, D.N.; Colmorn, L.; Strom, C.; Girach, A.; Hubbard, L.D.; Lund-Andersen, H.; Larsen, M. Progression of Diabetic Macular Edema: Correlation with Blood Retinal Barrier Permeability, Retinal Thickness, and Retinal Vessel Diameter. Investig. Opthalmology Vis. Sci. 2007, 48, 3983–3987. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Barber, A.J.; Khin, S.; Lieth, E.; Tarbell, J.M.; Gardner, T.W. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: Vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes 1998, 47, 1953–1959. [Google Scholar] [CrossRef]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Moss, M.L.; Jin, S.L.; Milla, M.E.; Bickett, D.M.; Burkhart, W.; Carter, H.L.; Chen, W.J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Garbers, C.; Rose-John, S. ADAM17: A molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011, 32, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Garton, K.J.; Gough, P.J.; Philalay, J.; Wille, P.T.; Blobel, C.P.; Whitehead, R.H.; Dempsey, P.J.; Raines, E.W. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17). J. Biol. Chem. 2003, 278, 37459–37464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walcheck, B.; Herrera, A.H.; Hill, C.S.; Mattila, P.E.; Whitney, A.R.; DeLeo, F.R. ADAM17 activity during human neutrophil activation and apoptosis. Eur. J. Immunol. 2006, 36, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Abel, S.; Hundhausen, C.; Mentlein, R.; Schulte, A.; A Berkhout, T.; Broadway, N.; Hartmann, D.; Sedlacek, R.; Dietrich, S.; Muetze, B.; et al. The transmembrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAM10. J. Immunol. 2004, 172, 6362–6372. [Google Scholar] [CrossRef] [Green Version]
- Koenen, R.R.; Pruessmeyer, J.; Soehnlein, O.; Fraemohs, L.; Zernecke, A.; Schwarz, N.; Reiss, K.; Sarabi, A.; Lindbom, L.; Hackeng, T.M.; et al. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood 2009, 113, 4799–4809. [Google Scholar] [CrossRef] [Green Version]
- Peschon, J.J. An Essential Role for Ectodomain Shedding in Mammalian Development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Dreymueller, D.; Martin, C.; Kogel, T.; Pruessmeyer, J.; Hess, F.M.; Horiuchi, K.; Uhlig, S.; Ludwig, A. Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol. Med. 2017, 4, 412–423. [Google Scholar] [CrossRef]
- McGowan, P.; Ryan, B.M.; Hill, A.D.; McDermott, E.; O’Higgins, N.; Duffy, M.J. ADAM-17 Expression in Breast Cancer Correlates with Variables of Tumor Progression. Clin. Cancer Res. 2007, 13, 2335–2343. [Google Scholar] [CrossRef] [Green Version]
- Hooper, N.M.; Turner, A.J. The search for alpha-secretase and its potential as a therapeutic approach to Alzheimer s disease. Curr. Med. Chem. 2002, 9, 1107–1119. [Google Scholar] [CrossRef]
- Moss, M.L.; Sklair-Tavron, L.; Nudelman, R. Drug Insight: Tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat. Clin. Pr. Rheumatol. 2008, 4, 300–309. [Google Scholar] [CrossRef]
- Canault, M.; Peiretti, F.; Kopp, F.; Bonardo, B.; Bonzi, M.-F.; Coudeyre, J.-C.; Alessi, M.-C.; Juhan-Vague, I.; Nalbone, G. The TNF alpha converting enzyme (TACE/ADAM17) is expressed in the atherosclerotic lesions of apolipoprotein E-deficient mice: Possible contribution to elevated plasma levels of soluble TNF alpha receptors. Atherosclerosis 2006, 187, 82–91. [Google Scholar] [CrossRef]
- Ford, B.M.; Eid, A.A.; Göõz, M.; Barnes, J.L.; Gorin, Y.C.; Abboud, H.E. ADAM17 mediates Nox4 expression and NADPH oxidase activity in the kidney cortex of OVE26 mice. Am. J. Physiol. Physiol. 2013, 305, F323–F332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Wang, T.; Walia, K.; Gao, B.; Krepinsky, J.C. Regulation of profibrotic responses by ADAM17 activation in high glucose requires its C-terminus and FAK. J. Cell Sci. 2018, 131, jcs208629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togashi, N.; Ura, N.; Higashiura, K.; Murakami, H.; Shimamoto, K. Effect of TNF-α–Converting Enzyme Inhibitor on Insulin Resistance in Fructose-Fed Rats. Hypertension 2002, 39, 578–580. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Purohit, S.; Sharma, A.; Hopkins, D.; Steed, L.; Bode, B.; Anderson, S.W.; Caldwell, R.; She, J.-X. Elevated Serum Levels of Soluble TNF Receptors and Adhesion Molecules Are Associated with Diabetic Retinopathy in Patients with Type-1 Diabetes. Mediat. Inflamm. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Limb, G.; Hollifield, R.D.; Webster, L.; Charteris, D.G.; Chignell, A.H. Soluble TNF receptors in vitreoretinal proliferative disease. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1586–1591. [Google Scholar]
- Arita, R.; Nakao, S.; Kita, T.; Kawahara, S.; Asato, R.; Yoshida, S.; Enaida, H.; Hafezi-Moghadam, A.; Ishibashi, T. A key role for ROCK in TNF-alpha-mediated diabetic microvascular damage. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2373–2383. [Google Scholar] [CrossRef]
- Takaguri, A.; Kimura, K.; Hinoki, A.; Bourne, A.M.; Autieri, M.V.; Eguchi, S. A disintegrin and metalloprotease 17 mediates neointimal hyperplasia in vasculature. Hypertension 2011, 57, 841–845. [Google Scholar] [CrossRef]
- Kawai, T.; Takayanagi, T.; Forrester, S.J.; Preston, K.J.; Obama, T.; Tsuji, T.; Kobayashi, T.; Boyer, M.J.; Cooper, H.A.; Kwok, H.F.; et al. Vascular ADAM17 (a Disintegrin and Metalloproteinase Domain 17) Is Required for Angiotensin II/β-Aminopropionitrile-Induced Abdominal Aortic Aneurysm. Hypertension 2017, 70, 959–963. [Google Scholar] [CrossRef]
- Dou, H.; Feher, A.; Davila, A.C.; Romero, M.J.; Patel, V.S.; Kamath, V.M.; Gooz, M.B.; Rudic, R.D.; Lucas, R.; Fulton, D.J.; et al. Role of Adipose Tissue Endothelial ADAM17 in Age-Related Coronary Microvascular Dysfunction. Arter. Thromb. Vasc. Boil. 2017, 37, 1180–1193. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, T.; Forrester, S.J.; Kawai, T.; Obama, T.; Tsuji, T.; Elliott, K.J.; Nuti, E.; Rossello, A.; Kwok, H.F.; Scalia, R.; et al. Vascular ADAM17 as a Novel Therapeutic Target in Mediating Cardiovascular Hypertrophy and Perivascular Fibrosis Induced by Angiotensin II. Hypertension 2016, 68, 949–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, D.; Arima, M.; Takubo, K.; Kimura, T.; Horiuchi, K.; Minagawa, T.; Matsuda, S.; Ikeda, E. ADAM12 and ADAM17 are essential molecules for hypoxia-induced impairment of neural vascular barrier function. Sci. Rep. 2015, 5, 12796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Steinle, J.J. IGFBP-3 inhibits TNF-alpha production and TNFR-2 signaling to protect against retinal endothelial cell apoptosis. Microvasc. Res. 2014, 95, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Gooz, P.; Gooz, M.; Baldys, A.; Hoffman, S. ADAM-17 regulates endothelial cell morphology, proliferation, and in vitro angiogenesis. Biochem. Biophys. Res. Commun. 2009, 380, 33–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weskamp, G.; Mendelson, K.; Swendeman, S.; Le Gall, S.; Ma, Y.; Lyman, S.; Hinoki, A.; Eguchi, S.; Guaiquil, V.; Horiuchi, K.; et al. Pathological neovascularization is reduced by inactivation of ADAM17 in endothelial cells but not in pericytes. Circ. Res. 2010, 106, 932–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewing, N.J.; Weskamp, G.; Vermaat, J.; Farage, E.; Glomski, K.; Swendeman, S.; Chan, R.V.P.; Chiang, M.F.; Khokha, R.; Anand-Apte, B.; et al. Intravitreal Injection of TIMP3 or the EGFR Inhibitor Erlotinib Offers Protection from Oxygen-Induced Retinopathy in Mice. Investig. Opthalmology Vis. Sci. 2013, 54, 864–870. [Google Scholar] [CrossRef] [Green Version]
- Thounaojam, M.C.; Powell, F.L.; Patel, S.; Gutsaeva, D.R.; Tawfik, A.; Smith, S.B.; Nussbaum, J.; Block, N.L.; Martin, P.M.; Schally, A.V.; et al. Protective effects of agonists of growth hormone-releasing hormone (GHRH) in early experimental diabetic retinopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 13248–13253. [Google Scholar] [CrossRef] [Green Version]
- Thounaojam, M.C.; Montemari, A.; Powell, F.L.; Malla, P.; Gutsaeva, D.R.; Bachettoni, A.; Ripandelli, G.; Repossi, A.; Tawfik, A.; Martin, P.M.; et al. Monosodium Urate Contributes to Retinal Inflammation and Progression of Diabetic Retinopathy. Diabetes 2019, 68, 1014–1025. [Google Scholar] [CrossRef] [Green Version]
- Gutsaeva, D.R.; Thounaojam, M.; Rajpurohit, S.; Powell, F.L.; Martin, P.M.; Goei, S.; Duncan, M.; Bartoli, M. STAT3-mediated activation of miR-21 is involved in down-regulation of TIMP3 and neovascularization in the ischemic retina. Oncotarget 2017, 8, 103568–103580. [Google Scholar] [CrossRef]
- Ye, J.; Yuen, S.M.; Murphy, G.; Xie, R.; Kwok, H.F. Anti-tumor effects of a ‘human & mouse cross-reactive’ anti-ADAM17 antibody in a pancreatic cancer model in vivo. Eur. J. Pharm. Sci. 2017, 110, 62–69. [Google Scholar]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alva, J.A.; Zovein, A.C.; Monvoisin, A.; Murphy, T.; Salazar, A.; Harvey, N.L.; Carmeliet, P.; Iruela-Arispe, M.L. VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 2006, 235, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Fouda, A.Y.; Xu, Z.; Shosha, E.; Lemtalsi, T.; Chen, J.; Toque, H.A.; Tritz, R.; Cui, X.; Stansfield, B.K.; Huo, Y.; et al. Arginase 1 promotes retinal neurovascular protection from ischemia through suppression of macrophage inflammatory responses. Cell Death Dis. 2018, 9, 1001. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Anderson, J.M. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann. N. Y. Acad. Sci. 2009, 1165, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Al-Shabrawey, M.; Rojas, M.; Sanders, T.; Behzadian, A.; El-Remessy, A.; Bartoli, M.; Parpia, A.K.; Liou, G.; Caldwell, R.B. Role of NADPH oxidase in retinal vascular inflammation. Investig. Opthalmology Vis. Sci. 2008, 49, 3239–3244. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.J.; Yu, Q.; Chen, K.; Mahadev, K.; Zhang, S.X. Inhibition of Reactive Oxygen Species by Lovastatin Downregulates Vascular Endothelial Growth Factor Expression and Ameliorates Blood-Retinal Barrier Breakdown in db/db Mice. Diabetes 2010, 59, 1528–1538. [Google Scholar] [CrossRef] [Green Version]
- Antonetti, D.A.; Barber, A.J.; Bronson, S.K.; Freeman, W.M.; Gardner, T.W.; Jefferson, L.S.; Kester, M.; Kimball, S.R.; Krady, J.K.; LaNoue, K.F.; et al. Diabetic retinopathy: Seeing beyond glucose-induced microvascular disease. Diabetes 2006, 55, 2401–2411. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Chan, P.-S. Oxidative Stress and Diabetic Retinopathy. Exp. Diabetes Res. 2007, 2007, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kern, T.S. Contributions of Inflammatory Processes to the Development of the Early Stages of Diabetic Retinopathy. Exp. Diabetes Res. 2007, 2007, 1–14. [Google Scholar] [CrossRef]
- Huang, H.; Gandhi, J.K.; Zhong, X.; Wei, Y.; Gong, J.; Duh, E.J.; Vinores, S.A. TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Investig. Opthalmology Vis. Sci. 2011, 52, 1336–1344. [Google Scholar] [CrossRef] [Green Version]
- Nagy, J.A.; Benjamin, L.; Zeng, H.; Dvorak, A.M.; Dvorak, H.F. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 2008, 11, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, D.S.; Lefer, D.J.; Merges, C.; Lutty, G.A. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am. J. Pathol. 1995, 147, 642–653. [Google Scholar] [PubMed]
- Shen, M.; Hu, M.; Fedak, P.W.; Oudit, G.Y.; Kassiri, Z. Cell-Specific Functions of ADAM17 Regulate the Progression of Thoracic Aortic Aneurysm. Circ. Res. 2018, 123, 372–388. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Khosrof, S.; Bursell, S.-E.; Rohan, R.; Murata, T.; Clermont, A.C.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc. Natl. Acad. Sci. USA 1999, 96, 10836–10841. [Google Scholar] [CrossRef] [Green Version]
- Tsakadze, N.L.; Sithu, S.D.; Sen, U.; English, W.R.; Murphy, G.; D’Souza, S.E. Tumor necrosis factor-alpha-converting enzyme (TACE/ADAM-17) mediates the ectodomain cleavage of intercellular adhesion molecule-1 (ICAM-1). J. Biol. Chem. 2006, 281, 3157–3164. [Google Scholar] [CrossRef] [Green Version]
- Morigi, M.; Angioletti, S.; Imberti, B.; Donadelli, R.; Micheletti, G.; Figliuzzi, M.; Remuzzi, A.; Zoja, C.; Remuzzi, G. Leukocyte-endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-kB-dependent fashion. J. Clin. Investig. 1998, 101, 1905–1915. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhang, X.; Li, J.J. The role of NF-κB in the regulation of cell stress responses. Int. Immunopharmacol. 2002, 2, 1509–1520. [Google Scholar] [CrossRef]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Forro, L.; Schlegel, W.; Krause, K.H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalaby, L.; Thounaojam, M.; Tawfik, A.; Li, J.; Hussein, K.; Jahng, W.J.; Al-Shabrawey, M.; Kwok, H.F.; Bartoli, M.; Gutsaeva, D. Role of Endothelial ADAM17 in Early Vascular Changes Associated with Diabetic Retinopathy. J. Clin. Med. 2020, 9, 400. https://doi.org/10.3390/jcm9020400
Shalaby L, Thounaojam M, Tawfik A, Li J, Hussein K, Jahng WJ, Al-Shabrawey M, Kwok HF, Bartoli M, Gutsaeva D. Role of Endothelial ADAM17 in Early Vascular Changes Associated with Diabetic Retinopathy. Journal of Clinical Medicine. 2020; 9(2):400. https://doi.org/10.3390/jcm9020400
Chicago/Turabian StyleShalaby, Lamiaa, Menaka Thounaojam, Amany Tawfik, Junnan Li, Khaled Hussein, Wan Jin Jahng, Mohamed Al-Shabrawey, Hang Fai Kwok, Manuela Bartoli, and Diana Gutsaeva. 2020. "Role of Endothelial ADAM17 in Early Vascular Changes Associated with Diabetic Retinopathy" Journal of Clinical Medicine 9, no. 2: 400. https://doi.org/10.3390/jcm9020400
APA StyleShalaby, L., Thounaojam, M., Tawfik, A., Li, J., Hussein, K., Jahng, W. J., Al-Shabrawey, M., Kwok, H. F., Bartoli, M., & Gutsaeva, D. (2020). Role of Endothelial ADAM17 in Early Vascular Changes Associated with Diabetic Retinopathy. Journal of Clinical Medicine, 9(2), 400. https://doi.org/10.3390/jcm9020400